Influence of Hyperglycemia on Dexmedetomidine-Induced Cardioprotection in the Isolated Perfused Rat Heart


Abstract
1. Introduction
2. Material and Methods
2.1. Surgical Preparation and Langendorff Model
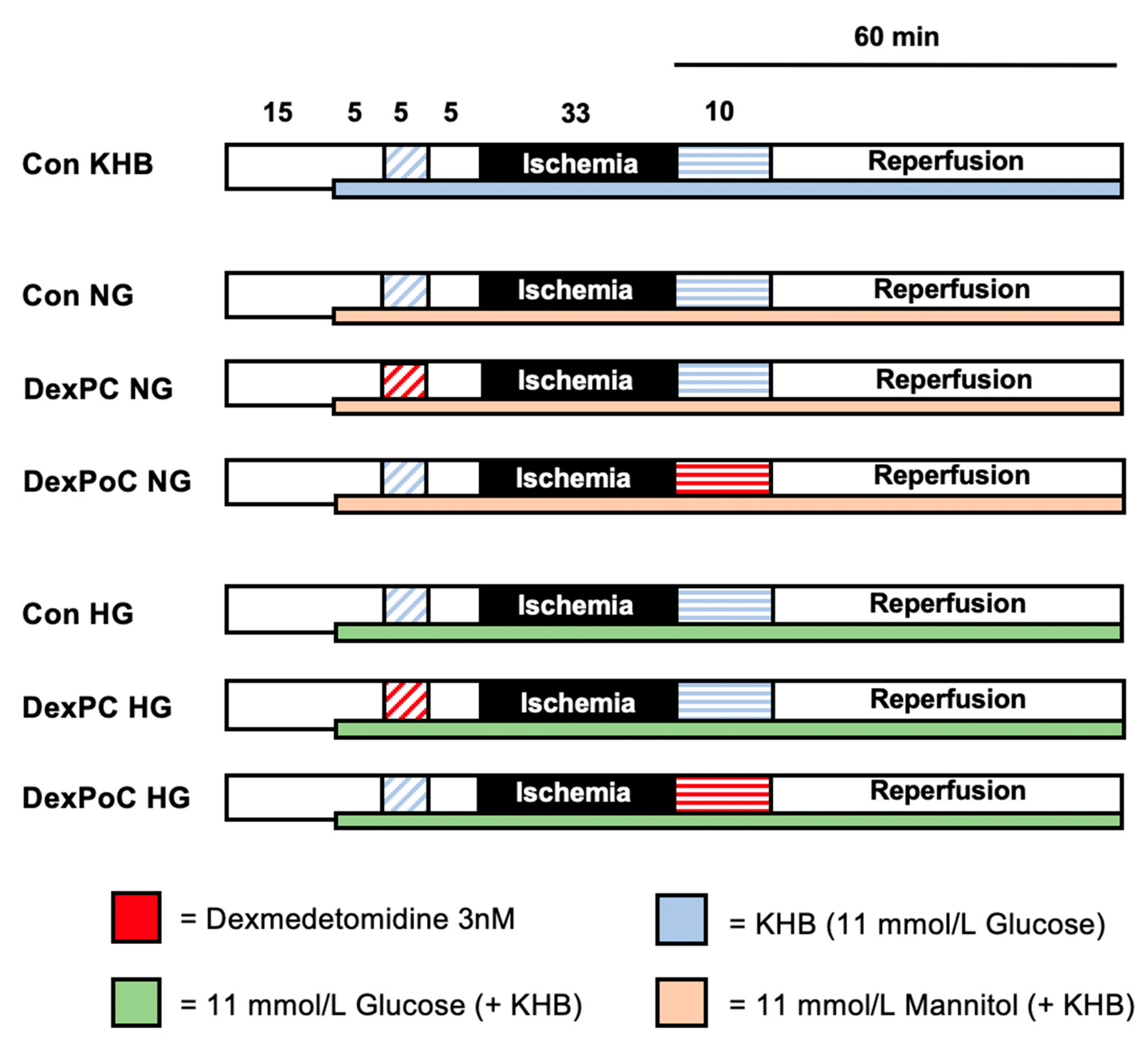
2.2. Experimental Setting
2.2.1. Subgroup—Normoglycemia (NG)
2.2.2. Subgroup—Hyperglycemia (HG)
2.3. Statistical Analysis
3. Results
3.1. Animal Characteristics
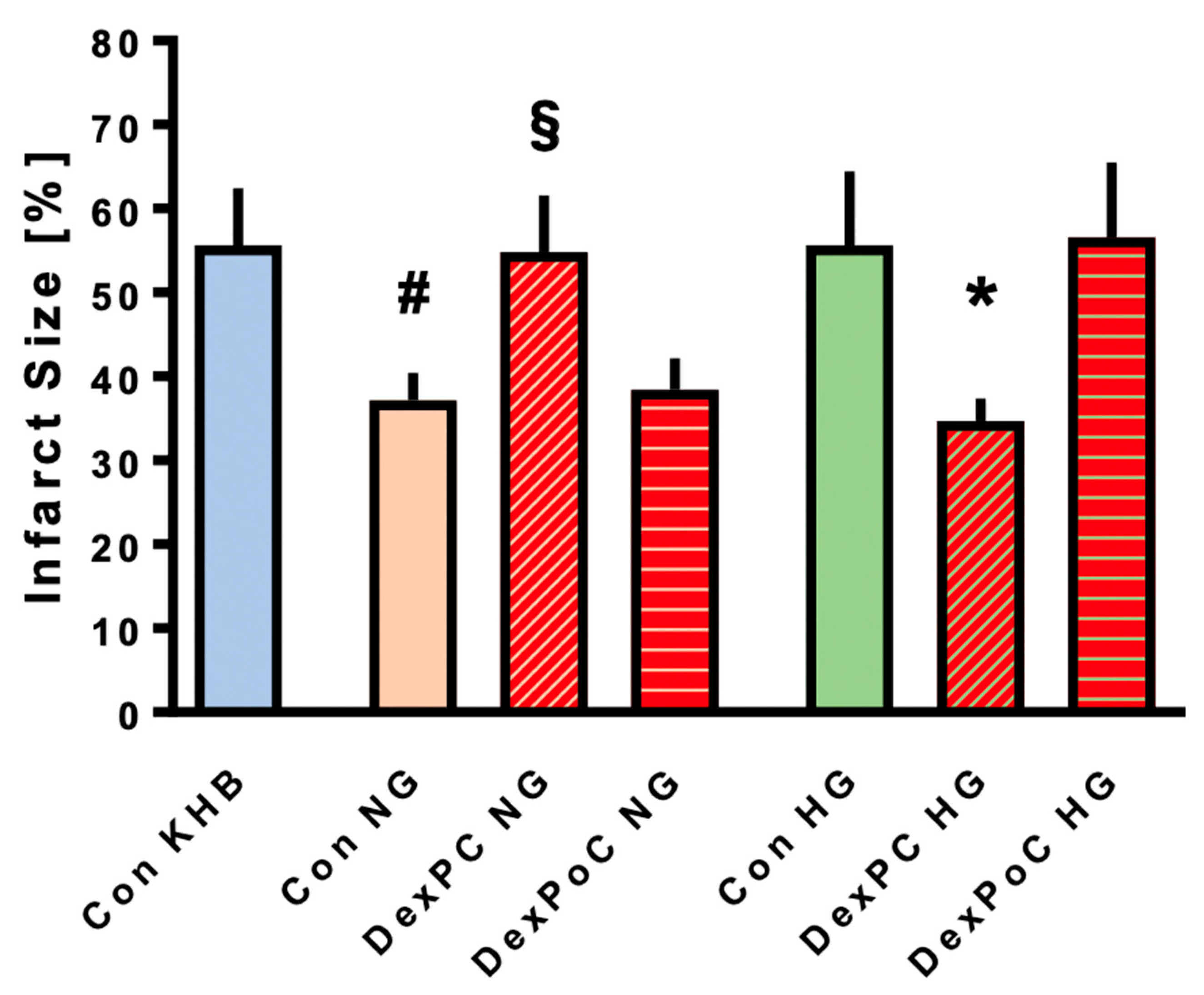
3.2. Infarct Size Measurements
3.3. Cardiac Function
3.4. Glucose Values
4. Discussion
4.1. Dexmedetomidine and Hyperglycemia
4.2. Dexmedetomidine and Mannitol
4.3. Limitations
Author Contributions
Funding
Conflicts of Interest
References
- Caricati-Neto, A.; Errante, P.R.; Menezes-Rodrigues, F.S. Recent Advances in Pharmacological and Non-Pharmacological Strategies of Cardioprotection. Int. J. Mol. Sci. 2019, 20, 4002. [Google Scholar] [CrossRef] [PubMed]
- Garratt, K.N.; Whittaker, P.; Przyklenk, K. Remote Ischemic Conditioning and the Long Road to Clinical Translation. Circ. Res. 2016, 118, 1052–1054. [Google Scholar] [CrossRef] [PubMed]
- Meybohm, P.; Bein, B.; Brosteanu, O.; Cremer, J.T.; Gruenewald, M.; Stoppe, C.; Coburn, M.; Schaelte, G.; Böning, A.; Niemann, B.; et al. A Multicenter Trial of Remote Ischemic Preconditioning for Heart Surgery. N. Engl. J. Med. 2015, 373, 1397–1407. [Google Scholar] [CrossRef] [PubMed]
- Meybohm, P.; Kohlhaas, M.; Stoppe, C.; Gruenewald, M.; Bein, B.; Albrecht, M.; Cremer, J.; Coburn, M.; Schaelte, G.; Boening, A.; et al. RIPHeart (Remote Ischemic Preconditioning for Heart Surgery) Study: Myocardial Dysfunction, Postoperative Neurocognitive Dysfunction, and 1 Year Follow-Up. J. Am. Heart Assoc. 2018, 7, e008077. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Candilio, L.; Evans, R.; Ariti, C.; Jenkins, D.P.; Kolvekar, S.; Knight, R.; Kunst, G.; Laing, C.; Nicholas, J.; et al. Effect of Remote Ischaemic preconditioning on Clinical outcomes in patients undergoing Coronary Artery bypass graft surgery (ERICCA study): A multicentre double-blind randomised controlled clinical trial. Effic. Mech. Eval. 2016, 3, 1–58. [Google Scholar] [CrossRef]
- Ferdinandy, P.; Schulz, R.; Baxter, G. Interaction of Cardiovascular Risk Factors with Myocardial Ischemia/Reperfusion Injury, Preconditioning, and Postconditioning. Pharmacol. Rev. 2007, 59, 418–458. [Google Scholar] [CrossRef]
- Heusch, G. Critical Issues for the Translation of Cardioprotection. Circ. Res. 2017, 120, 1477–1486. [Google Scholar] [CrossRef] [PubMed]
- Bellodi, G.; Manicardi, V.; Malavasi, V.; Veneri, L.; Bernini, G.; Bpssini, P.; Distefano, S.; Magnanini, G.; Muratori, L.; Rossi, G.; et al. Hyperglycemia and prognosis of acute myocardial infarction in patients without diabetes mellitus. Am. J. Cardiol. 1989, 64, 885–888. [Google Scholar] [CrossRef]
- Jelesoff, N.E.; Feinglos, M.; Granger, C.B.; Califf, R.M. Outcomes of diabetic patients following acute myocardial infarction: A review of the major thrombolytic trials. Coron. Artery Dis. 1996, 7, 732–743. [Google Scholar] [CrossRef]
- Ceriello, A. Acute hyperglycaemia: A ‘new’ risk factor during myocardial infarction. Eur. Heart J. 2004, 26, 328–331. [Google Scholar] [CrossRef]
- Kristiansen, S.B.; Pælestik, K.B.; Johnsen, J.; Nr, J.; Pryds, K.; Hjortbak, M.V.; Jensen, R.V.; Bøtker, H.E. Impact of hyperglycemia on myocardial ischemia-reperfusion susceptibility and ischemic preconditioning in hearts from rats with type 2 diabetes. Cardiovasc. Diabetol. 2019, 18, 66. [Google Scholar] [CrossRef] [PubMed]
- Zálešák, M.; Blažíček, P.; Pancza, D.; Ledvényiová, V.; Bartekova, M.; Nemčeková, M.; Čarnická, S.; Ziegelhöffer, A.; Ravingerová, T. Severity of lethal ischemia/reperfusion injury in rat hearts subjected to ischemic preconditioning is increased under conditions of simulated hyperglycemia. Physiol. Res. 2014, 63, 577–585. [Google Scholar] [PubMed]
- Kehl, F.; Krolikowski, J.G.; Mraovic, B.; Pagel, P.S.; Warltier, D.C.; Kersten, J.R. Hyperglycemia Prevents Isoflurane-induced Preconditioning against Myocardial Infarction. Anesthesiology 2002, 96, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.J.; McCormick, L.; Dutka, D.P. Optimising cardioprotection during myocardial ischaemia: Targeting potential intracellular pathways with glucagon-like peptide-1. Cardiovasc. Diabetol. 2014, 13, 12. [Google Scholar] [CrossRef] [PubMed]
- Keating, G.M. Dexmedetomidine: A Review of Its Use for Sedation in the Intensive Care Setting. Drugs 2015, 75, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Behmenburg, F.; Pickert, E.; Mathes, A.; Heinen, A.; Hollmann, M.W.; Huhn, R.; Berger, M.M. The Cardioprotective Effect of Dexmedetomidine in Rats Is Dose-Dependent and Mediated by BKCa Channels. J. Cardiovasc. Pharmacol. 2017, 69, 228–235. [Google Scholar] [CrossRef]
- Bunte, S.; Behmenburg, F.; Majewski, N.; Stroethoff, M.; Raupach, A.; Mathes, A.; Heinen, A.; Hollmann, M.W.; Huhn, R. Characteristics of Dexmedetomidine Postconditioning in the Field of Myocardial Ischemia–Reperfusion Injury. Anesthesia Analg. 2020, 130, 90–98. [Google Scholar] [CrossRef]
- Cheng, X.; Gu, X.Y.; Gao, Q.; Zong, Q.F.; Li, X.H.; Zhang, Y. Effects of dexmedetomidine postconditioning on myocardial ischemia and the role of the PI3K/Akt-dependent signaling pathway in reperfusion injury. Mol. Med. Rep. 2016, 14, 797–803. [Google Scholar] [CrossRef]
- Ibacache, M.; Sánchez, G.; Pedrozo, Z.; Galvez, F.; Humeres, C.; Echevarría, G.; Duaso, J.; Hassi, M.; Garcia, L.; Díaz-Araya, G.; et al. Dexmedetomidine preconditioning activates pro-survival kinases and attenuates regional ischemia/reperfusion injury in rat heart. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2012, 1822, 537–545. [Google Scholar] [CrossRef]
- Yuan, F.; Fu, H.; Sun, K.; Wu, S.; Dong, T. Effect of dexmedetomidine on cerebral ischemia-reperfusion rats by activating mitochondrial ATP-sensitive potassium channel. Metab. Brain Dis. 2016, 32, 539–546. [Google Scholar] [CrossRef]
- Ishihara, M.; Inoue, I.; Kawagoe, T.; Shimatani, Y.; Kurisu, S.; Nishioka, K.; Kouno, Y.; Umemura, T.; Nakamura, S.; Sato, H. Diabetes mellitus prevents ischemic preconditioning in patients with a first acute anterior wall myocardial infarction. J. Am. Coll. Cardiol. 2001, 38, 1007–1011. [Google Scholar] [CrossRef]
- Deng, L.; Chen, H.; Wei, N.; Zhang, Z.; Wang, G. The cardioprotective effect of dexmedetomidine on regional ischemia/reperfusion injury in type 2 diabetic rat hearts. Microvasc. Res. 2019, 123, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.H.; Litwin, S.E. Hyperglycemia and adverse outcomes in acute coronary syndromes: Is serum glucose the provocateur or innocent bystander? Diabetes 2014, 63, 2209–2212. [Google Scholar] [CrossRef] [PubMed]
- Raupach, A.; Reinle, J.; Stroethoff, M.; Mathes, A.; Heinen, A.; Hollmann, M.W.; Huhn, R.; Bunte, S. Milrinone-Induced Pharmacological Preconditioning in Cardioprotection: Hints for a Role of Mitochondrial Mechanisms. J. Clin. Med. 2019, 8, 507. [Google Scholar] [CrossRef]
- Behmenburg, F.; Dorsch, M.; Huhn, R.; Mally, D.; Heinen, A.; Hollmann, M.W.; Berger, M.M. Impact of Mitochondrial Ca2+-Sensitive Potassium (mBKCa) Channels in Sildenafil-Induced Cardioprotection in Rats. PLoS ONE 2015, 10, e0144737. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.M.; Mocanu, M.M.; Yellon, D.M. Retrograde heart perfusion: The Langendorff technique of isolated heart perfusion. J. Mol. Cell. Cardiol. 2011, 50, 940–950. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, Y.X.; Xiang, X.; Zhu, Y.; Men, J.; He, M. Estimation of the normal range of blood glucose in rats. J. Hyg. Res. 2010, 39, 133–142. [Google Scholar]
- Fang, W.; Lü, G. Effect of dexmedetomidine postconditioning on mitochondria injury during myocardial ischemiia-reperfusion in isolated rat hearts. Chin. J. Anesthesiol. 2011, 31, 1394–1396. [Google Scholar]
- Braunwald, E.; Kloner, R.A. The stunned myocardium: Prolonged, postischemic ventricular dysfunction. Circulation 1982, 66, 1146–1149. [Google Scholar] [CrossRef]
- Lindsey, M.L.; Bolli, R.; Canty, J.M.; Du, X.-J.; Frangogiannis, N.G.; Frantz, S.; Gourdie, R.G.; Holmes, J.W.; Jones, S.; Kloner, R.A.; et al. Guidelines for experimental models of myocardial ischemia and infarction. Am. J. Physiol. Circ. Physiol. 2018, 314, H812–H838. [Google Scholar] [CrossRef]
- Kersten, J.R.; Schmeling, T.J.; Orth, K.G.; Pagel, P.S.; Warltier, D.C. Acute hyperglycemia abolishes ischemic preconditioning in vivo. Am. J. Physiol. Content 1998, 275, H721–H725. [Google Scholar] [CrossRef] [PubMed]
- Kersten, J.R.; Toller, W.G.; Gross, E.R.; Pagel, P.S.; Warltier, D.C. Diabetes abolishes ischemic preconditioning: Role of glucose, insulin, and osmolality. Am. J. Physiol. Circ. Physiol. 2000, 278, H1218–H1224. [Google Scholar] [CrossRef] [PubMed]
- Kiss, A.; Tratsiakovich, Y.; Gonon, A.; Fedotovskaya, O.; Lanner, J.T.; Andersson, D.; Yang, J.; Pernow, J. The Role of Arginase and Rho Kinase in Cardioprotection from Remote Ischemic Perconditioning in Non-Diabetic and Diabetic Rat In Vivo. PLoS ONE 2014, 9, e104731. [Google Scholar] [CrossRef] [PubMed]
- Baranyai, T.; Nagy, C.T.; Koncsos, G.; Onódi, Z.; Károlyi-Szabó, M.; Makkos, A.; Varga, Z.; Ferdinandy, P.; Giricz, Z. Acute hyperglycemia abolishes cardioprotection by remote ischemic perconditioning. Cardiovasc. Diabetol. 2015, 14, 151. [Google Scholar] [CrossRef] [PubMed]
- Goergens, J.I.; Heinen, N.M.; Zoller, J.; Preckel, B.; Bauer, I.; Huhn, R.; Ebel, D.; Raupach, A. Influence of Hyperglycemia During Different Phases of Ischemic Preconditioning on Cardioprotection—A Focus on Apoptosis and Aggregation of Granulocytes. Shock 2020, 53, 637–645. [Google Scholar] [CrossRef]
- Weber, N.C.; Goletz, C.; Huhn, R.; Grueber, Y.; Preckel, B.; Schlack, W.; Ebel, D. Blockade of anaesthetic-induced preconditioning in the hyperglycaemic myocardium. Eur. J. Pharmacol. 2008, 592, 48–54. [Google Scholar] [CrossRef]
- Huhn, R.; Heinen, A.; Weber, N.C.; Hollmann, M.W.; Schlack, W.S.; Preckel, B. Hyperglycaemia blocks sevoflurane-induced postconditioning in the rat heart in vivo: Cardioprotection can be restored by blocking the mitochondrial permeability transition pore. Br. J. Anaesth. 2008, 100, 465–471. [Google Scholar] [CrossRef]
- Kersten, J.R.; Montgomery, M.W.; Ghassemi, T.; Gross, E.R.; Toller, W.G.; Pagel, P.S.; Warltier, D.C. Diabetes and hyperglycemia impair activation of mitochondrial K(ATP) channels. Am. J. Physiol. Circ. Physiol. 2001, 280, 1744–1750. [Google Scholar] [CrossRef]
- Toller, W.G.; Gross, E.R.; Kersten, J.R.; Pagel, P.S.; Gross, G.J.; Warltier, D.C. Sarcolemmal and mitochondrial adenosine triphosphate- dependent potassium channels: Mechanism of desflurane-induced cardioprotection. Anesthesiology 2000, 92, 1731–1739. [Google Scholar] [CrossRef]
- Raphael, J.; Gozal, Y.; Navot, N.; Zuo, Z. Hyperglycemia Inhibits Anesthetic-induced Postconditioning in the Rabbit Heart via Modulation of Phosphatidylinositol-3-kinase/Akt and Endothelial Nitric Oxide Synthase Signaling. J. Cardiovasc. Pharmacol. 2010, 55, 348–357. [Google Scholar] [CrossRef]
- Heusch, G. Molecular Basis of Cardioprotection. Circ. Res. 2015, 116, 674–699. [Google Scholar] [CrossRef] [PubMed]
- Riquelme, J.A.; Westermeier, F.; Hall, A.R.; Vicencio, J.M.; Pedrozo, Z.; Ibacache, M.; Fuenzalida, B.; Sobrevia, L.; Davidson, S.M.; Yellon, D.M.; et al. Dexmedetomidine protects the heart against ischemia-reperfusion injury by an endothelial eNOS/NO dependent mechanism. Pharmacol. Res. 2016, 103, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Xia, M.; Wang, M.; Chen, S. Dexmedetomidine preconditioning protects isolated rat hearts against ischemia/reperfusion injuries and its mechanism. J. Zhejiang Univ. Med Sci. 2013, 42, 326–330. [Google Scholar]
- Rossello, X.; Yellon, D.M. The RISK pathway and beyond. Basic Res. Cardiol. 2017, 113, 2. [Google Scholar] [CrossRef]
- Schulman, D.; Latchman, D.S.; Yellon, D.M. Urocortin protects the heart from reperfusion injury via upregulation of p42/p44 MAPK signaling pathway. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1481–H1488. [Google Scholar] [CrossRef]
- Varma, S.; Lal, B.K.; Zheng, R.; Breslin, J.W.; Saito, S.; Pappas, P.J.; Hobson, R.W.; Durán, W.N. Hyperglycemia alters PI3k and Akt signaling and leads to endothelial cell proliferative dysfunction. Am. J. Physiol. Circ. Physiol. 2005, 289, H1744–H1751. [Google Scholar] [CrossRef]
- Nakadate, Y.; Sato, H.; Oguchi, T.; Sato, T.; Kawakami, A.; Ishiyama, T.; Matsukawa, T.; Schricker, T. Glycemia and the cardioprotective effects of insulin pre-conditioning in the isolated rat heart. Cardiovasc. Diabetol. 2017, 16, 43. [Google Scholar] [CrossRef]
- Kehl, F.; Krolikowski, J.G.; Weihrauch, D.; Pagel, P.S.; Warltier, D.C.; Kersten, J.R. N-Acetylcysteine Restores Isoflurane-induced Preconditioning against Myocardial Infarction during Hyperglycemia. Anesthesiology 2003, 98, 1384–1390. [Google Scholar] [CrossRef]
- Tanaka, K.; Kehl, F.; Gu, W.; Krolikowski, J.G.; Pagel, P.S.; Warltier, D.C.; Kersten, J.R. Isoflurane-induced preconditioning is attenuated by diabetes. Am. J. Physiol. Circ. Physiol. 2002, 282, H2018–H2023. [Google Scholar] [CrossRef]
- Queliconi, B.B.; Wojtovich, A.P.; Nadtochiy, S.M.; Kowaltowski, A.J.; Brookes, P.S. Redox regulation of the mitochondrial KATP channel in cardioprotection. Biochim. Biophys. Acta (BBA) Bioenerg. 2010, 1813, 1309–1315. [Google Scholar] [CrossRef]
- Gross, G.J.; Peart, J.N. KATP channels and myocardial preconditioning: An update. Am. J. Physiol. Circ. Physiol. 2003, 285, H921–H930. [Google Scholar] [CrossRef] [PubMed]
- Ardehali, H.; O’Rourke, B. Mitochondrial K(ATP) channels in cell survival and death. J. Mol. Cell. Cardiol. 2005, 39, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wei, T.; Wang, X.-H.; Chen, H.-Z.; Gu, J.-Z.; Fu, J.; Ni, Y.-F.; Gao, P.-J.; Zhu, D.-L.; Higashino, H. Paradoxically enhanced heart tolerance to ischaemia in type 1 diabetes and role of increased osmolarity. Clin. Exp. Pharmacol. Physiol. 2006, 33, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Chiong, M.; Parra, V.; Eisner, V.; Ibarra, C.; Maldonado, C.; Criollo, A.; Bravo-Sagua, R.; Quiroga, C.; Contreras, A.; Vicencio, J.M.; et al. Parallel activation of Ca2+-induced survival and death pathways in cardiomyocytes by sorbitol-induced hyperosmotic stress. Apoptosis 2010, 15, 887–903. [Google Scholar] [CrossRef]
- Zálešák, M.; Blažíček, P.; Pancza, D.; Gablovský, I.; Štrbák, V.; Ravingerová, T. Hyperosmotic Environment Blunts Effectivity of Ischemic Preconditioning Against Ischemia-Reperfusion Injury and Improves Ischemic Tolerance in Non-Preconditioned Isolated Rat Hearts. Physiol. Res. 2016, 65, 1045–1051. [Google Scholar] [CrossRef]
- Pastukh, V.; Ricci, C.; Solodushko, V.; Mozaffari, M.; Schaffer, S.W. Contribution of the PI 3-kinase/Akt survival pathway toward osmotic preconditioning. Mol. Cell. Biochem. 2005, 269, 59–67. [Google Scholar] [CrossRef]
- Bunte, S.; Lill, T.; Falk, M.; Stroethoff, M.; Raupach, A.; Mathes, A.; Heinen, A.; Hollmann, M.W.; Huhn, R. Impact of Anesthetics on Cardioprotection Induced by Pharmacological Preconditioning. J. Clin. Med. 2019, 8, 396. [Google Scholar] [CrossRef]
- Ceriello, A.; Quagliaro, L.; D’Amico, M.; Di Filippo, C.; Marfella, R.; Nappo, F.; Berrino, L.; Rossi, F.; Giugliano, D. Acute hyperglycemia induces nitrotyrosine formation and apoptosis in perfused heart from rat. Diabetes 2002, 51, 1076–1082. [Google Scholar] [CrossRef]
- Ling, P.-R.; Mueller, C.; Smith, R.J.; Bistrian, B.R. Hyperglycemia induced by glucose infusion causes hepatic oxidative stress and systemic inflammation, but not STAT3 or MAP kinase activation in liver in rats. Metabolism 2003, 52, 868–874. [Google Scholar] [CrossRef]
- Filippo, C.D.; Marfella, R.; Cuzzocrea, S.; Piegari, E.; Petronella, P.; Giugliano, D.; Rossi, F.; D’Amico, M. Hyperglycemia in Streptozotocin-Induced Diabetic Rat Increases Infarct Size Associated With Low Levels of Myocardial HO-1 During Ischemia/Reperfusion. Diabetes 2005, 54, 803–810. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Subgroup | Treatment | n | Body Weight (g) | Heart Wet Weight (g) | Time of Max. Ischemic Contracture (min) | Level of Max. Ischemic Contracture (mmHg) |
|---|---|---|---|---|---|---|
| KHB | Con | 7 | 295 ± 14 | 1.17 ± 0.07 | 17 ± 3 | 107 ± 16 |
| NG | Con | 6 | 299 ± 17 | 1.23 ± 0.04 | 17 ± 1 | 76 ± 21 |
| DexPC | 7 | 299 ± 15 | 1.25 ± 0.09 | 16 ± 2 | 70 ± 14 | |
| DexPoC | 8 | 308 ± 26 | 1.21 ± 0.05 | 16 ± 3 | 71 ± 17 | |
| HG | Con | 8 | 301 ± 10 | 1.26 ± 0.04 | 15 ± 1 | 107 ± 26 |
| DexPC | 6 | 309 ± 21 | 1.28 ± 0.03 | 17 ± 2 | 98 ± 16 | |
| DexPoC | 8 | 306 ± 19 | 1.25 ± 0.06 | 14 ± 2 | 102 ± 14 |
| Subgroup | Treatment | Baseline | PC | Reperfusion | |
|---|---|---|---|---|---|
| 30 | 60 | ||||
| Heart Rate (bpm) | |||||
| KHB | Con | 319 ± 43 | 305 ± 48 | 260 ± 53 | 243 ± 50 |
| NG | Con | 286 ± 21 | 268 ± 16 | 317 ± 40 | 207 ± 66 |
| DexPC | 308 ± 23 | 288 ± 20 | 287 ± 54 | 232 ± 54 | |
| DexPoC | 314 ± 40 | 293 ± 42 | 298 ± 47 | 250 ± 71 | |
| HG | Con | 289 ± 41 | 286 ± 32 | 235 ± 71 | 209 ± 60 |
| DexPC | 302 ± 26 | 286 ± 26 | 265 ± 99 | 202 ± 40 | |
| DexPoC | 305 ± 44 | 293 ± 36 | 254 ± 60 | 217 ± 33 | |
| LVDP (mmHg) | |||||
| KHB | Con | 105 ± 14 | 110 ± 11 | 27 ± 16 * | 33 ± 14 * |
| NG | Con | 141 ± 31 # | 148 ± 28 # | 32 ± 15 * | 43 ± 19 * |
| DexPC | 129 ± 36 | 129 ± 32 | 24 ± 19 * | 30 ± 17 * | |
| DexPoC | 136 ± 30 | 133 ± 34 | 34 ± 19 * | 40 ± 18 * | |
| HG | Con | 141 ± 23 # | 145 ± 33 # | 20 ± 9 * | 32 ± 12 * |
| DexPC | 118 ± 27 | 126 ± 29 | 20 ± 6 * | 32 ± 6 * | |
| DexPoC | 140 ± 38 | 145 ± 30 | 28 ± 18 * | 36 ± 16 * | |
| LVEDP (mmHg) | |||||
| KHB | Con | 4 ± 2 | 4 ± 3 | 129 ± 19 * | 107 ± 13 * |
| NG | Con | 4 ± 1 | 3 ± 2 | 109 ± 28 * | 94 ± 22 * |
| DexPC | 4 ± 1 | 4 ± 2 | 106 ± 14 * | 93 ± 14 * | |
| DexPoC | 5 ± 2 | 5 ± 2 | 99 ± 18 * | 89 ± 18 * | |
| HG | Con | 4 ± 2 | 4 ± 2 | 127 ± 29 * | 109 ± 19 * |
| DexPC | 6 ± 2 | 7 ± 3 | 129 ± 17 * | 111 ± 16 * | |
| DexPoC | 4 ± 1 | 4 ± 3 | 123 ± 20 * | 112 ± 19 * | |
| dP/dt max. (mmHg/s) | |||||
| KHB | Con | 4357 ± 1047 | 4976 ± 1097 | 1256 ± 313 * | 1699 ± 625 * |
| NG | Con | 4793 ± 1337 | 5356 ± 1291 | 2559 ± 1429 * | 2230 ± 1681 * |
| DexPC | 4470 ± 1119 | 4805 ± 1143 | 1588 ± 611 * | 1603 ± 362 * | |
| DexPoC | 4974 ± 953 | 5055 ± 1063 | 2351 ± 966 * | 2093 ± 923 * | |
| HG | Con | 5312 ± 1299 | 5476 ± 859 | 1453 ± 310 * | 1726 ± 481 * |
| DexPC | 4337 ± 1089 | 5004 ± 1347 | 2242 ± 1991 * | 1461 ± 259 * | |
| DexPoC | 4834 ± 1470 | 5190 ± 1489 | 1696 ± 791 * | 1821 ± 1007 * | |
| Coronary flow (mL/min) | |||||
| KHB | DexPoC | 11 ± 2 | 10 ± 2 | 8 ± 1 | 9 ± 3 |
| NG | Con | 12 ± 2 | 11 ± 2 | 9 ± 2 | 9 ± 2 |
| DexPC | 14 ± 4 | 14 ± 5 | 10 ± 2 * | 9 ± 2 * | |
| DexPoC | 15 ± 5 | 14 ± 6 | 10 ± 1 * | 9 ± 3 * | |
| HG | Con | 13 ± 2 | 13 ± 3 | 8 ± 2 * | 7 ± 2 * |
| DexPC | 12 ± 3 | 12 ± 3 | 7 ± 2 * | 7 ± 2 * | |
| DexPoC | 14 ± 4 | 14 ± 4 | 6 ± 1 * | 6 ± 1 * | |
| Subgroup | Treatment | Baseline | PT | Reperfusion |
|---|---|---|---|---|
| KHB | Con | 11.0 ± 0.2 | 10.7 ± 0.4 | 10.7 ± 0.2 |
| NG | Con | 11.1 ± 0.4 | 10.9 ± 0.3 | 10.9 ± 0.3 |
| DexPC | 11.1 ± 0.3 | 10.9 ± 0.2 | 11.0 ± 0.5 | |
| DexPoC | 11.2 ± 0.2 | 11.1 ± 0.3 | 11.0 ± 0.2 | |
| HG | Con | 11.0 ± 0.3 | 21.6 ± 0.5 *,# | 23.1 ± 2.5 *,# |
| DexPC | 11.2 ± 0.3 | 22.1 ± 0.7 *,§ | 23.2 ± 1.1 *,§ | |
| DexPoC | 11.0 ± 0.3 | 22.0 ± 0.5 *,$ | 23.7 ± 0.8 *,$ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torregroza, C.; Feige, K.; Schneider, L.; Bunte, S.; Stroethoff, M.; Heinen, A.; Hollmann, M.W.; Huhn, R.; Raupach, A. Influence of Hyperglycemia on Dexmedetomidine-Induced Cardioprotection in the Isolated Perfused Rat Heart. J. Clin. Med. 2020, 9, 1445. https://doi.org/10.3390/jcm9051445
Torregroza C, Feige K, Schneider L, Bunte S, Stroethoff M, Heinen A, Hollmann MW, Huhn R, Raupach A. Influence of Hyperglycemia on Dexmedetomidine-Induced Cardioprotection in the Isolated Perfused Rat Heart. Journal of Clinical Medicine. 2020; 9(5):1445. https://doi.org/10.3390/jcm9051445
Chicago/Turabian StyleTorregroza, Carolin, Katharina Feige, Laura Schneider, Sebastian Bunte, Martin Stroethoff, André Heinen, Markus W. Hollmann, Ragnar Huhn, and Annika Raupach. 2020. "Influence of Hyperglycemia on Dexmedetomidine-Induced Cardioprotection in the Isolated Perfused Rat Heart" Journal of Clinical Medicine 9, no. 5: 1445. https://doi.org/10.3390/jcm9051445
APA StyleTorregroza, C., Feige, K., Schneider, L., Bunte, S., Stroethoff, M., Heinen, A., Hollmann, M. W., Huhn, R., & Raupach, A. (2020). Influence of Hyperglycemia on Dexmedetomidine-Induced Cardioprotection in the Isolated Perfused Rat Heart. Journal of Clinical Medicine, 9(5), 1445. https://doi.org/10.3390/jcm9051445