Breakpoint Mapping of Symptomatic Balanced Translocations Links the EPHA6, KLF13 and UBR3 Genes to Novel Disease Phenotype

 , , , , , , ,
, , , , , , ,
Abstract
1. Introduction
1.1. Proband 1
1.2. Proband 2
1.3. Proband 3
1.4. Proband 4
1.5. Proband 5
1.6. Proband 6
1.7. Proband 7
2. Materials and Methods
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fernandez-Marmiesse, A.; Gouveia, S.; Couce, M.L. NGS Technologies as a Turning Point in Rare Disease Research, Diagnosis and Treatment. Curr. Med. Chem. 2018, 25, 404–432. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdottir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant Review with the Integrative Genomics Viewer. Cancer Res. 2017, 77, e31–e34. [Google Scholar] [CrossRef]
- Halgren, C.; Nielsen, N.M.; Nazaryan-Petersen, L.; Silahtaroglu, A.; Collins, R.L.; Lowther, C.; Kjaergaard, S.; Frisch, M.; Kirchhoff, M.; Brondum-Nielsen, K.; et al. Risks and Recommendations in Prenatally Detected De Novo Balanced Chromosomal Rearrangements from Assessment of Long-Term Outcomes. Am. J. Hum. Genet. 2018, 102, 1090–1103. [Google Scholar] [CrossRef]
- Harewood, L.; Chaignat, E.; Reymond, A. Structural variation and its effect on expression. Methods Mol. Biol. 2012, 838, 173–186. [Google Scholar] [CrossRef]
- Mitelman, F.; Johansson, B.; Mertens, F. The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer 2007, 7, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Nothwang, H.G.; Kim, H.G.; Aoki, J.; Geisterfer, M.; Kubart, S.; Wegner, R.D.; van Moers, A.; Ashworth, L.K.; Haaf, T.; Bell, J.; et al. Functional hemizygosity of PAFAH1B3 due to a PAFAH1B3-CLK2 fusion gene in a female with mental retardation, ataxia and atrophy of the brain. Hum. Mol. Genet. 2001, 10, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Harewood, L.; Schutz, F.; Boyle, S.; Perry, P.; Delorenzi, M.; Bickmore, W.A.; Reymond, A. The effect of translocation-induced nuclear reorganization on gene expression. Genome Res. 2010, 20, 554–564. [Google Scholar] [CrossRef]
- Murcia Pienkowski, V.; Kucharczyk, M.; Mlynek, M.; Szczaluba, K.; Rydzanicz, M.; Poszewiecka, B.; Skorka, A.; Sykulski, M.; Biernacka, A.; Koppolu, A.A.; et al. Mapping of breakpoints in balanced chromosomal translocations by shallow whole-genome sequencing points to EFNA5, BAHD1 and PPP2R5E as novel candidates for genes causing human Mendelian disorders. J. Med. Genet. 2019, 56, 104–112. [Google Scholar] [CrossRef] [PubMed]
- TADeus a Web Service Dedicated for Clinical Evaluation of Variants in Genes Whose Expression May Be Affected by Chromosomal Rearrangements. Available online: http://bioputer.mimuw.edu.pl/tadeus (accessed on 5 February 2020).
- Mendelian Inheritance in Man. Available online: https://www.omim.org (accessed on 5 January 2020).
- Konrad, J.; Karczewski, L.C.F.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Kristen, M.L.; Ganna, A.; Daniel, P.B.; et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv 2020. [Google Scholar] [CrossRef]
- Beach, K.Q.H.; Kiyama, T.; Wang, S.; Otteson, D. Expression of the Axonal Guidance Receptors EPHA5 and EPHA6 Changes Across Retinal Development. IOVS 2013, 54, 5147. [Google Scholar]
- Marcelli, F.; Boisset, G.; Schorderet, D.F. A dimerized HMX1 inhibits EPHA6/epha4b in mouse and zebrafish retinas. PLoS ONE 2014, 9, e100096. [Google Scholar] [CrossRef] [PubMed]
- Das, G.; Yu, Q.; Hui, R.; Reuhl, K.; Gale, N.W.; Zhou, R. EphA5 and EphA6: Regulation of neuronal and spine morphology. Cell Biosci. 2016, 6, 48. [Google Scholar] [CrossRef] [PubMed]
- Savelieva, K.V.; Rajan, I.; Baker, K.B.; Vogel, P.; Jarman, W.; Allen, M.; Lanthorn, T.H. Learning and memory impairment in Eph receptor A6 knockout mice. Neurosci. Lett. 2008, 438, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Bult, C.J.; Blake, J.A.; Smith, C.L.; Kadin, J.A.; Richardson, J.E.; the Mouse Genome Database Group. Mouse Genome Database (MGD) 2019. Nucleic Acids Res. 2019, 47, D801–D806. [Google Scholar] [CrossRef]
- Tissue-Based Map of the Human Proteome. Available online: https://www.proteinatlas.org (accessed on 10 January 2020).
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic. Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Kim, T.Y.; Healy, K.D.; Der, C.J.; Sciaky, N.; Bang, Y.J.; Juliano, R.L. Effects of structure of Rho GTPase-activating protein DLC-1 on cell morphology and migration. J. Biol. Chem. 2008, 283, 32762–32770. [Google Scholar] [CrossRef]
- Ashraf, S.; Kudo, H.; Rao, J.; Kikuchi, A.; Widmeier, E.; Lawson, J.A.; Tan, W.; Hermle, T.; Warejko, J.K.; Shril, S.; et al. Mutations in six nephrosis genes delineate a pathogenic pathway amenable to treatment. Nat. Commun. 2018, 9, 1960. [Google Scholar] [CrossRef]
- Lin, B.; Wang, Y.; Wang, Z.; Tan, H.; Kong, X.; Shu, Y.; Zhang, Y.; Huang, Y.; Zhu, Y.; Xu, H.; et al. Uncovering the rare variants of DLC1 isoform 1 and their functional effects in a Chinese sporadic congenital heart disease cohort. PLoS ONE 2014, 9, e90215. [Google Scholar] [CrossRef]
- Lemay, P.; De Marco, P.; Traverso, M.; Merello, E.; Dionne-Laporte, A.; Spiegelman, D.; Henrion, E.; Diallo, O.; Audibert, F.; Michaud, J.L.; et al. Whole exome sequencing identifies novel predisposing genes in neural tube defects. Mol. Genet. Genomic Med. 2019, 7, e00467. [Google Scholar] [CrossRef]
- Shi, S.H.; Jan, L.Y.; Jan, Y.N. Hippocampal neuronal polarity specified by spatially localized mPar3/mPar6 and PI 3-kinase activity. Cell 2003, 112, 63–75. [Google Scholar] [CrossRef]
- Zhang, H.; Macara, I.G. The polarity protein PAR-3 and TIAM1 cooperate in dendritic spine morphogenesis. Nat. Cell Biol. 2006, 8, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Guo, H.; Xiong, B.; Stessman, H.A.; Wu, H.; Coe, B.P.; Turner, T.N.; Liu, Y.; Zhao, W.; Hoekzema, K.; et al. De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat. Commun. 2016, 7, 13316. [Google Scholar] [CrossRef] [PubMed]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Griswold, A.J.; Dueker, N.D.; Van Booven, D.; Rantus, J.A.; Jaworski, J.M.; Slifer, S.H.; Schmidt, M.A.; Hulme, W.; Konidari, I.; Whitehead, P.L.; et al. Targeted massively parallel sequencing of autism spectrum disorder-associated genes in a case control cohort reveals rare loss-of-function risk variants. Mol. Autism. 2015, 6, 43. [Google Scholar] [CrossRef]
- Kosmicki, J.A.; Samocha, K.E.; Howrigan, D.P.; Sanders, S.J.; Slowikowski, K.; Lek, M.; Karczewski, K.J.; Cutler, D.J.; Devlin, B.; Roeder, K.; et al. Refining the role of de novo protein-truncating variants in neurodevelopmental disorders by using population reference samples. Nat. Genet. 2017, 49, 504–510. [Google Scholar] [CrossRef]
- Chen, C.H.; Chen, H.I.; Chien, W.H.; Li, L.H.; Wu, Y.Y.; Chiu, Y.N.; Tsai, W.C.; Gau, S.S. High resolution analysis of rare copy number variants in patients with autism spectrum disorder from Taiwan. Sci. Rep. 2017, 7, 11919. [Google Scholar] [CrossRef]
- Chen, X.; An, Y.; Gao, Y.; Guo, L.; Rui, L.; Xie, H.; Sun, M.; Lam Hung, S.; Sheng, X.; Zou, J.; et al. Rare Deleterious PARD3 Variants in the aPKC-Binding Region are Implicated in the Pathogenesis of Human Cranial Neural Tube Defects Via Disrupting Apical Tight Junction Formation. Hum. Mutat. 2017, 38, 378–389. [Google Scholar] [CrossRef]
- Rao, A.; O’Donnell, S.; Bain, N.; Meldrum, C.; Shorter, D.; Goel, H. An intragenic deletion of the NFIA gene in a patient with a hypoplastic corpus callosum, craniofacial abnormalities and urinary tract defects. Eur. J. Med. Genet. 2014, 57, 65–70. [Google Scholar] [CrossRef]
- Nyboe, D.; Kreiborg, S.; Kirchhoff, M.; Hove, H.B. Familial craniosynostosis associated with a microdeletion involving the NFIA gene. Clin. Dysmorphol. 2015, 24, 109–112. [Google Scholar] [CrossRef]
- Bizet, A.A.; Becker-Heck, A.; Ryan, R.; Weber, K.; Filhol, E.; Krug, P.; Halbritter, J.; Delous, M.; Lasbennes, M.C.; Linghu, B.; et al. Mutations in TRAF3IP1/IFT54 reveal a new role for IFT proteins in microtubule stabilization. Nat. Commun. 2015, 6, 8666. [Google Scholar] [CrossRef]
- O’Roak, B.J.; Vives, L.; Girirajan, S.; Karakoc, E.; Krumm, N.; Coe, B.P.; Levy, R.; Ko, A.; Lee, C.; Smith, J.D.; et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012, 485, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Study, D.D.; McRae, J.F.; Clayton, S.; Fitzgerald, T.W.; Kaplanis, J.; Prigmore, E.; Rajan, D.; Sifrim, A.; Aitken, S.; Akawi, N.; et al. Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017, 542, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Fan, J.; Blanco-Sanchez, B.; Giagtzoglou, N.; Lin, G.; Yamamoto, S.; Jaiswal, M.; Chen, K.; Zhang, J.; Wei, W.; et al. Ubr3, a Novel Modulator of Hh Signaling Affects the Degradation of Costal-2 and Kif7 through Poly-ubiquitination. PLoS Genet. 2016, 12, e1006054. [Google Scholar] [CrossRef] [PubMed]
- Fritzen, D.; Kuechler, A.; Grimmel, M.; Becker, J.; Peters, S.; Sturm, M.; Hundertmark, H.; Schmidt, A.; Kreiss, M.; Strom, T.M.; et al. De novo FBXO11 mutations are associated with intellectual disability and behavioural anomalies. Hum. Genet. 2018, 137, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Meisenberg, C.; Tait, P.S.; Dianova, I.I.; Wright, K.; Edelmann, M.J.; Ternette, N.; Tasaki, T.; Kessler, B.M.; Parsons, J.L.; Kwon, Y.T.; et al. Ubiquitin ligase UBR3 regulates cellular levels of the essential DNA repair protein APE1 and is required for genome stability. Nucleic. Acids Res. 2012, 40, 701–711. [Google Scholar] [CrossRef]
- Gerard-Blanluet, M.; Birk-Moller, L.; Caubel, I.; Gelot, A.; Billette de Villemeur, T.; Horn, N. Early development of occipital horns in a classical Menkes patient. Am. J. Med. Genet. A 2004, 130A, 211–213. [Google Scholar] [CrossRef]
- Kaler, S.G.; Tang, J.; Donsante, A.; Kaneski, C.R. Translational read-through of a nonsense mutation in ATP7A impacts treatment outcome in Menkes disease. Ann. Neurol. 2009, 65, 108–113. [Google Scholar] [CrossRef]
- Tang, J.; Robertson, S.; Lem, K.E.; Godwin, S.C.; Kaler, S.G. Functional copper transport explains neurologic sparing in occipital horn syndrome. Genet. Med. 2006, 8, 711–718. [Google Scholar] [CrossRef]
- Kennerson, M.L.; Nicholson, G.A.; Kaler, S.G.; Kowalski, B.; Mercer, J.F.; Tang, J.; Llanos, R.M.; Chu, S.; Takata, R.I.; Speck-Martins, C.E.; et al. Missense mutations in the copper transporter gene ATP7A cause X-linked distal hereditary motor neuropathy. Am. J. Hum. Genet. 2010, 86, 343–352. [Google Scholar] [CrossRef]
- Smpokou, P.; Samanta, M.; Berry, G.T.; Hecht, L.; Engle, E.C.; Lichter-Konecki, U. Menkes disease in affected females: The clinical disease spectrum. Am. J. Med. Genet. A 2015, 167A, 417–420. [Google Scholar] [CrossRef]
- Sirleto, P.; Surace, C.; Santos, H.; Bertini, E.; Tomaiuolo, A.C.; Lombardo, A.; Boenzi, S.; Bevivino, E.; Dionisi-Vici, C.; Angioni, A. Lyonization effects of the t(X;16) translocation on the phenotypic expression in a rare female with Menkes disease. Pediatr. Res. 2009, 65, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Ogoh, H.; Yamagata, K.; Nakao, T.; Sandell, L.L.; Yamamoto, A.; Yamashita, A.; Tanga, N.; Suzuki, M.; Abe, T.; Kitabayashi, I.; et al. Mllt10 knockout mouse model reveals critical role of Af10-dependent H3K79 methylation in midfacial development. Sci. Rep. 2017, 7, 11922. [Google Scholar] [CrossRef] [PubMed]
- An, J.Y.; Cristino, A.S.; Zhao, Q.; Edson, J.; Williams, S.M.; Ravine, D.; Wray, J.; Marshall, V.M.; Hunt, A.; Whitehouse, A.J.; et al. Towards a molecular characterization of autism spectrum disorders: An exome sequencing and systems approach. Transl. Psychiatry 2014, 4, e394. [Google Scholar] [CrossRef] [PubMed]
- de Ligt, J.; Willemsen, M.H.; van Bon, B.W.; Kleefstra, T.; Yntema, H.G.; Kroes, T.; Vulto-van Silfhout, A.T.; Koolen, D.A.; de Vries, P.; Gilissen, C.; et al. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 2012, 367, 1921–1929. [Google Scholar] [CrossRef]
- Tang, J.; Ip, J.P.; Ye, T.; Ng, Y.P.; Yung, W.H.; Wu, Z.; Fang, W.; Fu, A.K.; Ip, N.Y. Cdk5-dependent Mst3 phosphorylation and activity regulate neuronal migration through RhoA inhibition. J. Neurosci. 2014, 34, 7425–7436. [Google Scholar] [CrossRef]
- Halawa, A.A.; Rees, K.A.; McCamy, K.M.; Winzer-Serhan, U.H. Central and peripheral immune responses to low-dose lipopolysaccharide in a mouse model of the 15q13.3 microdeletion. Cytokine 2019, 126, 154879. [Google Scholar] [CrossRef]
- Roelfsema, J.H.; White, S.J.; Ariyurek, Y.; Bartholdi, D.; Niedrist, D.; Papadia, F.; Bacino, C.A.; den Dunnen, J.T.; van Ommen, G.J.; Breuning, M.H.; et al. Genetic heterogeneity in Rubinstein-Taybi syndrome: Mutations in both the CBP and EP300 genes cause disease. Am. J. Hum. Genet. 2005, 76, 572–580. [Google Scholar] [CrossRef]
- Witteveen, J.S.; Willemsen, M.H.; Dombroski, T.C.; van Bakel, N.H.; Nillesen, W.M.; van Hulten, J.A.; Jansen, E.J.; Verkaik, D.; Veenstra-Knol, H.E.; van Ravenswaaij-Arts, C.M.; et al. Haploinsufficiency of MeCP2-interacting transcriptional co-repressor SIN3A causes mild intellectual disability by affecting the development of cortical integrity. Nat. Genet. 2016, 48, 877–887. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Weisburd, B.; Thomas, B.; Solomonson, M.; Ruderfer, D.M.; Kavanagh, D.; Hamamsy, T.; Lek, M.; Samocha, K.E.; Cummings, B.B.; et al. The ExAC browser: displaying reference data information from over 60 000 exomes. Nucleic Acids Res. 2017, 45, D840–D845. [Google Scholar] [CrossRef]
- The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Available online: https://www.biorxiv.org/content/10.1101/531210v4.full (accessed on 10 January 2020).



{kind=link}
{kind=link}
| Proband | Karyotyping Results | Break-Point Positions | Phenotype | Disrupted Gene(s) |
|---|---|---|---|---|
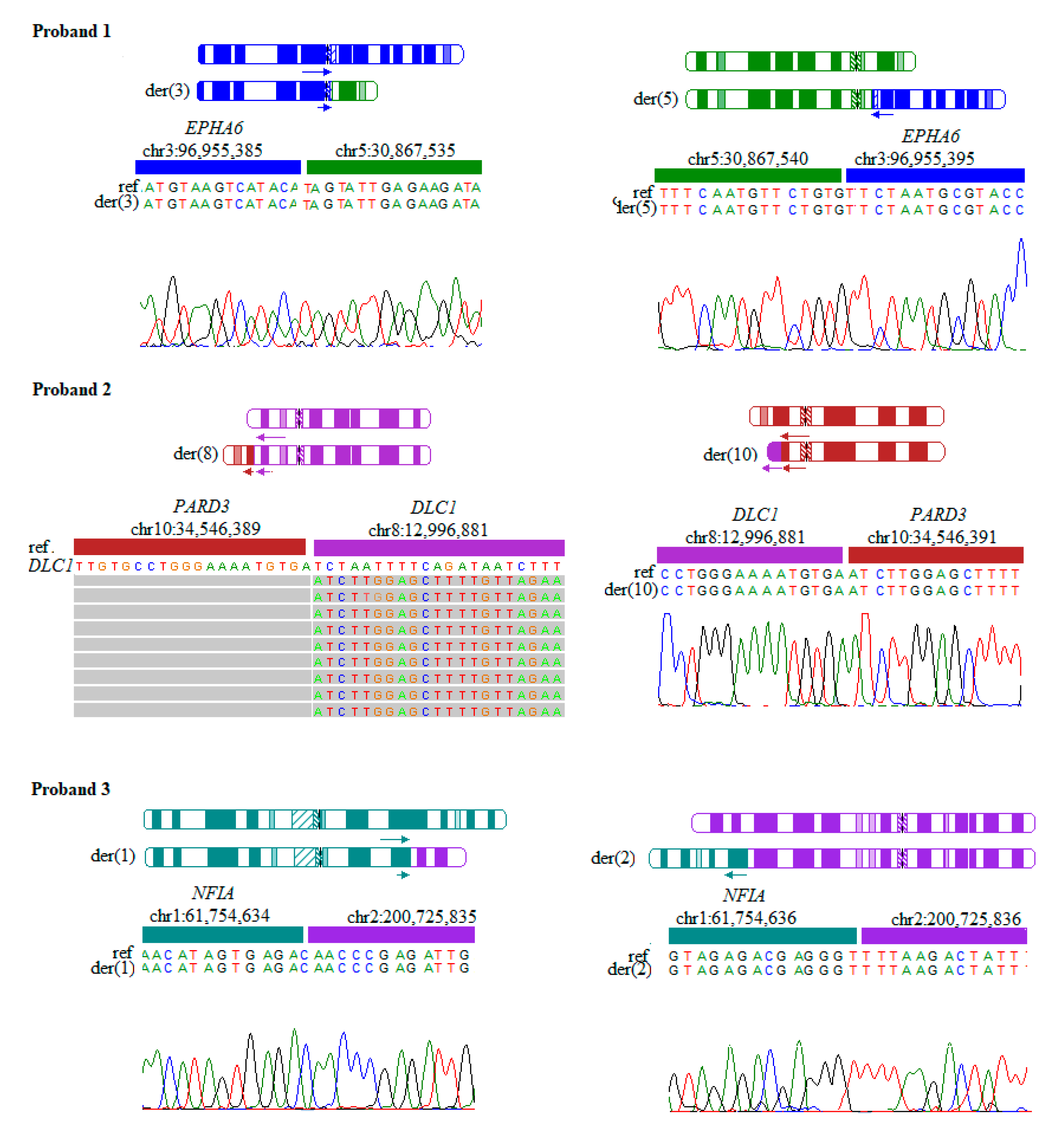
| 1 | 46,XY,t(3;5)(q11.2;p13.1) | chr3:96,955,385/chr5:30,867,535 chr3:96,955,395/chr5:30,867,540 | Developmental delay, anophthalmia | EPHA6 (MIM: 600066) |
| 2 | 46,XY, t(8;10)(p22;p11.21) | chr10:34,546,389/chr8:12,996,881 chr8:12,996,881/chr10:34,546,391 | Hypotonia, dysmorphic features | PARD3 (MIM: 606745) DLC1 (MIM: 604258) |
| 3 | 46,XY,t(1;2)(p31.3;q33.1) | chr1:61,754,634/chr2:200,725,835 chr1:61,754,636/chr2:200,725,835 | Developmental delay | NFIA (MIM: 600727) |
| 4 | 46,XX,inv(2)(q31.1;q37.3) | chr2:170,714,752/chr2:239,263,182 | Severe mental retardation, cured brain dysembryoplastic neuroepithelial tumor | UBR3 (MIM: 613831) TRAF3IP1 (MIM: 607380) |
| 5 | 46,X,t(X;1)(q21.1;p21.1) | chrX:77,213,561/chr1:106,152,109 chrX:77,213,557/chr1:106,152,110 | Menkes disease | ATP7A (MIM: 300011) |
| 6 | 46,XX,t(10;15)(p12.31;q23) | chr10:21,972,544/chr15:70,361,328 chr10:21,972,539/chr15:70,361,330 | Speech delay | MLLT10 (MIM: 602409) TLE3 (MIM: 600190) |
| 7 | 46,XX,t(13;15)(q32.2;q13.3) | chr13:99,169,136/chr15:31,620,350 chr15:31,620,348/chr13:99,169,137 | Developmental delay | STK24 (MIM: 604984) KLF13 (MIM: 605328) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murcia Pienkowski, V.; Kucharczyk, M.; Rydzanicz, M.; Poszewiecka, B.; Pachota, K.; Młynek, M.; Stawiński, P.; Pollak, A.; Kosińska, J.; Wojciechowska, K.; et al. Breakpoint Mapping of Symptomatic Balanced Translocations Links the EPHA6, KLF13 and UBR3 Genes to Novel Disease Phenotype. J. Clin. Med. 2020, 9, 1245. https://doi.org/10.3390/jcm9051245
Murcia Pienkowski V, Kucharczyk M, Rydzanicz M, Poszewiecka B, Pachota K, Młynek M, Stawiński P, Pollak A, Kosińska J, Wojciechowska K, et al. Breakpoint Mapping of Symptomatic Balanced Translocations Links the EPHA6, KLF13 and UBR3 Genes to Novel Disease Phenotype. Journal of Clinical Medicine. 2020; 9(5):1245. https://doi.org/10.3390/jcm9051245
Chicago/Turabian StyleMurcia Pienkowski, Victor, Marzena Kucharczyk, Małgorzata Rydzanicz, Barbara Poszewiecka, Katarzyna Pachota, Marlena Młynek, Piotr Stawiński, Agnieszka Pollak, Joanna Kosińska, Katarzyna Wojciechowska, and et al. 2020. "Breakpoint Mapping of Symptomatic Balanced Translocations Links the EPHA6, KLF13 and UBR3 Genes to Novel Disease Phenotype" Journal of Clinical Medicine 9, no. 5: 1245. https://doi.org/10.3390/jcm9051245
APA StyleMurcia Pienkowski, V., Kucharczyk, M., Rydzanicz, M., Poszewiecka, B., Pachota, K., Młynek, M., Stawiński, P., Pollak, A., Kosińska, J., Wojciechowska, K., Lejman, M., Cieślikowska, A., Wicher, D., Stembalska, A., Matuszewska, K., Materna-Kiryluk, A., Gambin, A., Chrzanowska, K., Krajewska-Walasek, M., & Płoski, R. (2020). Breakpoint Mapping of Symptomatic Balanced Translocations Links the EPHA6, KLF13 and UBR3 Genes to Novel Disease Phenotype. Journal of Clinical Medicine, 9(5), 1245. https://doi.org/10.3390/jcm9051245