Possible Role of Mitochondrial DNA Mutations in Chronification of Inflammation: Focus on Atherosclerosis

 ,
,  and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
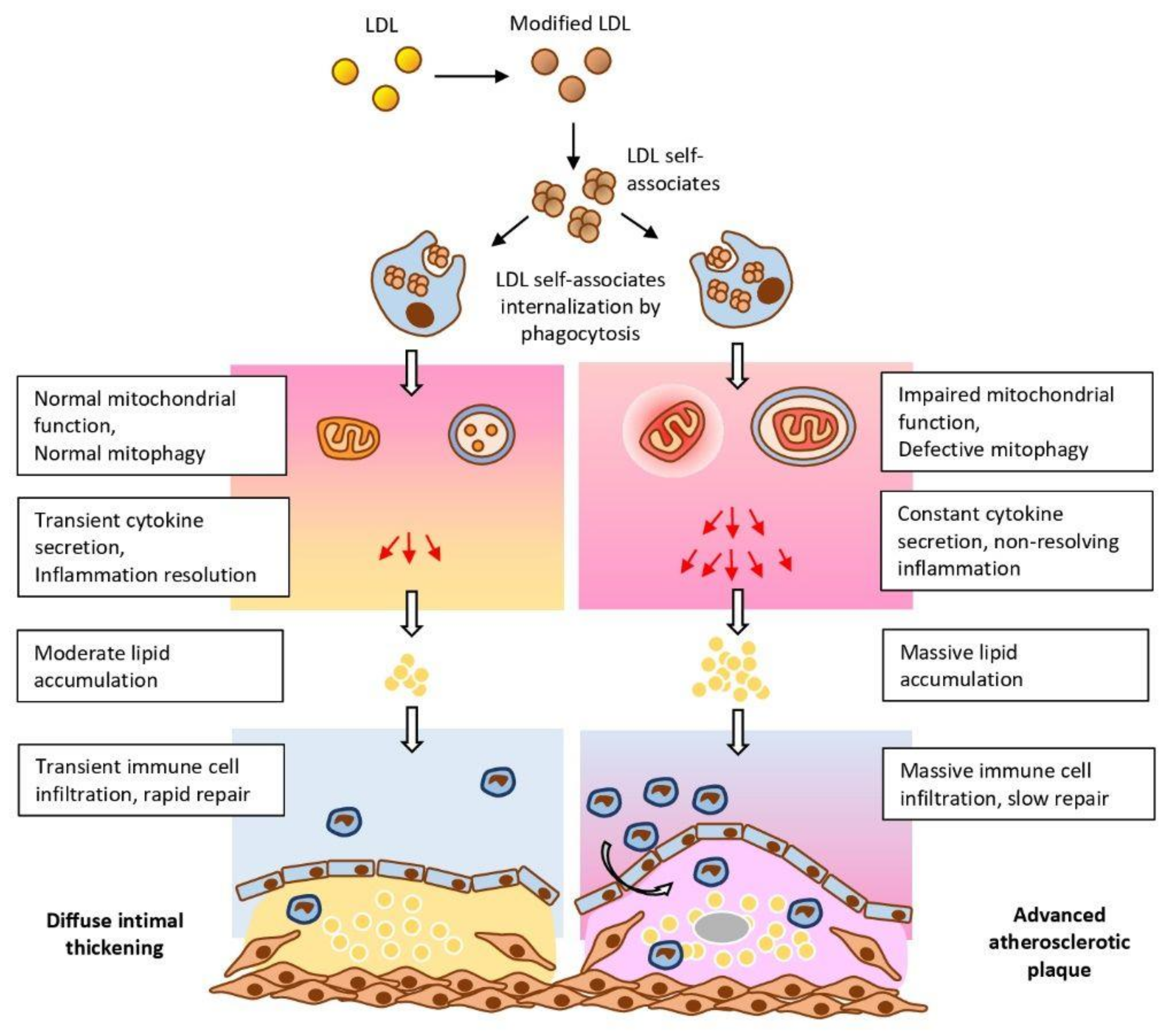{kind=link}
1. Introduction
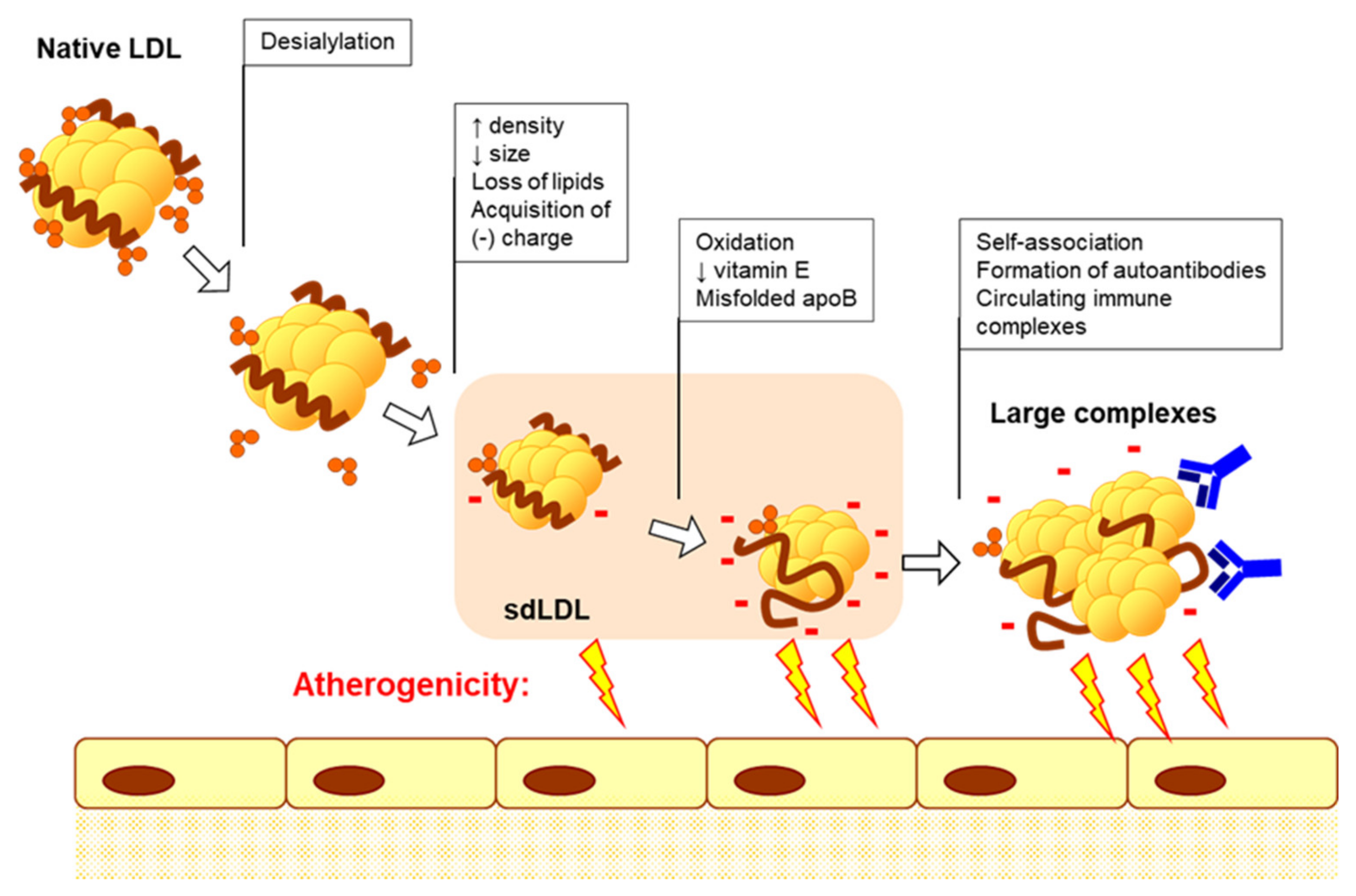
2. Atherosclerosis as Inflammatory Disease
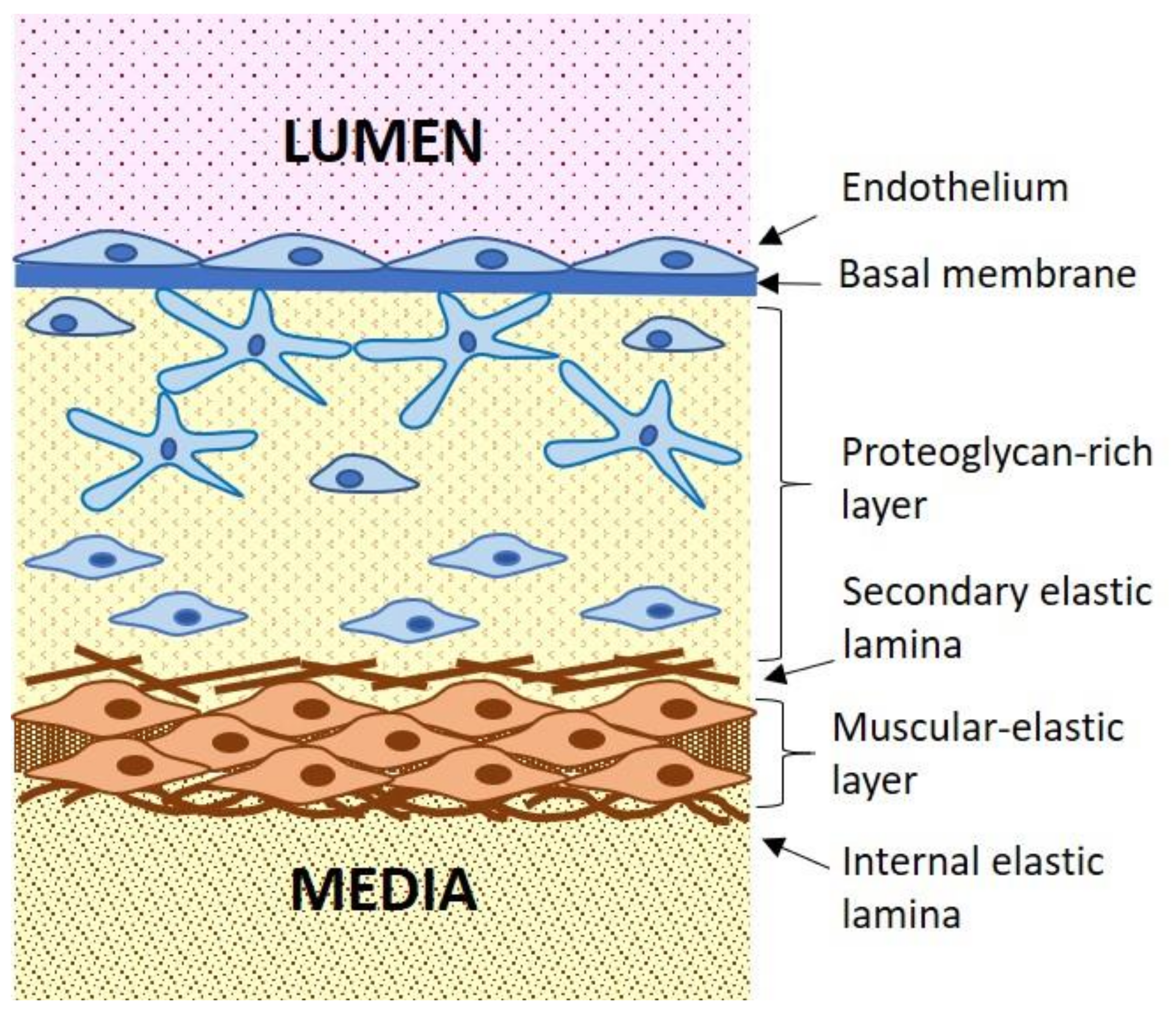
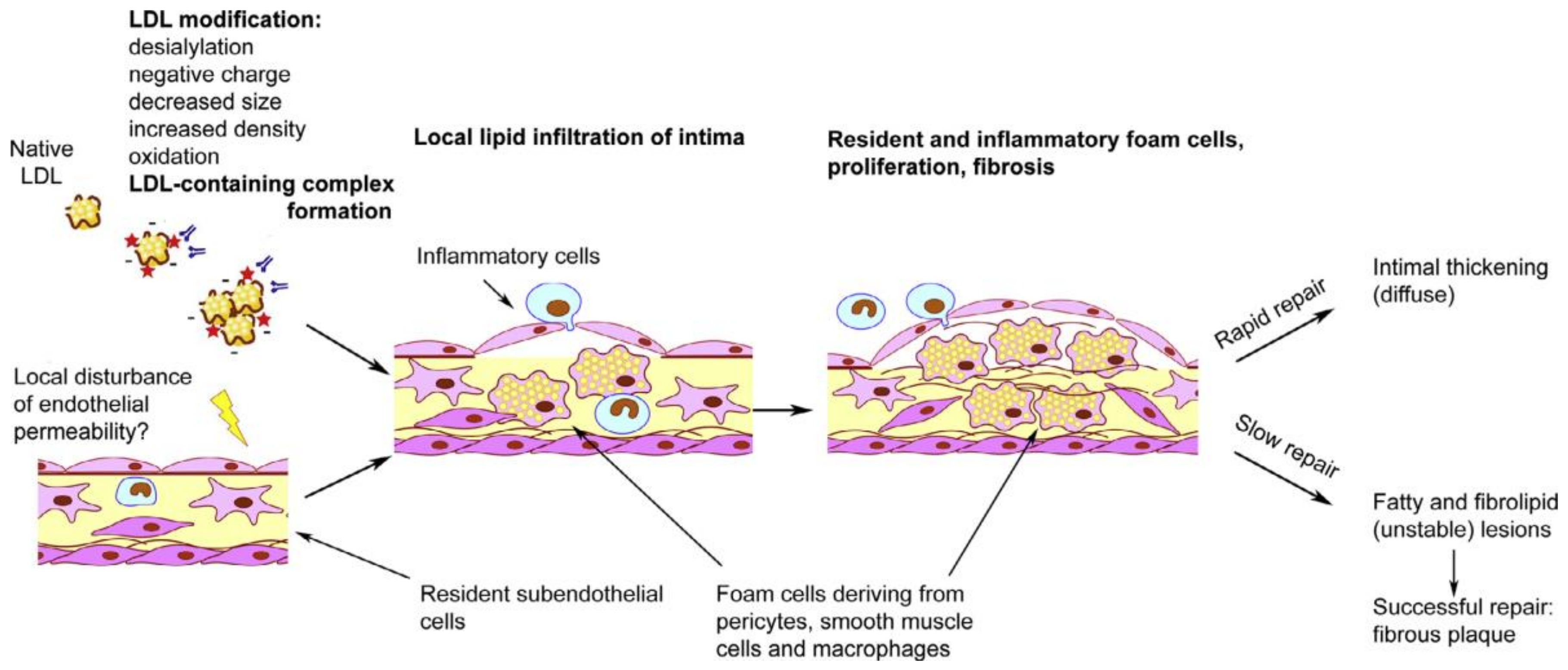
3. Cellular Mechanisms of Atherogenesis
4. Innate Immunity Response and Cellular Mechanisms of Inflammation
5. Innate Immunity and Mitochondrial Dysfunction
6. Mitochondrial Dysfunction and mtDNA Mutations in Chronic Pathologies
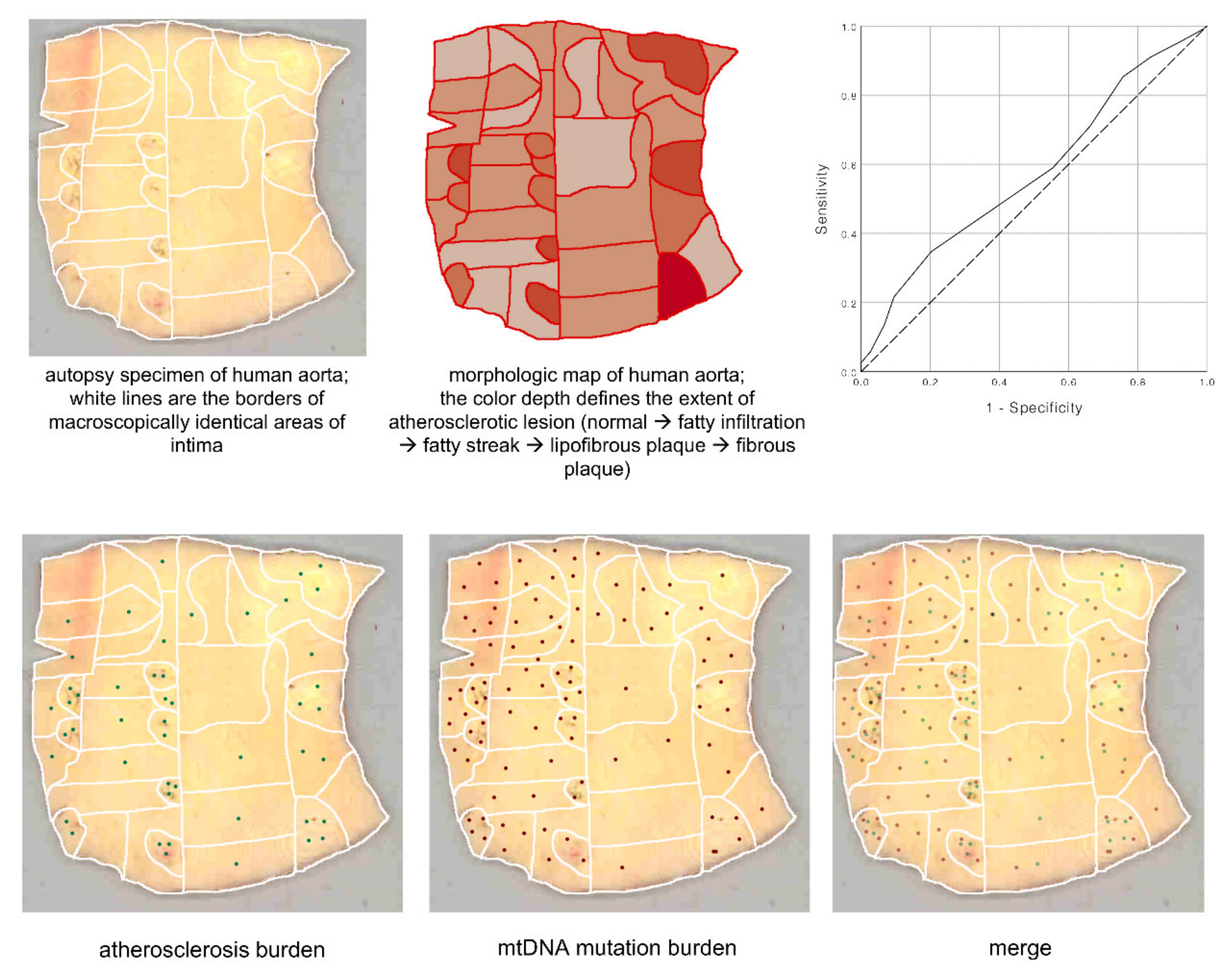
7. Mosaicism of Atherosclerosis-Associated mtDNA Mutations in the Vascular Wall
8. Innate Immunity and mtDNA Mutations
9. Mitophagy and mtDNA Mutations
10. The Role of Mitophagy in the Innate Immunity
11. Possible Role of mtDNA Mutations in Chronification of Inflammation
12. Future Directions
13. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Taubert, K.A. Cardiology patient pages. Can patients with cardiovascular disease take nonsteroidal antiinflammatory drugs? Circulation 2008, 117, 322–324. [Google Scholar] [CrossRef]
- Kirkby, N.S.; Lundberg, M.H.; Wright, W.R.; Warner, T.D.; Paul-Clark, M.J.; Mitchell, J.A. COX-2 protects against atherosclerosis independently of local vascular prostacyclin: Identification of COX-2 associated pathways implicate Rgl1 and lymphocyte networks. PLoS ONE 2014, 9, e98165. [Google Scholar] [CrossRef]
- Fava, C.; Montagnana, M. Atherosclerosis Is an Inflammatory Disease which Lacks a Common Anti-inflammatory Therapy: How Human Genetics Can Help to This Issue. A Narrative Review. Front. Pharmacol. 2018, 9, 55. [Google Scholar] [CrossRef]
- Liberale, L.; Carbone, F.; Montecucco, F.; Sahebkar, A. Statins reduce vascular inflammation in atherogenesis: A review of underlying molecular mechanisms. Int. J. Biochem. Cell Biol. 2020, 122, 105735. [Google Scholar] [CrossRef]
- Schwartz, S.M.; Galis, Z.S.; Rosenfeld, M.E.; Falk, E. Plaque rupture in humans and mice. Arter. Thromb. Vasc. Biol. 2007, 27, 705–713. [Google Scholar] [CrossRef]
- Aliev, G.; Castellani, R.J.; Petersen, R.B.; Burnstock, G.; Perry, G.; Smith, M.A. Pathobiology of familial hypercholesterolemic atherosclerosis. J. Submicrosc. Cytol. Pathol. 2004, 36, 225–240. [Google Scholar]
- Vancheri, F.; Longo, G.; Vancheri, S.; Danial, J.S.H.; Henein, M.Y. Coronary artery microcalcification: Imaging and clinical implications. Diagnostics 2019, 9, 125. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Kashirskikh, D.A.; Khotina, V.A.; Grechko, A.V.; Orekhov, A.N. Immune-inflammatory responses in atherosclerosis: The role of myeloid cells. J. Clin. Med. 2019, 8, 1798. [Google Scholar] [CrossRef]
- Weber, C.; Noels, H. Atherosclerosis: Current pathogenesis and therapeutic options. Nat. Med. 2011, 17, 1410–1422. [Google Scholar] [CrossRef]
- Loppnow, H.; Werdan, K.; Buerke, M. Vascualr cells contribute to atherosclerosis by cytokine-and innate-immunity-related inflammatory mechanisms. Innate Immun. 2008, 17, 63–87. [Google Scholar] [CrossRef]
- Fioranelli, M.; Bottaccioli, A.G.; Bottaccioli, F.; Bianchi, M.; Rovesti, M.; Roccia, M.G. Stress and inflammation in coronary artery disease: A review psychoneuroendocrinology-based. Front. Immunol. 2018, 9, 2031. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, B.; Toss, H.; Siegbahn, A.; Venge, P.; Wallentin, L. Markers of myocardial damage and inflammation in relation to long-term mortality in unstable coronary artery disease: FRISC Study Group: Fragmin During Instability in Coronary artery disease. N. Engl. J. Med. 2000, 343, 1139–1147. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Rifai, N.; Rose, L.; Buring, J.E.; Cook, N.R. Comparison of C-reactive protein and low-density’ lipoprotein cholesterol levels in the prediction of first cardiovascular events. N. Engl. J. Med. 2002, 347, 1557–1565. [Google Scholar] [CrossRef]
- Stitham, J.; Hwa, J. Prostacyclin, atherothrombosis and diabetes mellitus: Physiologic and clinical considerations. Curr. Mol. Med. 2016, 16, 328–342. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Thompson, P.L.; Nidorf, S.M. Colchicine: An affordable anti-inflammatory agent for atherosclerosis. Curr. Opin. Lipidol. 2018, 29, 467–473. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Pradhan, A.; MacFadyen, J.G.; Solomon, D.H.; Zaharris, E.; Mam, V.; Hasan, A.; Rosenberg, Y.; Iturriaga, E.; et al. Low-Dose Methotrexate for the Prevention of Atherosclerotic Events. N. Engl. J. Med. 2019, 380, 752–762. [Google Scholar] [CrossRef]
- Cecconi, A.; Vilchez-Tschischke, J.P.; Mateo, J.; Sanchez-Gonzalez, J.; España, S.; Fernandez-Jimenez, R.; Lopez-Melgar, B.; Fernández Friera, L.; López-Martín, G.J.; Fuster, V.; et al. Effects of Colchicine on Atherosclerotic Plaque Stabilization: A Multimodality Imaging Study in an Animal Model. J. Cardiovasc. Transl. Res. 2020. [Google Scholar] [CrossRef]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef]
- Romanov, Y.A.; Balyasnikova, I.V.; Bystrevskaya, V.B.; Byzova, T.V.; Ilyinskaya, O.P.; Krushinsky, A.V.; Latsis, R.V.; Soboleva, E.L.; Tararak, E.M.; Smirnov, V.N. Endothelial heterogeneity and intimal blood-borne cells. Relation to human atherosclerosis. Ann. N. Y. Acad. Sci. 1995, 748, 12–37. [Google Scholar] [CrossRef]
- Rekhter, M.D.; Andreeva, E.R.; Mironov, A.A.; Orekhov, A.N. Three-dimensional cytoarchitecture of normal and atherosclerotic intima of human aorta. Am. J. Pathol. 1991, 138, 569–580. [Google Scholar]
- Ivanova, E.A.; Bobryshev, Y.V.; Orekhov, A.N. Intimal pericytes as the second line of immune defence in atherosclerosis. World J. Cardiol. 2015, 7, 583–593. [Google Scholar] [CrossRef]
- Summerhill, V.; Orekhov, A.N. Pericytes in atherosclerosis. Adv. Exp. Med. Biol. 2019, 1147, 279–297. [Google Scholar]
- Orekhov, A.N.; Bobryshev, Y.V.; Chistiakov, D.A. The complexity of cell composition of the intima of large arteries: Focus on pericyte-like cells. Cardiovasc. Res. 2014, 103, 438–451. [Google Scholar] [CrossRef]
- Ivanova, E.A.; Orekhov, A.N. Cellular Model of Atherogenesis Based on Pluripotent Vascular Wall Pericytes. Stem Cells Int. 2016, 2016, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Geer, J.C.; Haust, M.D. Smooth muscle cells in atherosclerosis. Monogr. Atheroscler. 1972, 2, 1–140. [Google Scholar] [PubMed]
- Summerhill, V.I.; Grechko, A.V.; Yet, S.F.; Sobenin, I.A.; Orekhov, A.N. The atherogenic role of circulating modified lipids in atherosclerosis. Int. J. Mol. Sci. 2019, 20, 3561. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Sobenin, I.A. Modified lipoproteins as biomarkers of atherosclerosis. Front. Biosci. 2018, 23, 1422–1444. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Sobenin, I.A. Modified and Dysfunctional Lipoproteins in Atherosclerosis: Effectors or Biomarkers? Curr. Med. Chem. 2019, 26, 1512–1524. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. The LDL receptor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 431–438. [Google Scholar] [CrossRef]
- Kontush, A. HDL particle number and size as predictors of cardiovascular disease. Front. Pharmacol. 2015, 6, 276. [Google Scholar] [CrossRef]
- Buono, C.; Li, Y.; Waldo, S.W.; Kruth, H.S. Liver X receptors inhibit human monocyte-derived macrophage foam cell formation by inhibiting fluid-phase pinocytosis of LDL. J. Lipid Res. 2007, 48, 2411–2418. [Google Scholar] [CrossRef]
- Reiss, A.B.; Patel, C.A.; Rahman, M.M.; Chan, E.S.; Hasneen, K.; Montesinos, M.C.; Trachman, J.D.; Cronstein, B.N. Interferon-gamma impedes reverse cholesterol transport and promotes foam cell transformation in THP-1 human monocytes/macrophages. Med. Sci Monit. 2004, 10, 420–425. [Google Scholar]
- Hashizume, M.; Mihara, M. Atherogenic effects of TNF-α and IL-6 via up-regulation of scavenger receptors. Cytokine 2012, 58, 424–430. [Google Scholar] [CrossRef]
- Xu, Z.; Dong, A.; Feng, Z.; Li, J. Interleukin-32 promotes lipid accumulation through inhibition of cholesterol efflux. Exp. Ther. Med. 2017, 14, 947–952. [Google Scholar] [CrossRef][Green Version]
- Liu, Q.; Fan, J.; Bai, J.; Peng, L.; Zhang, T.; Deng, L.; Wang, G.; Zhao, Y.; Nong, J.; Zhang, M.; et al. IL-34 promotes foam cell formation by enhancing CD36 expression through p38 MAPK pathway. Sci. Rep. 2018, 8, 17347. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Wu, W.K.; Melnichenko, A.A.; Wetzker, R.; Sukhorukov, V.; Markin, A.M.; Khotina, V.A.; Orekhov, A.N. Signaling pathways and key genes involved in regulation of foam cell formation in atherosclerosis. Cells 2020, 9, 584. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Andreeva, E.R.; Bobryshev, Y.V. Cellular mechanisms of human atherosclerosis: Role of cell-to-cell communications in subendothelial cell functions. Tissue Cell 2016, 48, 25–34. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Tertov, V.V.; Kudryashov, S.A.; Smirnov, V.N. Triggerlike stimulation of cholesterol accumulation and DNA and extracellular matrix synthesis induced by atherogenic serum or low-density lipoprotein in cultured cells. Circ. Res. 1990, 66, 311–320. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Ivanova, E.A. Cellular models of atherosclerosis and their implication for testing natural substances with anti-atherosclerotic potential. Phytomedicine 2016, 23, 1190–1197. [Google Scholar] [CrossRef]
- Ishii, K.J.; Koyama, S.; Nakagawa, A.; Coban, C.; Akira, S. Host innate immune receptors and beyond: Making sense of microbial infections. Cell Host Microbe 2008, 3, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.T. Macrophage phagocytosis of neutrophils at inflammatory/infectious foci: A cooperative mechanism in the control of infection and infectious inflammation. J. Leukoc. Biol. 2011, 89, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Mayor, A.; Tschopp, J. The inflammasomes: Guardians of the body. Annu Rev. Immunol. 2009, 27, 229–265. [Google Scholar] [CrossRef]
- Yang, K.; Wang, X.; Liu, Z.; Lu, L.; Mao, J.; Meng, H.; Wang, Y.; Hu, Y.; Zeng, Y.; Zhang, X.; et al. Oxidized low-density lipoprotein promotes macrophage lipid accumulation via the toll-like receptor 4-Src pathway. Circ. J. 2015, 79, 2509–2516. [Google Scholar] [CrossRef]
- Wang, J.G.; Aikawa, M. Toll-like receptors and Src-family kinases in atherosclerosis—Focus on macrophages. Circ. J. 2015, 79, 2332–2334. [Google Scholar] [CrossRef]
- Hovland, A.; Jonasson, L.; Garred, P.; Yndestad, A.; Aukrust, P.; Lappegård, K.T.; Espevik, T.; Mollnes, T.E. The complement system and toll-like receptors as integrated players in the pathophysiology of atherosclerosis. Atherosclerosis 2015, 241, 480–494. [Google Scholar] [CrossRef]
- Karasawa, T.; Takahashi, M. Role of NLRP3 Inflammasomes in Atherosclerosis. J. Atheroscler. Thromb. 2017, 24, 443–451. [Google Scholar] [CrossRef]
- Matsuura, E.; Lopez, L.R.; Shoenfeld, Y.; Ames, P.R. β2-glycoprotein I and oxidative inflammation in early atherogenesis: A progression from innate to adaptive immunity? Autoimmun. Rev. 2012, 12, 241–249. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef]
- Afonina, I.S.; Zhong, Z.; Karin, M.; Beyaert, R. Limiting inflammation-the negative regulation of NF-kappaB and the NLRP3 inflammasome. Nat. Immunol. 2017, 18, 861–869. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef]
- Davis, B.K.; Wen, H.; Ting, J.P. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 2011, 29, 707–735. [Google Scholar] [CrossRef]
- De Zoete, M.R.; Palm, N.W.; Zhu, S.; Flavell, R.A. Inflammasomes. Cold Spring Harb. Perspect. Biol. 2014, 6, a016287. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. The role of mitochondria in NLRP3 inflammasome activation. Mol. Immunol. 2018, 103, 115–124. [Google Scholar] [CrossRef]
- Hoseini, Z.; Sepahvand, F.; Rashidi, B.; Sahebkar, A.; Masoudifar, A.; Mirzaei, H. NLRP3 inflammasome: Its regulation and involvement in atherosclerosis. J. Cell Physiol. 2018, 233, 2116–2132. [Google Scholar] [CrossRef]
- Gurung, P.; Lukens, J.R.; Kanneganti, T.D. Mitochondria: Diversity in the regulation of the NLRP3 inflammasome. Trends Mol. Med. 2015, 21, 193–201. [Google Scholar] [CrossRef]
- Kim, Y.G.; Kim, S.M.; Kim, K.P.; Lee, S.H.; Moon, J.Y. The Role of Inflammasome-Dependent and Inflammasome-Independent NLRP3 in the Kidney. Cells 2019, 8, 1389. [Google Scholar] [CrossRef]
- Vaamonde-García, C.; López-Armada, M.J. Role of mitochondrial ysfunction on rheumatic diseases. Biochem. Pharmacol. 2019, 165, 181–195. [Google Scholar] [CrossRef]
- Dela Cruz, C.S.; Kang, M.J. Mitochondrial dysfunction and damage associated molecular patterns (DAMPs) in chronic inflammatory diseases. Mitochondrion 2018, 41, 37–44. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, M. Phylogenomic Reconstruction Indicates Mitochondrial Ancestor Was an Energy Parasite. PLoS ONE 2014, 9, e110685. [Google Scholar] [CrossRef]
- Nicholls, T.J.; Minczuk, M. In D-loop: 40 years of mitochondrial 7S DNA. Exp. Gerontol. 2014, 56, 175–181. [Google Scholar] [CrossRef]
- Schroeder, P.; Gremmel, T.; Berneburg, M.; Krutmann, J. Partial depletion of mitochondrial DNA from human skin fibroblasts induces a gene expression profile reminiscent of photoaged skin. J. Investig. Dermatol. 2008, 128, 2297–2303. [Google Scholar] [CrossRef]
- Fukui, H.; Moraes, C.T. Mechanisms of formation and accumulation of mitochondrial DNA deletions in aging neurons. Hum. Mol. Genet. 2009, 18, 1028–1036. [Google Scholar] [CrossRef]
- Vermulst, M.; Wanagat, J.; Kujoth, G.C.; Bielas, J.H.; Rabinovitch, P.S.; Prolla, T.A.; Loeb, L.A. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat. Genet. 2008, 40, 392–394. [Google Scholar] [CrossRef]
- Larsson, N.G. Somatic mitochondrial DNA mutations in mammalian aging. Annu. Rev. Biochem. 2010, 79, 683–706. [Google Scholar] [CrossRef]
- Twig, G.; Hyde, B.; Shirihai, O.S. Mitochondrial fusion, fission and autophagy as a quality control axis: The bioenergetic view. Biochim. Biophys. Acta 2008, 1777, 1092–1097. [Google Scholar] [CrossRef]
- Osman, C.; Voelker, D.R.; Langer, T. Making heads or tails of phospholipids in mitochondria. J. Cell Biol. 2011, 192, 7–16. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef]
- Krysko, D.V.; Agostinis, P.; Krysko, O.; Garg, A.D.; Bachert, C.; Lambrecht, B.N.; Vandenabeele, P. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 2011, 32, 157–164. [Google Scholar] [CrossRef]
- Ghayur, T.; Banerjee, S.; Hugunin, M.; Butler, D.; Herzog, L.; Carter, A.; Quintal, L.; Sekut, L.; Talanian, R.; Paskind, M.; et al. Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature 1997, 386, 619–623. [Google Scholar] [CrossRef]
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 2013, 39, 311–323. [Google Scholar] [CrossRef]
- Yu, J.; Nagasu, H.; Murakami, T.; Hoang, H.; Broderick, L.; Hoffman, H.M.; Horng, T. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 15514–15519. [Google Scholar] [CrossRef]
- Fischer, F.; Hamann, A.; Osiewacz, H.D. Mitochondrial quality control: An integrated network of pathways. Trends Biochem. Sci. 2012, 37, 284–292. [Google Scholar] [CrossRef]
- Szklarczyk, R.; Nooteboom, M.; Osiewacz, H.D. Control of mitochondrial integrity in ageing and disease. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130439. [Google Scholar] [CrossRef]
- Tan, T.; Zimmermann, M.; Reichert, A.S. Controlling quality and amount of mitochondria by mitophagy: Insights into the role of ubiquitination and deubiquitination. Biol. Chem. 2016, 397, 637–647. [Google Scholar] [CrossRef]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-κB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef]
- Wu, N.N.; Zhang, Y.; Ren, J. Mitophagy, Mitochondrial Dynamics, and Homeostasis in Cardiovascular Aging. Oxid. Med. Cell. Longev. 2019, 2019, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Shirakabe, A.; Zhai, P.; Ikeda, Y.; Saito, T.; Maejima, Y.; Hsu, C.P.; Nomura, M.; Egashira, K.; Levine, B.; Sadoshima, J. Drp1-dependent mitochondrial autophagy plays a protective role against pressure overload-induced mitochondrial dysfunction and heart failure. Circulation 2016, 133, 1249–1263. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Feng, N.; Tang, D.; Feng, J.; Li, Z.; Jia, M.; Liu, Z.; Gu, X.; Wang, Y.; Fu, F.; et al. Melatonin prevents Drp1-mediated mitochondrial fission in diabetic hearts through SIRT1-PGC1α pathway. J. Pineal Res. 2018, 65, e12491. [Google Scholar] [CrossRef] [PubMed]
- Gkikas, I.; Palikaras, K.; Tavernarakis, N. The role of mitophagy in innate immunity. Front. Immunol. 2018, 9, 1283. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Shkurat, T.P.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. The role of mitochondrial dysfunction in cardiovascular disease: A brief review. Ann. Med. 2018, 50, 121–127. [Google Scholar] [CrossRef]
- Lee, H.C.; Yin, P.H.; Lin, J.C.; Wu, C.C.; Chen, C.Y.; Wu, C.W.; Chi, C.W.; Tam, T.N.; Wei, Y.H. Mitochondrial genome instability and mtDNA depletion in human cancers. Ann. N. Y. Acad. Sci. 2005, 1042, 109–122. [Google Scholar] [CrossRef]
- Petros, J.A.; Baumann, A.K.; Ruiz-Pesini, E.; Amin, M.B.; Sun, C.Q.; Hall, J.; Lim, S.; Issa, M.M.; Flanders, W.D.; Hosseini, S.H.; et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 719–724. [Google Scholar] [CrossRef]
- Shidara, Y.; Yamagata, K.; Kanamori, T.; Nakano, K.; Kwong, J.Q.; Manfredi, G.; Oda, H.; Ohta, S. Positive contribution of pathogenic mutations in the mitochondrial genome to the promotion of cancer by prevention from apoptosis. Cancer Res. 2005, 65, 1655–1663. [Google Scholar] [CrossRef]
- Higuchi, M.; Kudo, T.; Suzuki, S.; Evans, T.T.; Sasaki, R.; Wada, Y.; Shirakawa, T.; Sawyer, J.R.; Gotoh, A. Mitochondrial DNA determines androgen dependence in prostate cancer cell lines. Oncogene 2006, 25, 1437–1445. [Google Scholar] [CrossRef]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef]
- Kang, M.J.; Shadel, G.S. A mitochondrial perspective of chronic obstructive pulmonary disease pathogenesis. Tuberc. Respir. Dis. 2016, 79, 207–213. [Google Scholar] [CrossRef]
- Kang, M.J.; Yoon, C.M.; Kim, B.H.; Lee, C.M.; Zhou, Y.; Sauler, M.; Homer, R.; Dhamija, A.; Boffa, D.; West, A.P.; et al. Suppression of NLRX1 in chronic obstructive pulmonary disease. J. Clin. Investig. 2015, 125, 2458–2462. [Google Scholar] [CrossRef]
- Yoon, C.M.; Nam, M.; Oh, Y.M.; Dela Cruz, C.S.; Kang, M.J. Mitochondrial Regulation of Inflammasome Activation in Chronic Obstructive Pulmonary Disease. J. Innate Immun. 2016, 8, 121–128. [Google Scholar] [CrossRef]
- Puente-Maestu, L.; Pérez-Parra, J.; Godoy, R.; Moreno, N.; Tejedor, A.; González-Aragoneses, F.; Bravo, J.L.; Alvarez, F.V.; Camaño, S.; Agustí, A. Abnormal mitochondrial function in locomotor and respiratory muscles of COPD patients. Eur. Respir. J. 2009, 33, 1045–1052. [Google Scholar] [CrossRef]
- Rabinovich, R.A.; Bastos, R.; Ardite, E.; Llinàs, L.; Orozco-Levi, M.; Gea, J.; Vilaró, J.; Barberà, J.A.; Rodríguez-Roisin, R.; Fernández-Checa, J.C.; et al. Mitochondrial dysfunction in COPD patients with low body mass index. Eur. Respir. J. 2007, 29, 643–650. [Google Scholar] [CrossRef]
- Zhang, X.; Shan, P.; Homer, R.; Zhang, Y.; Petrache, I.; Mannam, P.; Lee, P.J. Cathepsin E promotes pulmonary emphysema via mitochondrial fission. Am. J. Pathol. 2014, 184, 2730–2741. [Google Scholar] [CrossRef]
- Cho, D.-H.; Kim, J.K.; Jo, E.-K. Mitophagy and Innate Immunity in Infection. Mol. Cells 2020, 43, 10–22. [Google Scholar]
- Mora, A.L.; Bueno, M.; Rojas, M. Mitochondria in the spotlight of aging and idiopathic pulmonary fibrosis. J. Clin. Investig. 2017, 127, 405–414. [Google Scholar] [CrossRef]
- Iyer, D.; Mishra, N.; Agrawal, A. Mitochondrial Function in Allergic Disease. Curr. Allergy Asthma Rep. 2017, 17, 865. [Google Scholar] [CrossRef]
- Kauppila, T.E.S.; Kauppila, J.H.K.; Larsson, N.G. Mammalian mitochondria and aging: An update. Cell Metab. 2017, 25, 57–71. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Inflammaging: Disturbed interplay between autophagy and inflammasomes. Aging 2012, 4, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Fivenson, E.M.; Lautrup, S.; Sun, N.; Scheibye-Knudsen, M.; Stevnsner, T.; Nilsen, H.; Bohr, V.A.; Fang, E.F. Mitophagy in neurodegeneration and aging. Neurochem. Int. 2017, 109, 202–209. [Google Scholar] [CrossRef]
- Lerner, C.A.; Sundar, I.K.; Rahman, I. Mitochondrial redox system, dynamics, and dysfunction in lung inflammaging and COPD. Int. J. Biochem. Cell Biol. 2016, 81, 294–306. [Google Scholar] [CrossRef]
- Fang, E.F.; Scheibye-Knudsen, M.; Brace, L.E.; Kassahun, H.; SenGupta, T.; Nilsen, H.; Mitchell, J.R.; Croteau, D.L.; Bohr, V.A. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell 2014, 157, 882–896. [Google Scholar] [CrossRef]
- Li, H.; Shen, L.; Hu, P.; Huang, R.; Cao, Y.; Deng, J.; Yuan, W.; Liu, D.; Yang, J.; Gu, H.; et al. Aging-associated mitochondrial DNA mutations alter oxidative phosphorylation machinery and cause mitochondrial dysfunctions. Biochem. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2266–2273. [Google Scholar] [CrossRef]
- Ding, Y.; Leng, J.; Fan, F.; Xia, B.; Xu, P. The role of mitochondrial DNA mutations in hearing loss. Biochem. Genet. 2013, 51, 588–602. [Google Scholar] [CrossRef]
- Gong, S.; Wang, X.; Meng, F.; Cui, L.; Yi, Q.; Zhao, Q.; Cang, X.; Cai, Z.; Mo, J.Q.; Liang, Y.; et al. Overexpression of mitochondrial histidyl-tRNA synthetase restores mitochondrial dysfunction caused by a deafness-associated tRNA(His) mutation. J. Biol. Chem. 2020, 295, 940–954. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Poznyak, A.V.; Sobenin, I.A.; Nikifirov, N.N.; Ivanova, E.A. Mitochondrion as a selective target for treatment of atherosclerosis: Role of mitochondrial DNA mutations and defective mitophagy in the pathogenesis of atherosclerosis and chronic inflammation. Curr. Neuropharmacol. 2019, 17, 1. [Google Scholar] [CrossRef]
- Sazonova, M.A.; Sinyov, V.V.; Ryzhkova, A.I.; Galitsyna, E.V.; Khasanova, Z.B.; Postnov, A.Y.; Yarygina, E.I.; Orekhov, A.N.; Sobenin, I.A. Role of Mitochondrial Genome Mutations in Pathogenesis of Carotid Atherosclerosis. Oxid. Med. Cell. Longev. 2017, 2017, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sobenin, I.A.; Zhelankin, A.V.; Mitrofanov, K.Y.; Sinyov, V.V.; Sazonova, M.A.; Postnov, A.Y.; Orekhov, A.N. Mutations of mitochondrial DNA in atherosclerosis and atherosclerosis-related diseases. Curr. Pharm. Des. 2015, 21, 1158–1163. [Google Scholar] [CrossRef] [PubMed]
- Sobenin, I.A.; Sazonova, M.A.; Postnov, A.Y.; Salonen, J.T.; Bobryshev, Y.V.; Orekhov, A.N. Association of mitochondrial genetic variation with carotid atherosclerosis. PLoS ONE 2013, 8, e68070. [Google Scholar] [CrossRef] [PubMed]
- Andreassi, M.G. Coronary atherosclerosis and somatic mutations: An overview of the contributive factors for oxidative DNA damage. Mutat. Res. 2003, 543, 67–86. [Google Scholar] [CrossRef]
- Avital, G.; Buchshtav, M.; Zhidkov, I.; Tuval Feder, J.; Dadon, S.; Rubin, E.; Glass, D.; Spector, T.D.; Mishmar, D. Mitochondrial DNA heteroplasmy in diabetes and normal adults: Role of acquired and inherited mutationalpatterns in twins. Hum. Mol. Genet. 2012, 21, 4214–4224. [Google Scholar] [CrossRef]
- Marian, A.J. Mitochondrial genetics and human systemic hypertension. Circ. Res. 2011, 108, 784–786. [Google Scholar] [CrossRef]
- Nomiyama, T.; Tanaka, Y.; Piao, L.; Hattori, N.; Uchino, H.; Watada, H.; Kawamori, R.; Ohta, S. Accumulation of somatic mutation in mitochondrial DNA and atherosclerosis in diabetic patients. Ann. N. Y. Acad. Sci. 2004, 1011, 193–204. [Google Scholar] [CrossRef]
- Yu, E.; Foote, K.; Bennett, M. Mitochondrial function in thoracic aortic aneurysms. Cardiovasc. Res. 2018, 114, 1696–1698. [Google Scholar] [CrossRef]
- Sazonova, M.A.; Sinyov, V.V.; Barinova, V.A.; Ryzhkova, A.I.; Zhelankin, A.V.; Postnov, A.Y.; Sobenin, I.A.; Bobryshev, Y.V.; Orekhov, A.N. Mosaicism of mitochondrial genetic variation in atherosclerotic lesions of the human aorta. BioMed Res. Int. 2015, 2015, 1–9. [Google Scholar] [CrossRef]
- Han, C.-B.; Ma, J.-M.; Xin, Y.; Mao, X.Y.; Zhao, Y.J.; Wu, D.Y.; Zhang, S.M.; Zhang, Y.K. Mutations of mitochondrial 12S rRNA in gastric carcinoma and their significance. World J. Gastroenterol. 2005, 11, 31–35. [Google Scholar] [CrossRef]
- Li, R.; Xing, G.; Yan, M.; Cao, X.; Liu, X.Z.; Bu, X.; Guan, M.X. Cosegregation of C-insertion at position 961 with the A1555G mutation of the mitochondrial 12S rRNA gene in a large Chinese family with maternally inherited hearing loss. Am. J. Med. Genet. 2004, 124, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Giordano, C.; Pallotti, F.; Walker, W.F.; Checcarelli, N.; Musumeci, O.; Santorelli, F.; d’Amati, G.; Schon, E.A.; DiMauro, S.; Hirano, M.; et al. Pathogenesis of the deafness-associated A1555G mitochondrial DNA mutation. Biochem. Biophys. Res. Commun. 2002, 293, 521–529. [Google Scholar] [CrossRef]
- Bykhovskaya, Y.; Shohat, M.; Ehrenman, K.; Johnson, D.; Hamon, M.; Cantor, R.M.; Aouizerat, B.; Bu, X.; Rotter, J.I.; Jaber, L.; et al. Evidence for complex nuclear inheritance in a pedigree with nonsyndromic deafness due to a homoplasmic mitochondrial mutation. Am. J. Med. Genet. 1998, 77, 421–426. [Google Scholar] [CrossRef]
- Jeppesen, D.; Schwartz, M.; Hansen, K.; Danielsen, E.R.; Wibrand, F.; Vissing, J. Late onset of stroke-like episode associated with a 3256C-T point mutation of mitochondrial DNA. J. Neurol. Sci. 2003, 214, 17–20. [Google Scholar] [CrossRef]
- Matsunaga, H.; Tanaka, Y.; Tanaka, M.; Gong, J.S.; Zhang, J.; Nomiyama, T.; Ogawa, O.; Ogihara, T.; Yamada, Y.; Yagi, K.; et al. Antiatherogenic mitochondrial genotype in patients with type 2 diabetes. Diabetes Care 2001, 24, 500–503. [Google Scholar] [CrossRef]
- Mukae, S.; Aoki, S.; Itoh, S.; Sato, R.; Nishio, K.; Iwata, T.; Katagiri, T. Mitochondrial 5178A/C genotype is associated with acute myocardial infarction. Circ. J. 2003, 67, 16–20. [Google Scholar] [CrossRef]
- Fu, K.; Hartlen, R.; Johns, T.; Genge, A.; Karpati, G.; Shoubridge, E.A. A novel heteroplasmic tRNA(leu(CUN)) mtDNA point mutation in a sporadic patient with mitochondrial encephalomyopathy segregates rapidly in skeletal muscle and suggests an approach to therapy. Hum. Mol. Genet. 1996, 5, 1835–1840. [Google Scholar] [CrossRef]
- Chol, M.; Lebon, S.; Benit, P.; Chretien, D.; de Lonlay, P.; Goldenberg, A.; Odent, S.; Hertz-Pannier, L.; Vincent-Delorme, C.; Cornier-Daire, V.; et al. The mitochondrial DNA ’G13513A MELAS mutation in the NADH dehydrogenase 5 gene is a frequent cause of Leigh-like syndrome with isolated complex I deficiency. J. Med. Genet. 2003, 40, 188–191. [Google Scholar] [CrossRef]
- Ruiter, E.M.; Siers, M.H.; van den Elzen, C.; van Engelen, B.G.; Smeitink, J.A.; Rodenburg, R.J.; Hol, F.A. The mitochondrial 13513G>A mutation is most frequent in Leigh syndrome combined with reduced complex I activity, optic atrophy and/or Wolff-Parkinson-White. Eur. J. Hum. Genet. 2007, 15, 155–161. [Google Scholar] [CrossRef]
- Brown, M.D.; Voljavec, A.S.; Lott, M.T.; Torroni, A.; Yang, C.C.; Wallace, D.C. Mitochondrial DNA complex I and III mutations associated with Leber’s hereditary optic neuropathy. Genetics 1992, 130, 163–173. [Google Scholar]
- Andreu, A.L.; Bruno, C.; Shanske, S.; Shtilbans, A.; Hirano, M.; Krishna, S.; Hayward, L.; Systrom, D.S.; Brown, R.H., Jr.; DiMauro, S. Missense mutation in the mtDNA cytochrome b gene in a patient with myopathy. Neurology 1998, 51, 1444–1447. [Google Scholar] [CrossRef] [PubMed]
- Sazonova, M.; Budnikov, E.; Khasanova, Z.; Sobenin, I.; Postnov, A.; Orekhov, A. Studies of the human aortic intima by a direct quantitative assay of mutant alleles in the mitochondrial genome. Atherosclerosis 2009, 204, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Orekhov, A.N.; Zhelankin, A.V.; Kolmychkova, K.I.; Mitrofanov, K.Y.; Kubekina, M.V.; Ivanova, E.A.; Sobenin, I.A. Susceptibility of monocytes to activation correlates with atherogenic mitochondrial DNA mutations. Exp. Mol. Pathol. 2015, 99, 672–676. [Google Scholar] [CrossRef]
- Dominguez-Andres, J.; Netea, M.G. Long-term reprogramming of the innate immune system. J. Leukoc. Biol. 2019, 105, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G. Training innate immunity: The changing concept of immunological memory in innate host defence. Eur. J. Clin. Investig. 2013, 43, 881–884. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 2015, 521, 525–528. [Google Scholar] [CrossRef]
- Xu, Y.; Shen, J.; Ran, Z. Emerging views of mitophagy in immunity and autoimmune diseases. Autophagy 2019, 16, 3–17. [Google Scholar] [CrossRef]
- Montava-Garriga, L.; Ganley, I.G. Outstanding questions in mitophagy; what we do and do not know. J. Mol. Biol. 2020, 432, 206–230. [Google Scholar] [CrossRef]
- Zhang, J.; Ji, Y.; Lu, Y.; Fu, R.; Xu, M.; Liu, X.; Guan, M.X. Leber’s hereditary optic neuropathy (LHON)-associated ND5 12338T > C mutation altered the assembly and function of complex I, apoptosis and mitophagy. Hum. Mol. Genet. 2018, 27, 1999–2011. [Google Scholar] [CrossRef]
- Sharma, L.K.; Tiwari, M.; Rai, N.K.; Bai, Y. Mitophagy activation repairs Leber’s hereditary optic neuropathy-associated mitochondrial dysfunction and improves cell survival. Hum. Mol. Genet. 2019, 28, 422–433. [Google Scholar] [CrossRef]
- Orogo, A.M.; Gonzalez, E.R.; Kubli, D.A.; Baptista, I.L.; Ong, S.B.; Prolla, T.A.; Sussman, M.A.; Murphy, A.N.; Gustafsson, Å.B. Accumulation of Mitochondrial DNA Mutations Disrupts Cardiac Progenitor Cell Function and Reduces Survival. J. Biol. Chem. 2015, 290, 22061–22075. [Google Scholar] [CrossRef] [PubMed]
- Granatiero, V.; Giorgio, V.; Calì, T.; Patron, M.; Brini, M.; Bernardi, P.; Tiranti, V.; Zeviani, M.; Pallafacchina, G.; De Stefani, D.; et al. Reduced mitochondrial Ca(2+) transients stimulate autophagy in human fibroblasts carrying the 13514A>G mutation of the ND5 subunit of NADH dehydrogenase. Cell Death Differ. 2016, 23, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Gilkerson, R.W.; De Vries, R.L.; Lebot, P.; Wikstrom, J.D.; Torgyekes, E.; Shirihai, O.S.; Przedborski, S.; Schon, E.A. Mitochondrial autophagy in cells with mtDNA mutations results from synergistic loss of transmembrane potential and mTORC1 inhibition. Hum. Mol. Genet. 2012, 21, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.H.; Luo, S.F.; Ho, L.J. Operation of mitochondrial machinery in viral infection-induced immune responses. Biochem. Pharmacol. 2018, 156, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Rongvaux, A. Innate immunity and tolerance toward mitochondria. Mitochondrion 2018, 41, 14–20. [Google Scholar] [CrossRef]
- Zhang, L.; Qin, Y.; Chen, M. Viral strategies for triggering and manipulating mitophagy. Autophagy 2018, 14, 1665–1673. [Google Scholar] [CrossRef]
- Mohanty, A.; Tiwari-Pandey, R.; Pandey, N.R. Mitochondria: The indispensable players in innate immunity and guardians of the inflammatory response. J. Cell Commun. Signal. 2019, 13, 303–318. [Google Scholar] [CrossRef]
- Tertov, V.V.; Sobenin, I.A.; Gabbasov, Z.A.; Popov, E.G.; Jaakkola, O.; Solakivi, T.; Nikkari, T.; Smirnov, V.N.; Orekhov, A.N. Multiple-modified desialylated low density lipoproteins that cause intracellular lipid accumulation. Isolation, fractionation and characterization. Lab. Investig. 1992, 67, 665–675. [Google Scholar]
- Orekhov, A.N.; Nikiforov, N.G.; Elizova, N.V.; Korobov, G.A.; Aladinskaya, A.V.; Sobenin, I.A.; Bobryshev, Y.V. Tumor Necrosis Factor-α and C-C Motif Chemokine Ligand 18 Associate with Atherosclerotic Lipid Accumulation In situ and In vitro. Curr. Pharm. Des. 2018, 24, 2883–2889. [Google Scholar] [CrossRef]
- Meyer, A.; Laverny, G.; Bernardi, L.; Charles, A.L.; Alsaleh, G.; Pottecher, J.; Sibilia, J.; Geny, B. Mitochondria: An Organelle of Bacterial Origin Controlling Inflammation. Front. Immunol. 2018, 9, 536. [Google Scholar] [CrossRef]
- Kiyuna, L.A.; Albuquerque, R.P.E.; Chen, C.H.; Mochly-Rosen, D.; Ferreira, J.C.B. Targeting mitochondrial dysfunction and oxidative stress in heart failure: Challenges and opportunities. Free. Radic. Biol. Med. 2018, 129, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Sinyov, V.V.; Sazonova, M.A.; Ryzhkova, A.I.; Galitsyna, E.V.; Melnichenko, A.A.; Postnov, A.Y.; Orekhov, A.N.; Grechko, A.V.; Sobenin, I.A. Potential use of buccal epithelium for genetic diagnosis of atherosclerosis using mtDNA mutations. Vessel Plus 2017, 1, 145–150. [Google Scholar] [CrossRef][Green Version]











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orekhov, A.N.; Nikiforov, N.N.; Ivanova, E.A.; Sobenin, I.A. Possible Role of Mitochondrial DNA Mutations in Chronification of Inflammation: Focus on Atherosclerosis. J. Clin. Med. 2020, 9, 978. https://doi.org/10.3390/jcm9040978
Orekhov AN, Nikiforov NN, Ivanova EA, Sobenin IA. Possible Role of Mitochondrial DNA Mutations in Chronification of Inflammation: Focus on Atherosclerosis. Journal of Clinical Medicine. 2020; 9(4):978. https://doi.org/10.3390/jcm9040978
Chicago/Turabian StyleOrekhov, Alexander N., Nikita N. Nikiforov, Ekaterina A. Ivanova, and Igor A. Sobenin. 2020. "Possible Role of Mitochondrial DNA Mutations in Chronification of Inflammation: Focus on Atherosclerosis" Journal of Clinical Medicine 9, no. 4: 978. https://doi.org/10.3390/jcm9040978
APA StyleOrekhov, A. N., Nikiforov, N. N., Ivanova, E. A., & Sobenin, I. A. (2020). Possible Role of Mitochondrial DNA Mutations in Chronification of Inflammation: Focus on Atherosclerosis. Journal of Clinical Medicine, 9(4), 978. https://doi.org/10.3390/jcm9040978