Auditory Neuropathy Spectrum Disorders: From Diagnosis to Treatment: Literature Review and Case Reports

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Prevalence of ANSD
3. Etiologies
3.1. Genetic Etiologies
3.1.1. Presynaptic Synaptopathies
3.1.2. Postsynaptic Synaptopathies
3.1.3. Auditory Neuropathy
3.1.4. Synaptopathy and Neuropathy
4. Psychoacoustic Tests
4.1. Tonal and Speech Audiometry Thresholds
4.2. Supraliminal Tests
5. Objective Assessment of ANSD
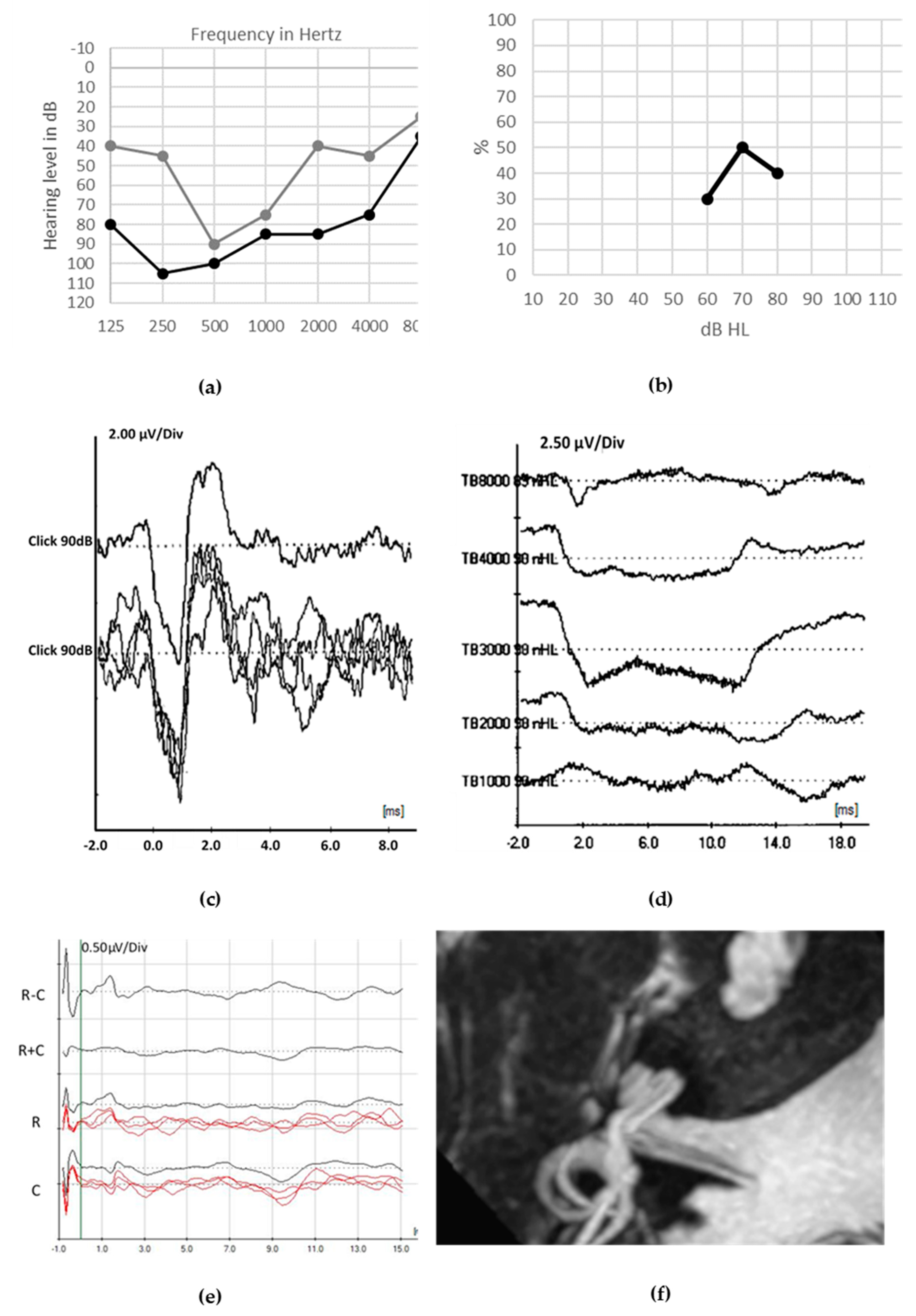
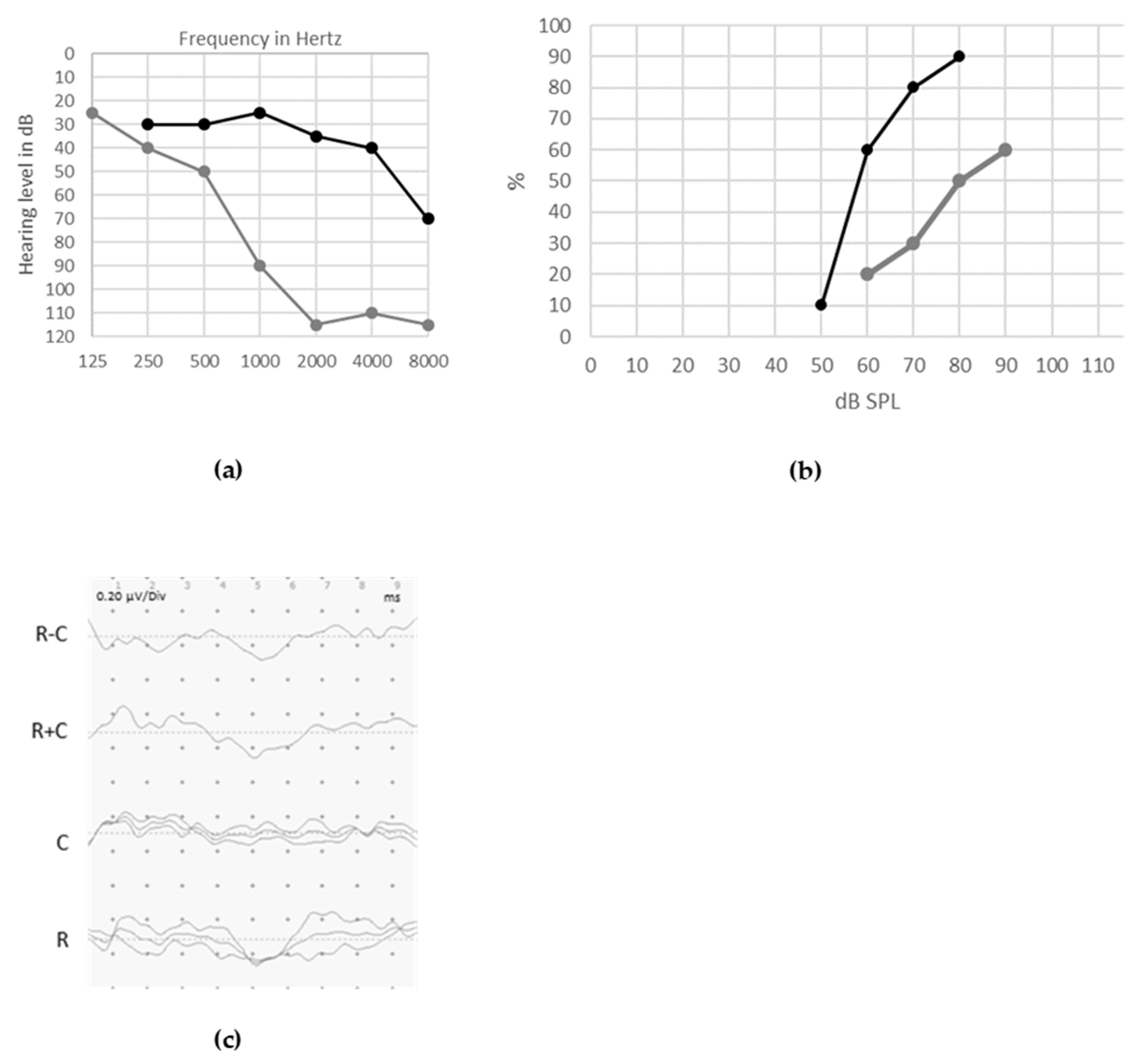
6. Therapy and Outcomes
- Signal-to-noise ratio maximization
- Amplification: Hearing aids and cochlear implant
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABRs | Auditory Brainstem Responses |
| AN/AD | Auditory Neuropathy/Auditory Dys-synchrony |
| ANN | Auditory Nerve Neurophonic |
| ANSD | Auditory Neuropathy Spectrum Disorder |
| ASSRs | Auditory Steady-State Responses |
| C | Condensation |
| CAEPs | Cortical Auditory Evoked Potentials |
| CAP | Compound Action Potential |
| CM | Cochlear Microphonic |
| CMT | Charcot–Marie–Tooth disease |
| CNS | Central Nervous System |
| DOA | Dominant Optic Atrophy |
| DPOAEs | Distortion-Product Otoacoustic Emissions |
| ECochG | Electrocochleography |
| HHL | Hidden Hearing Loss |
| HSMN | Hereditary Sensori-Motor Neuropathy |
| IHCs | Inner Hair Cells |
| MMN | Mismatch Negativity |
| NICU | Neonatal Intensive Care Unit |
| OAEs | Otoacoustic Emissions |
| OHCs | Outer Hair Cells |
| R | Rarefaction |
| SANDD | Sinoatrial Node Dysfunction and Deafness |
| SGNs | Spiral Ganglion Neurons |
| SNHL | Sensorineural Hearing Loss |
| SP | Summating Potential |
| TEOAEs | Transient-Evoked Otoacoustic Emissions |
References
- Rance, G. Auditory Neuropathy/Dys-Synchrony and Its Perceptual Consequences. Trends Amplif. 2005, 9, 1–43. [Google Scholar] [CrossRef] [PubMed]
- Berlin, C.I.; Morlet, T.; Hood, L.J. Auditory Neuropathy/Dyssynchrony: Its Diagnosis and Management. Pediatr. Clin. N. Am. 2003, 50, 331–340. [Google Scholar] [CrossRef]
- Berlin, C.I.; Hood, L.J.; Morlet, T.; Wilensky, D.; Li, L.; Mattingly, K.R.; Taylor-Jeanfreau, J.; Keats, B.J.B.; John, P.S.; Montgomery, E.; et al. Multi-Site Diagnosis and Management of 260 Patients with Auditory Neuropathy/Dys-Synchrony (Auditory Neuropathy Spectrum Disorder). Int. J. Audiol. 2010, 49, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.G.; Kong, Y.Y.; Michalewski, H.J.; Starr, A. Perceptual Consequences of Disrupted Auditory Nerve Activity. J. Neurophysiol. 2005, 93, 3050–3063. [Google Scholar] [CrossRef]
- Fontenot, T.E.; Giardina, C.K.; Teagle, H.F.; Park, L.R.; Adunka, O.F.; Buchman, C.A.; Brown, K.D.; Fitzpatrick, D.C. Clinical Role of Electrocochleography in Children with Auditory Neuropathy Spectrum Disorder. Int. J. Pediatr. Otorhinolaryngol. 2017, 99, 120–127. [Google Scholar] [CrossRef]
- Kitao, K.; Mutai, H.; Namba, K.; Morimoto, N.; Nakano, A.; Arimoto, Y.; Sugiuchi, T.; Masuda, S.; Okamoto, Y.; Morita, N.; et al. Deterioration in Distortion Product Otoacoustic Emissions in Auditory Neuropathy Patients with Distinct Clinical and Genetic Backgrounds. Ear Hear. 2019, 40, 184–191. [Google Scholar] [CrossRef]
- Starr, A.; Picton, T.W.; Sininger, Y.; Hood, L.J.; Berlin, C.I. Auditory Neuropathy. Brain 1996, 119, 741–753. [Google Scholar] [CrossRef]
- Starr, A.; Dong, C.J.; Michalewski, H.J. Brain Potentials before and during Memory Scanning. Electroencephalogr. Clin. Neurophysiol. 1996, 99, 28–37. [Google Scholar] [CrossRef][Green Version]
- Zeng, F.G.; Oba, S.; Garde, S.; Sininger, Y.; Starr, A. Temporal and Speech Processing Deficits in Auditory Neuropathy. Neuroreport 1999, 10, 3429–3435. [Google Scholar] [CrossRef]
- Berlin, C.I.; Hood, L.J.; Rose, K. On Renaming Audi- Tory Neuropathy as Auditory Dys-Synchrony. Audiol. Today 2001, 13, 15–17. [Google Scholar]
- Santarelli, R.; del Castillo, I.; Cama, E.; Scimemi, P.; Starr, A. Audibility, Speech Perception and Processing of Temporal Cues in Ribbon Synaptic Disorders Due to OTOF Mutations. Hear. Res. 2015, 200–212. [Google Scholar] [CrossRef]
- Moser, T.; Starr, A. Auditory Neuropathy-Neural and Synaptic Mechanisms. In Nature Reviews Neurology; Nature Publishing Group: Berlin, Germany, 2016; pp. 135–149. [Google Scholar]
- Shearer, A.E.; Hansen, M.R. Auditory Synaptopathy, Auditory Neuropathy, and Cochlear Implantation. Laryngoscope Investig. Otolaryngol. 2019, 4, 429–440. [Google Scholar] [CrossRef]
- Foerst, A.; Beutner, D.; Lang-Roth, R.; Huttenbrink, K.B.; von Wedel, H.; Walger, M. Prevalence of Auditory Neuropathy/Synaptopathy in a Population of Children with Profound Hearing Loss. Int. J. Pediatr. Otorhinolaryngol. 2006, 70, 1415–1422. [Google Scholar] [CrossRef]
- Sergeyenko, Y.; Lall, K.; Charles Liberman, M.; Kujawa, S.G. Age-Related Cochlear Synaptopathy: An Early-Onset Contributor to Auditory Functional Decline. J. Neurosci. 2013, 33, 13686–13694. [Google Scholar] [CrossRef]
- Guest, H.; Munro, K.J.; Prendergast, G.; Millman, R.E.; Plack, C.J. Impaired Speech Perception in Noise with a Normal Audiogram: No Evidence for Cochlear Synaptopathy and No Relation to Lifetime Noise Exposure. Hear. Res. 2018, 364, 142–151. [Google Scholar] [CrossRef]
- Kujawa, S.G.; Liberman, M.C. Adding Insult to Injury: Cochlear Nerve Degeneration after “Temporary” Noise-Induced Hearing Loss. J. Neurosci. 2009, 29, 14077–14085. [Google Scholar] [CrossRef]
- Chen, G.D. Hidden Cochlear Impairments. J. Otol. 2018, 13, 37–43. [Google Scholar] [CrossRef]
- Xiong, B.; Liu, Z.; Liu, Q.; Peng, Y.; Wu, H.; Lin, Y.; Zhao, X.; Sun, W. Missed Hearing Loss in Tinnitus Patients with Normal Audiograms. Hear. Res. 2019, 384, 107826. [Google Scholar] [CrossRef]
- Iliadou, V.V.; Ptok, M.; Grech, H.; Pedersen, E.R.; Brechmann, A.A.; Deggouj, N.N.; Kiese-Himmel, C.; Sliwinska-Kowalska, M.; Nickisch, A.; Demanez, L.; et al. A European Perspective on Auditory Processing Disorder-Current Knowledge and Future Research Focus. Front. Neurol. 2017, 8, 622. [Google Scholar] [CrossRef]
- Vignesh, S.S.; Jaya, V.; Muraleedharan, A. Prevalence and Audiological Characteristics of Auditory Neuropathy Spectrum Disorder in Pediatric Population: A Retrospective Study. Indian J. Otolaryngol. Head Neck Surg. 2016, 68, 196–201. [Google Scholar] [CrossRef]
- Boudewyns, A.; Declau, F.; van den Ende, J.; Hofkens, A.; Dirckx, S.; Van de Heyning, P. Auditory Neuropathy Spectrum Disorder (ANSD) in Referrals from Neonatal Hearing Screening at a Well-Baby Clinic. Eur. J. Pediatr. 2016, 175, 993–1000. [Google Scholar] [CrossRef]
- Penido, R.C.; Isaac, M.L. Prevalence of Auditory Neuropathy Spectrum Disorder in an Auditory Health Care Service. Braz. J. Otorhinolaryngol. 2013, 79, 429–433. [Google Scholar] [CrossRef]
- Robertson, C.M.T.; Howarth, T.M.; Bork, D.L.R.; Dinu, I.A. Permanent Bilateral Sensory and Neural Hearing Loss of Children after Neonatal Intensive Care Because of Extreme Prematurity: A Thirty-Year Study. Pediatrics 2009, 123, e797–e807. [Google Scholar] [CrossRef]
- Vohr, B.R.; Widen, J.E.; Cone-Wesson, B.; Sininger, Y.S.; Gorga, M.P.; Folsom, R.C.; Norton, S.J. Identification of Neonatal Hearing Impairment: Characteristics of Infants in the Neonatal Intensive Care Unit and Well-Baby Nursery. Ear Hear. 2000, 21, 373–382. [Google Scholar] [CrossRef]
- Schulman Galambos, C.; Galambos, R. Brain Stem Evoked Response Audiometry in Newborn Hearing Screening. Arch. Otolaryngol. 1979, 105, 86–90. [Google Scholar] [CrossRef]
- Amatuzzi, M.; Liberman, M.C.; Northrop, C. Selective Inner Hair Cell Loss in Prematurity: A Temporal Bone Study of Infants from a Neonatal Intensive Care Unit. JARO J. Assoc. Res. Otolaryngol. 2011, 12, 595–604. [Google Scholar] [CrossRef]
- Berg, A.L.; Spitzer, J.B.; Towers, H.M.; Bartosiewicz, C.; Diamond, B.E. Newborn Hearing Screening in the NICU: Profile of Failed Auditory Brainstem Response/Passed Otoacoustic Emission. Pediatrics 2005, 116, 933–938. [Google Scholar] [CrossRef]
- Xoinis, K.; Weirather, Y.; Mavoori, H.; Shaha, S.H.; Iwamoto, L.M. Extremely Low Birth Weight Infants Are at High Risk for Auditory Neuropathy. J. Perinatol. 2007, 27, 718–723. [Google Scholar] [CrossRef]
- Bielecki, I.; Horbulewicz, A.; Wolan, T. Prevalence and Risk Factors for Auditory Neuropathy Spectrum Disorder in a Screened Newborn Population at Risk for Hearing Loss. Int. J. Pediatr. Otorhinolaryngol. 2012, 76, 1668–1670. [Google Scholar] [CrossRef]
- Fuchs, P.A. Time and Intensity Coding at the Hair Cell’s Ribbon Synapse. J. Physiol. 2005, 566, 7–12. [Google Scholar] [CrossRef]
- Moser, T.; Neef, A.; Khimich, D. Mechanisms Underlying the Temporal Precision of Sound Coding at the Inner Hair Cell Ribbon Synapse. J. Physiol. 2006, 576, 55–62. [Google Scholar] [CrossRef]
- Kim, K.X.; Rutherford, M.A. Maturation of Nav and Kv Channel Topographies in the Auditory Nerve Spike Initiator before and after Developmental Onset of Hearing Function. J. Neurosci. 2016, 36, 2111–2118. [Google Scholar] [CrossRef]
- Nayagam, B.A.; Muniak, M.A.; Ryugo, D.K. The Spiral Ganglion: Connecting the Peripheral and Central Auditory Systems. Hear. Res. 2011, 278, 2–20. [Google Scholar] [CrossRef]
- Jeon, E.J.; Xu, N.; Xu, L.; Hansen, M.R. Influence of Central Glia on Spiral Ganglion Neuron Neurite Growth. Neuroscience 2011, 177, 321–334. [Google Scholar] [CrossRef]
- Jung, S.; Maritzen, T.; Wichmann, C.; Jing, Z.; Neef, A.; Revelo, N.H.; Al-Moyed, H.; Meese, S.; Wojcik, S.M.; Panou, I.; et al. Disruption of Adaptor Protein 2μ (AP -2μ) in Cochlear Hair Cells Impairs Vesicle Reloading of Synaptic Release Sites and Hearing. EMBO J. 2015, 34, 2686–2702. [Google Scholar] [CrossRef]
- Pangšrič, T.; Lasarow, L.; Reuter, K.; Takago, H.; Schwander, M.; Riedel, D.; Frank, T.; Tarantino, L.M.; Bailey, J.S.; Strenzke, N.; et al. Hearing Requires Otoferlin-Dependent Efficient Replenishment of Synaptic Vesicles in Hair Cells. Nat. Neurosci. 2010, 13, 869–876. [Google Scholar] [CrossRef]
- Khimich, D.; Nouvtan, R.; Pujol, R.; Diesk, S.T.; Egner, A.; Gundelfinger, E.D.; Moser, T. Hair Cell Synaptic Ribbons Are Essential for Synchronous Auditory Signalling. Nature 2005, 434, 889–894. [Google Scholar] [CrossRef]
- Parkinson, N.J.; Olsson, C.L.; Hallows, J.L.; Mckee-Johnson, J.; Keogh, B.P.; Noben-Trauth, K.; Kujawa, S.G.; Tempel, B.L. Mutant β-Spectrin 4 Causes Auditory and Motor Neuropathies in Quivering Mice. Nat. Genet. 2001, 29, 61–65. [Google Scholar] [CrossRef]
- Buran, B.N.; Strenzke, N.; Neef, A.; Gundelfinger, E.D.; Moser, T.; Liberman, M.C. Onset Coding Is Degraded in Auditory Nerve Fibers from Mutant Mice Lacking Synaptic Ribbons. J. Neurosci. 2010, 30, 7587–7597. [Google Scholar] [CrossRef]
- Starr, A.; Sininger, Y.; Nguyen, T.; Michalewski, H. Cochlear Receptor and Auditory Pathway Activity in Auditory Neuropathy. Ear Hear. 2001, 22, 91–99. [Google Scholar] [CrossRef]
- Ehrmann-Müller, D.; Cebulla, M.; Rak, K.; Scheich, M.; Back, D.; Hagen, R.; Shehata-Dieler, W. Evaluation and Therapy Outcome in Children with Auditory Neuropathy Spectrum Disorder (ANSD). Int. J. Pediatr. Otorhinolaryngol. 2019, 127, 109618. [Google Scholar] [CrossRef]
- Riggs, W.J.; Roche, J.P.; Giardina, C.K.; Harris, M.S.; Bastian, Z.J.; Fontenot, T.E.; Buchman, C.A.; Brown, K.D.; Adunka, O.F.; Fitzpatrick, D.C. Intraoperative Electrocochleographic Characteristics of Auditory Neuropathy Spectrum Disorder in Cochlear Implant Subjects. Front. Neurosci. 2017, 11, 416. [Google Scholar] [CrossRef] [PubMed]
- Attias, J.; Raveh, E.; Aizer-Dannon, A.; Bloch-Mimouni, A.; Fattal-Valevski, A. Auditory System Dysfunction Due to Infantile Thiamine Deficiency: Long-Term Auditory Sequelae. Audiol. Neurotol. 2012, 17, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Yasunaga, S.; Grati, M.; Cohen-Salmon, M.; El-Amraoui, A.; Mustapha, M.; Salem, N.; El-Zir, E.; Loiselet, J.; Petit, C. A Mutation in OTOF, Encoding Otoferlin, a FER-1-like Protein, Causes DFNB9, a Nonsyndromic Form of Deafness. Nat. Genet. 1999, 21, 363–369. [Google Scholar] [CrossRef]
- PangrŠič, T.; Reisinger, E.; Moser, T. Otoferlin: A Multi-C 2 Domain Protein Essential for Hearing. Trends Neurosci. 2012, 35, 671–680. [Google Scholar] [CrossRef]
- Roux, I.; Safieddine, S.; Nouvian, R.; Grati, M.; Simmler, M.C.; Bahloul, A.; Perfettini, I.; Le Gall, M.; Rostaing, P.; Hamard, G.; et al. Otoferlin, Defective in a Human Deafness Form, Is Essential for Exocytosis at the Auditory Ribbon Synapse. Cell 2006, 127, 277–289. [Google Scholar] [CrossRef]
- Johnson, C.P.; Chapman, E.R. Otoferlin Is a Calcium Sensor That Directly Regulates SNARE-Mediated Membrane Fusion. J. Cell Biol. 2010, 191, 187–197. [Google Scholar] [CrossRef]
- Padmanarayana, M.; Hams, N.; Speight, L.C.; Petersson, E.J.; Mehl, R.A.; Johnson, C.P. Characterization of the Lipid Binding Properties of Otoferlin Reveals Specific Interactions between PI(4,5)P2 and the C2C and C2F Domains. Biochemistry 2014, 53, 5023–5033. [Google Scholar] [CrossRef]
- Fuson, K.; Rice, A.; Mahling, R.; Snow, A.; Nayak, K.; Shanbhogue, P.; Meyer, A.G.; Redpath, G.M.I.; Hinderliter, A.; Cooper, S.T.; et al. Alternate Splicing of Dysferlin C2A Confers Ca2+-Dependent and Ca2+-Independent Binding for Membrane Repair. Structure 2014, 22, 104–115. [Google Scholar] [CrossRef]
- Jiménez, J.L.; Bashir, R. In Silico Functional and Structural Characterisation of Ferlin Proteins by Mapping Disease-Causing Mutations and Evolutionary Information onto Three-Dimensional Models of Their C2 Domains. J. Neurol. Sci. 2007, 260, 114–123. [Google Scholar] [CrossRef]
- Dulon, D.; Safieddine, S.; Jones, S.M.; Petit, C. Otoferlin Is Critical for a Highly Sensitive and Linear Calcium-Dependent Exocytosis at Vestibular Hair Cell Ribbon Synapses. J. Neurosci. 2009, 29, 10474–10487. [Google Scholar] [CrossRef]
- Varga, R.; Avenarius, M.R.; Kelley, P.M.; Keats, B.J.; Berlin, C.I.; Hood, L.J.; Morlet, T.G.; Brashears, S.M.; Starr, A.; Cohn, E.S.; et al. OTOF Mutations Revealed by Genetic Analysis of Hearing Loss Families Including a Potential Temperature Sensitive Auditory Neuropathy Allele. J. Med. Genet. 2006, 43, 576–581. [Google Scholar] [CrossRef]
- Wynne, D.P.; Zeng, F.G.; Bhatt, S.; Michalewski, H.J.; Dimitrijevic, A.; Starr, A. Loudness Adaptation Accompanying Ribbon Synapse and Auditory Nerve Disorders. Brain 2013, 136, 1626–1638. [Google Scholar] [CrossRef]
- Marlin, S.; Feldmann, D.; Nguyen, Y.; Rouillon, I.; Loundon, N.; Jonard, L.; Bonnet, C.; Couderc, R.; Garabedian, E.N.; Petit, C.; et al. Temperature-Sensitive Auditory Neuropathy Associated with an Otoferlin Mutation: Deafening Fever! Biochem. Biophys. Res. Commun. 2010, 394, 737–742. [Google Scholar] [CrossRef]
- Wang, D.Y.; Wang, Y.C.; Weil, D.; Zhao, Y.L.; Rao, S.Q.; Zong, L.; Ji, Y.B.; Liu, Q.; Li, J.Q.; Yang, H.M.; et al. Screening Mutations of OTOF Gene in Chinese Patients with Auditory Neuropathy, Including a Familial Case of Temperature-Sensitive Auditory Neuropathy. BMC Med. Genet. 2010, 11, 79. [Google Scholar] [CrossRef]
- Romanos, J.; Kimura, L.; Fávero, M.L.; Izarra, F.A.R.; De Mello Auricchio, M.T.B.; Batissoco, A.C.; Lezirovitz, K.; Abreu-Silva, R.S.; Mingroni-Netto, R.C. Novel OTOF Mutations in Brazilian Patients with Auditory Neuropathy. J. Hum. Genet. 2009, 54, 382–385. [Google Scholar] [CrossRef]
- Matsunaga, T.; Mutai, H.; Kunishima, S.; Namba, K.; Morimoto, N.; Shinjo, Y.; Arimoto, Y.; Kataoka, Y.; Shintani, T.; Morita, N.; et al. A Prevalent Founder Mutation and Genotype-Phenotype Correlations of OTOF in Japanese Patients with Auditory Neuropathy. Clin. Genet. 2012, 82, 425–432. [Google Scholar] [CrossRef]
- Runge, C.L.; Erbe, C.B.; Mcnally, M.T.; Van Dusen, C.; Friedland, D.R.; Kwitek, A.E.; Kerschner, J.E. A Novel Otoferlin Splice-Site Mutation in Siblings with Auditory Neuropathy Spectrum Disorder. Audiol. Neurotol. 2015, 18, 374–382. [Google Scholar] [CrossRef]
- Iwasa, Y.; Nishio, S.; Sugaya, A.; Kataoka, Y.; Kanda, Y.; Taniguchi, M.; Nagai, K.; Naito, Y.; Ikezono, T.; Horie, R.; et al. OTOF Mutation Analysis with Massively Parallel DNA Sequencing in 2,265 Japanese Sensorineural Hearing Loss Patients. PLoS ONE 2019, 14, e021593. [Google Scholar] [CrossRef]
- Deafness Variation Database. Available online: http://deafnessvariationdatabase.org/ (accessed on 10 January 2020).
- Sloan-Heggen, C.M.; Bierer, A.O.; Shearer, A.E.; Kolbe, D.L.; Nishimura, C.J.; Frees, K.L.; Ephraim, S.S.; Shibata, S.B.; Booth, K.T.; Campbell, C.A.; et al. Comprehensive Genetic Testing in the Clinical Evaluation of 1119 Patients with Hearing Loss. Hum. Genet. 2016, 135, 441–450. [Google Scholar] [CrossRef]
- Tertrais, M.; Bouleau, Y.; Emptoz, A.; Belleudy, S.; Sutton, R.B.; Petit, C.; Safieddine, S.; Dulon, D. Viral Transfer of Mini-Otoferlins Partially Restores the Fast Component of Exocytosis and Uncovers Ultrafast Endocytosis in Auditory Hair Cells of Otoferlin Knock-out Mice. J. Neurosci. 2019, 39, 3394–3411. [Google Scholar] [CrossRef]
- Baig, S.M.; Koschak, A.; Lieb, A.; Gebhart, M.; Dafinger, C.; Nürnberg, G.; Ali, A.; Ahmad, I.; Sinnegger-Brauns, M.J.; Brandt, N.; et al. Loss of Ca v 1.3 (CACNA1D) Function in a Human Channelopathy with Bradycardia and Congenital Deafness. Nat. Neurosci. 2011, 14, 77–86. [Google Scholar] [CrossRef]
- Brandt, A.; Striessnig, J.; Moser, T. Cav1.3 Channels Are Essential for Development and Presynaptic Activity of Cochlear Inner Hair Cells. J. Neurosci. 2003, 23, 10832–10840. [Google Scholar] [CrossRef]
- Platzer, J.; Engel, J.; Schrott-Fischer, A.; Stephan, K.; Bova, S.; Chen, H.; Zheng, H.; Striessnig, J. Congenital Deafness and Sinoatrial Node Dysfunction in Mice Lacking Class D L-Type Ca2+ Channels. Cell 2000, 102, 89–97. [Google Scholar] [CrossRef]
- Qi, F.; Zhang, R.; Chen, J.; Zhao, F.; Sun, Y.; Du, Z.; Bing, D.; Li, P.; Shao, S.; Zhu, H.; et al. Down-Regulation of Cav1.3 in Auditory Pathway Promotes Age-Related Hearing Loss by Enhancing Calcium-Mediated Oxidative Stress in Male Mice. Aging 2019, 11, 6490–6502. [Google Scholar] [CrossRef]
- Eckrich, S.; Hecker, D.; Sorg, K.; Blum, K.; Fischer, K.; Münkner, S.; Wenzel, G.; Schick, B.; Engel, J. Cochlea-Specific Deletion of Cav1.3 Calcium Channels Arrests Inner Hair Cell Differentiation and Unravels Pitfalls of Conditional Mouse Models. Front. Cell. Neurosci. 2019, 13, 225. [Google Scholar] [CrossRef]
- Haeseleer, F.; Imanishi, Y.; Sokal, I.; Filipek, S.; Palczewski, K. Calcium-Binding Proteins: Intracellular Sensors from the Calmodulin Superfamily. In Biochemical and Biophysical Research Communications; Academic Press Inc.: Cambridge, MA, USA, 2002; pp. 615–623. [Google Scholar]
- Christel, C.; Lee, A. Ca 2 +-Dependent Modulation of Voltage-Gated Ca 2 + Channels. Biochim. Biophys. Acta Gen. Subj. 2012, 1820, 1243–1252. [Google Scholar] [CrossRef]
- Schrauwen, I.; Helfmann, S.; Inagaki, A.; Predoehl, F.; Tabatabaiefar, M.A.; Picher, M.M.; Sommen, M.; Seco, C.Z.; Oostrik, J.; Kremer, H.; et al. A Mutation in CABP2, Expressed in Cochlear Hair Cells, Causes Autosomal-Recessive Hearing Impairment. Am. J. Hum. Genet. 2012, 91, 636–645. [Google Scholar] [CrossRef]
- Seal, R.P.; Akil, O.; Yi, E.; Weber, C.M.; Grant, L.; Yoo, J.; Clause, A.; Kandler, K.; Noebels, J.L.; Glowatzki, E.; et al. Sensorineural Deafness and Seizures in Mice Lacking Vesicular Glutamate Transporter 3. Neuron 2008, 57, 263–275. [Google Scholar] [CrossRef]
- Ruel, J.; Emery, S.; Nouvian, R.; Bersot, T.; Amilhon, B.; Van Rybroek, J.M.; Rebillard, G.; Lenoir, M.; Eybalin, M.; Delprat, B.; et al. Impairment of SLC17A8 Encoding Vesicular Glutamate Transporter-3, VGLUT3, Underlies Nonsyndromic Deafness DFNA25 and Inner Hair Cell Dysfunction in Null Mice. Am. J. Hum. Genet. 2008, 83, 278–292. [Google Scholar] [CrossRef]
- Petek, E.; Windpassinger, C.; Mach, M.; Rauter, L.; Scherer, S.W.; Wagner, K.; Kroisel, P.M. Molecular Characterization of a 12q22-Q24 Deletion Associated with Congenital Deafness: Confirmation and Refinement of the DFNA25 Locus. Am. J. Med. Genet. Part A 2002, 117, 122–126. [Google Scholar] [CrossRef]
- Ryu, N.; Sagong, B.; Park, H.J.; Kim, M.A.; Lee, K.Y.; Choi, J.Y.; Kim, U.K. Screening of the SLC17A8 Gene as a Causative Factor for Autosomal Dominant Non-Syndromic Hearing Loss in Koreans. BMC Med. Genet. 2016, 17, 6. [Google Scholar] [CrossRef]
- Ryu, N.; Lee, S.; Park, H.J.; Lee, B.; Kwon, T.J.; Bok, J.; Park, C.I.; Lee, K.Y.; Baek, J.I.; Kim, U.K. Identification of a Novel Splicing Mutation within SLC17A8 in a Korean Family with Hearing Loss by Whole-Exome Sequencing. Gene 2017, 627, 233–238. [Google Scholar] [CrossRef]
- Shearer, A.E.; Eppsteiner, R.W.; Frees, K.; Tejani, V.; Sloan-Heggen, C.M.; Brown, C.; Abbas, P.; Dunn, C.; Hansen, M.R.; Gantz, B.J.; et al. Genetic Variants in the Peripheral Auditory System Significantly Affect Adult Cochlear Implant Performance. Hear. Res. 2017, 348, 138–142. [Google Scholar] [CrossRef]
- Akil, O.; Lustig, L. AAV-Mediated Gene Delivery to the Inner Ear. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2019; Volume 1950, pp. 271–282. [Google Scholar]
- Alexander, C.; Votruba, M.; Pesch, U.E.A.; Thiselton, D.L.; Mayer, S.; Moore, A.; Rodriguez, M.; Kellner, U.; Leo-Kottler, B.; Auburger, G.; et al. OPA1, Encoding a Dynamin-Related GTPase, Is Mutated in Autosomal Dominant Optic Atrophy Linked to Chromosome 3q28. Nat. Genet. 2000, 26, 211–215. [Google Scholar] [CrossRef]
- Delettre, C.; Lenaers, G.; Griffoin, J.M.; Gigarel, N.; Lorenzo, C.; Belenguer, P.; Pelloquin, L.; Grosgeorge, J.; Turc-Carel, C.; Perret, E.; et al. Nuclear Gene OPA1, Encoding a Mitochondrial Dynamin-Related Protein, Is Mutated in Dominant Optic Atrophy. Nat. Genet. 2000, 26, 207–210. [Google Scholar] [CrossRef]
- Kasahara, A.; Scorrano, L. Mitochondria: From Cell Death Executioners to Regulators of Cell Differentiation. Trends Cell Biol. 2014, 24, 761–770. [Google Scholar] [CrossRef]
- Bansal, D.; Miyake, K.; Vogel, S.S.; Groh, S.; Chen, C.C.; Williamson, R.; McNeil, P.L.; Campbell, K.P. Defective Membrane Repair in Dysferlin-Deficient Muscular Dystrophy. Nature 2003, 423, 168–172. [Google Scholar] [CrossRef]
- Ferré, M.; Bonneau, D.; Milea, D.; Chevrollier, A.; Verny, C.; Dollfus, H.; Ayuso, C.; Defoort, S.; Vignal, C.; Zanlonghi, X.; et al. Molecular Screening of 980 Cases of Suspected Hereditary Optic Neuropathy with a Report on 77 Novel OPA1 Mutations. Hum. Mutat. 2009, 30, E692–E705. [Google Scholar] [CrossRef]
- Davies, V.J.; Hollins, A.J.; Piechota, M.J.; Yip, W.; Davies, J.R.; White, K.E.; Nicols, P.P.; Boulton, M.E.; Votruba, M. Opa1 Deficiency in a Mouse Model of Autosomal Dominant Optic Atrophy Impairs Mitochondrial Morphology, Optic Nerve Structure and Visual Function. Hum. Mol. Genet. 2007, 16, 1307–1318. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Griffiths, P.G.; Gorman, G.S.; Lourenco, C.M.; Wright, A.F.; Auer-Grumbach, M.; Toscano, A.; Musumeci, O.; Valentino, M.L.; Caporali, L.; et al. Multi-System Neurological Disease Is Common in Patients with OPA1 Mutations. Brain 2010, 133, 771–786. [Google Scholar] [CrossRef]
- Santarelli, R.; Rossi, R.; Scimemi, P.; Cama, E.; Valentino, M.L.; La Morgia, C.; Caporali, L.; Liguori, R.; Magnavita, V.; Monteleone, A.; et al. OPA1-Related Auditory Neuropathy: Site of Lesion and Outcome of Cochlear Implantation. Brain 2015, 138, 563–576. [Google Scholar] [CrossRef]
- Huang, T.; Santarelli, R.; Starr, A. Mutation of OPA1 Gene Causes Deafness by Affecting Function of Auditory Nerve Terminals. Brain Res. 2009, 1300, 97–104. [Google Scholar] [CrossRef]
- Diaz-Horta, O.; Abad, C.; Sennaroglu, L.; Ii, J.F.; DeSmidt, A.; Bademci, G.; Tokgoz-Yilmaz, S.; Duman, D.; Cengiz, F.B.; Grati, M.; et al. ROR1 Is Essential for Proper Innervation of Auditory Hair Cells and Hearing in Humans and Mice. Proc. Natl. Acad. Sci. USA 2016, 113, 5993–5998. [Google Scholar] [CrossRef]
- Holm, T.H.; Lykke-Hartmann, K. Insights into the Pathology of the A3 Na+/K+-ATPase Ion Pump in Neurological Disorders; Lessons from Animal Models. Front. Physiol. 2016, 7, 209. [Google Scholar] [CrossRef]
- Nicolaides, P.; Appleton, R.E.; Fryer, A. Cerebellar Ataxia, Areflexia, Pes Cavus, Optic Atrophy, and Sensorineural Hearing Loss (CAPOS): A New Syndrome. J. Med. Genet. 1996, 33, 419–421. [Google Scholar] [CrossRef]
- Demos, M.K.; van Karnebeek, C.D.; Ross, C.J.; Adam, S.; Shen, Y.; Zhan, S.H.; Shyr, C.; Horvath, G.; Suri, M.; Fryer, A.; et al. A Novel Recurrent Mutation in ATP1A3 Causes CAPOS Syndrome. Orphanet J. Rare Dis. 2014, 9, 15. [Google Scholar] [CrossRef]
- Rosewich, H.; Weise, D.; Ohlenbusch, A.; Gärtner, J.; Brockmann, K. Phenotypic Overlap of Alternating Hemiplegia of Childhood and CAPOS Syndrome. Neurology 2014, 83, 861–863. [Google Scholar] [CrossRef]
- Tranebjærg, L.; Strenzke, N.; Lindholm, S.; Rendtorff, N.D.; Poulsen, H.; Khandelia, H.; Kopec, W.; Lyngbye, T.J.B.; Hamel, C.; Delettre, C.; et al. Correction to: The CAPOS Mutation in ATP1A3 Alters Na/K-ATPase Function and Results in Auditory Neuropathy Which Has Implications for Management. Hum. Genet. 2018, 137, 111–127. [Google Scholar] [CrossRef]
- Heimer, G.; Sadaka, Y.; Israelian, L.; Feiglin, A.; Ruggieri, A.; Marshall, C.R.; Scherer, S.W.; Ganelin-Cohen, E.; Marek-Yagel, D.; Tzadok, M.; et al. CAOS-Episodic Cerebellar Ataxia, Areflexia, Optic Atrophy, and Sensorineural Hearing Loss: A Third Allelic Disorder of the ATP1A3 Gene. J. Child Neurol. 2015, 30, 1749–1756. [Google Scholar] [CrossRef]
- Paquay, S.; Wiame, E.; Deggouj, N.; Boschi, A.; De Siati, R.D.; Sznajer, Y.; Nassogne, M.-C. Childhood Hearing Loss Is a Key Feature of CAPOS Syndrome: A Case Report. Int. J. Pediatr. Otorhinolaryngol. 2018, 104, 191–194. [Google Scholar] [CrossRef]
- Potic, A.; Nmezi, B.; Padiath, Q.S. CAPOS Syndrome and Hemiplegic Migraine in a Novel Pedigree with the Specific ATP1A3 Mutation. J. Neurol. Sci. 2015, 358, 453–456. [Google Scholar] [CrossRef]
- Duat Rodriguez, A.; Prochazkova, M.; Santos Santos, S.; Rubio Cabezas, O.; Cantarin Extremera, V.; Gonzalez-Gutierrez-Solana, L. Early Diagnosis of CAPOS Syndrome Before Acute-Onset Ataxia—Review of the Literature and a New Family. Pediatr. Neurol. 2017, 71, 60–64. [Google Scholar] [CrossRef]
- Maas, R.P.P.W.M.; Schieving, J.H.; Schouten, M.; Kamsteeg, E.J.; Van De Warrenburg, B.P.C. The Genetic Homogeneity of CAPOS Syndrome: Four New Patients with the c.2452G>A (p.Glu818Lys) Mutation in the ATP1A3 Gene. Pediatr. Neurol. 2016, 59, 71–75. [Google Scholar] [CrossRef]
- Han, K.H.; Oh, D.Y.; Lee, S.; Lee, C.; Han, J.H.; Kim, M.Y.; Park, H.R.; Park, M.K.; Kim, N.K.D.; Lee, J.; et al. ATP1A3 Mutations Can Cause Progressive Auditory Neuropathy: A New Gene of Auditory Synaptopathy. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Schoen, C.J.; Burmeister, M.; Lesperance, M.M. Diaphanous Homolog 3 (Diap3) Overexpression Causes Progressive Hearing Loss and Inner Hair Cell Defects in a Transgenic Mouse Model of Human Deafness. PLoS ONE 2013, 8, e56520. [Google Scholar] [CrossRef]
- Schoen, C.J.; Emery, S.B.; Thorne, M.C.; Ammana, H.R.; Śliwerska, E.; Arnett, J.; Hortsch, M.; Hannan, F.; Burmeister, M.; Lesperance, M.M. Increased Activity of Diaphanous Homolog 3 (DIAPH3)/Diaphanous Causes Hearing Defects in Humans with Auditory Neuropathy and in Drosophila. Proc. Natl. Acad. Sci. USA 2010, 107, 13396–13401. [Google Scholar] [CrossRef]
- Kim, T.B.; Isaacson, B.; Sivakumaran, T.A.; Starr, A.; Keats, B.J.B.; Lesperance, M.M. A Gene Responsible for Autosomal Dominant Auditory Neuropathy (AUNA1) Maps to 13q14-21. J. Med. Genet. 2004, 41, 872–876. [Google Scholar] [CrossRef]
- Starr, A.; Isaacson, B.; Michalewski, H.J.; Zeng, F.G.; Kong, Y.Y.; Beale, P.; Paulson, G.W.; Keats, B.J.B.; Lesperance, M.M. A Dominantly Inherited Progressive Deafness Affecting Distal Auditory Nerve and Hair Cells. JARO J. Assoc. Res. Otolaryngol. 2004, 5, 411–426. [Google Scholar] [CrossRef]
- Charcot-Marie-Tooth Disease Fact Sheet|National Institute of Neurological Disorders and Stroke. Available online: https://www.ninds.nih.gov/Disorders/Patient-caregiver-education/Fact-sheets/Charcot-Marie-Tooth-Disease-Fact-Sheet (accessed on 31 December 2019).
- Goswamy, J.; Bruce, I.A.; Green, K.M.J.; O’Driscoll, M.P. Cochlear Implantation in a Patient with Sensori-Neural Deafness Secondary to Charcot-Marie-Tooth Disease. Cochlear Implants Int. 2012, 13, 184–187. [Google Scholar] [CrossRef]
- Verhagen, W.I.M.; Huygen, P.L.M.; Gabreëls-Festen, A.A.W.M.; Engelhart, M.; Van Mierlo, P.J.W.B.; Van Engelen, B.G.M. Sensorineural Hearing Impairment in Patients with Pmp22 Duplication, Deletion, and Frameshift Mutations. Otol. Neurotol. 2005, 26, 405–414. [Google Scholar] [CrossRef]
- Varga, R.; Kelley, P.M.; Keats, B.J.; Starr, A.; Leal, S.M.; Cohn, E.; Kimberling, W.J. Non-Syndromic Recessive Auditory Neuropathy Is the Result of Mutations in the Otoferlin (OTOF) Gene [2]. J. Med. Genet. 2003, 40, 45–50. [Google Scholar] [CrossRef]
- Kabzińska, D.; Korwin-Piotrowska, T.; Drechsler, H.; Drac, H.; Hausmanowa-Petrusewicz, I.; Kochański, A. Late-Onset Charcot-Marie-Tooth Type 2 Disease with Hearing Impairment Associated with a Novel Pro105Thr Mutation in the MPZ Gene. Am. J. Med. Genet. Part A 2007, 143, 2196–2199. [Google Scholar] [CrossRef]
- Rance, G.; Fava, R.; Baldock, H.; Chong, A.; Barker, E.; Corben, L.; Delatycki, M.B. Speech Perception Ability in Individuals with Friedreich Ataxia. Brain 2008, 131, 2002–2012. [Google Scholar] [CrossRef]
- Tranebjærg, L. Deafness-Dystonia-Optic Neuronopathy Syndrome. In GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2003. [Google Scholar]
- Bahmad, F.; Merchant, S.N.; Nadol, J.B.; Tranebjærg, L. Otopathology in Mohr-Tranebjærg Syndrome. Laryngoscope 2007, 117, 1202–1208. [Google Scholar] [CrossRef]
- Brookes, J.T.; Kanis, A.B.; Tan, L.Y.; Tranebjærg, L.; Vore, A.; Smith, R.J.H. Cochlear Implantation in Deafness-Dystonia-Optic Neuronopathy (DDON) Syndrome. Int. J. Pediatr. Otorhinolaryngol. 2008, 72, 121–126. [Google Scholar] [CrossRef]
- Kawarai, T.; Yamazaki, H.; Yamakami, K.; Tsukamoto-Miyashiro, A.; Kodama, M.; Rumore, R.; Caltagirone, C.; Nishino, I.; Orlacchio, A. A Novel AIFM1 Missense Mutation in a Japanese Patient with Ataxic Sensory Neuronopathy and Hearing Impairment. J. Neurol. Sci. 2020, 409, 116584. [Google Scholar] [CrossRef]
- Joza, N.; Pospisilik, J.A.; Hangen, E.; Hanada, T.; Modjtahedi, N.; Penninger, J.M.; Kroemer, G. AIF: Not Just an Apoptosis-Inducing Factor. Ann. N. Y. Acad. Sci. 2009, 1171, 2–11. [Google Scholar] [CrossRef]
- Zong, L.; Guan, J.; Ealy, M.; Zhang, Q.; Wang, D.; Wang, H.; Zhao, Y.; Shen, Z.; Campbell, C.A.; Wang, F.; et al. Mutations in Apoptosis-Inducing Factor Cause X-Linked Recessive Auditory Neuropathy Spectrum Disorder. J. Med. Genet. 2015, 52, 523–531. [Google Scholar] [CrossRef]
- Simon, M.; Richard, E.M.; Wang, X.; Shahzad, M.; Huang, V.H.; Qaiser, T.A.; Potluri, P.; Mahl, S.E.; Davila, A.; Nazli, S.; et al. Mutations of Human NARS2, Encoding the Mitochondrial Asparaginyl-TRNA Synthetase, Cause Nonsyndromic Deafness and Leigh Syndrome. PLoS Genet. 2015, 11, e1005097. [Google Scholar] [CrossRef]
- Defourny, J.; Aghaie, A.; Perfettini, I.; Avan, P.; Delmaghani, S.; Petit, C. Pejvakin-Mediated Pexophagy Protects Auditory Hair Cells against Noise-Induced Damage. Proc. Natl. Acad. Sci. USA 2019, 116, 8010–8017. [Google Scholar] [CrossRef]
- Delmaghani, S.; Del Castillo, F.J.; Michel, V.; Leibovici, M.; Aghaie, A.; Ron, U.; Van Laer, L.; Ben-Tal, N.; Van Camp, G.; Weil, D.; et al. Mutations in the Gene Encoding Pejvakin, a Newly Identified Protein of the Afferent Auditory Pathway, Cause DFNB59 Auditory Neuropathy. Nat. Genet. 2006, 38, 770–778. [Google Scholar] [CrossRef]
- Schwander, M.; Sczaniecka, A.; Grillet, N.; Bailey, J.S.; Avenarius, M.; Najmabadi, H.; Steffy, B.M.; Federe, G.C.; Lagler, E.A.; Banan, R.; et al. A Forward Genetics Screen in Mice Identifies Recessive Deafness Traits and Reveals That Pejvakin Is Essential for Outer Hair Cell Function. J. Neurosci. 2007, 27, 2163–2175. [Google Scholar] [CrossRef]
- Borck, G.; Rainshtein, L.; Hellman-Aharony, S.; Volk, A.; Friedrich, K.; Taub, E.; Magal, N.; Kanaan, M.; Kubisch, C.; Shohat, M.; et al. High Frequency of Autosomal-Recessive DFNB59 Hearing Loss in an Isolated Arab Population in Israel. Clin. Genet. 2012, 82, 271–276. [Google Scholar] [CrossRef]
- Ebermann, I.; Walger, M.; Scholl, H.P.N.; Issa, P.C.; Lüke, C.; Nürnberg, G.; Lang-Roth, R.; Becker, C.; Nürnberg, P.; Bolz, H.J. Truncating Mutation of the DFNB59 Gene Causes Cochlear Hearing Impairment and Central Vestibular Dysfunction. Hum. Mutat. 2007, 28, 571–577. [Google Scholar] [CrossRef]
- Chaleshtori, M.H.; Simpson, M.A.; Farrokhi, E.; Dolati, M.; Hoghooghi Rad, L.; Geshnigani, S.A.; Crosby, A.H. Novel Mutations in the Pejvakin Gene Are Associated with Autosomal Recessive Non-Syndromic Hearing Loss in Iranian Families. Clin. Genet. 2007, 72, 261–263. [Google Scholar] [CrossRef]
- Collin, R.W.J.; Kalay, E.; Oostrik, J.; Çaylan, R.; Wollnik, B.; Arslan, S.; Den Hollander, A.I.; Birinci, Y.; Lichtner, P.; Strom, T.M.; et al. Involvement of DFNB59 Mutations in Autosomal Recessive Nonsyndromic Hearing Impairment. Hum. Mutat. 2007, 28, 718–723. [Google Scholar] [CrossRef]
- Delmaghani, S.; Defourny, J.; Aghaie, A.; Beurg, M.; Dulon, D.; Thelen, N.; Perfettini, I.; Zelles, T.; Aller, M.; Meyer, A.; et al. Hypervulnerability to Sound Exposure through Impaired Adaptive Proliferation of Peroxisomes. Cell 2015, 163, 894–906. [Google Scholar] [CrossRef]
- Guipponi, M. The Transmembrane Serine Protease (TMPRSS3) Mutated in Deafness DFNB8/10 Activates the Epithelial Sodium Channel (ENaC) in Vitro. Hum. Mol. Genet. 2002, 11, 2829–2836. [Google Scholar] [CrossRef]
- Fasquelle, L.; Scott, H.S.; Lenoir, M.; Wang, J.; Rebillard, G.; Gaboyard, S.; Venteo, S.; François, F.; Mausset-Bonnefont, A.L.; Antonarakis, S.E.; et al. Tmprss3, a Transmembrane Serine Protease Deficient in Human DFNB8/10 Deafness, Is Critical for Cochlear Hair Cell Survival at the Onset of Hearing. J. Biol. Chem. 2011, 286, 17383–17397. [Google Scholar] [CrossRef]
- Shrestha, B.R.; Chia, C.; Wu, L.; Kujawa, S.G.; Liberman, M.C.; Goodrich, L.V. Sensory Neuron Diversity in the Inner Ear Is Shaped by Activity. Cell 2018, 174, 1229–1246. [Google Scholar] [CrossRef]
- Li, Y.; Peng, A.; Ge, S.; Wang, Q.; Liu, J. MiR-204 Suppresses Cochlear Spiral Ganglion Neuron Survival Invitro by Targeting TMPRSS3. Hear. Res. 2014, 314, 60–64. [Google Scholar] [CrossRef]
- Scott, H.S.; Kudoh, J.; Wattenhofer, M.; Shibuya, K.; Berry, A.; Chrast, R.; Guipponi, M.; Wang, J.; Kawasaki, K.; Asakawa, S.; et al. Insertion of β-Satellite Repeats Identifies a Transmembrane Protease Causing Both Congenital and Childhood Onset Autosomal Recessive Deafness. Nat. Genet. 2001, 27, 59–63. [Google Scholar] [CrossRef]
- Bonne-Tamir, B.; DeStefano, A.L.; Briggs, C.E.; Adair, R.; Franklyn, B.; Weiss, S.; Korostishevsky, M.; Frydman, M.; Baldwin, C.T.; Farrer, L.A. Linkage of Congenital Recessive Deafness (Gene DFNB10) to Chromosome 21q22.3. Am. J. Hum. Genet. 1996, 58, 1254–1259. [Google Scholar]
- Weegerink, N.J.D.; Schraders, M.; Oostrik, J.; Huygen, P.L.M.; Strom, T.M.; Granneman, S.; Pennings, R.J.E.; Venselaar, H.; Hoefsloot, L.H.; Elting, M.; et al. Genotype-Phenotype Correlation in DFNB8/10 Families with TMPRSS3 Mutations. JARO J. Assoc. Res. Otolaryngol. 2011, 12, 753–766. [Google Scholar] [CrossRef]
- Miyagawa, M.; Nishio, S.Y.; Sakurai, Y.; Hattori, M.; Tsukada, K.; Moteki, H.; Kojima, H.; Usami, S.I. The Patients Associated with TMPRSS3 Mutations Are Good Candidates for Electric Acoustic Stimulation. Ann. Otol. Rhinol. Laryngol. 2015, 124, 193S–204S. [Google Scholar] [CrossRef]
- Eppsteiner, R.W.; Shearer, A.E.; Hildebrand, M.S.; DeLuca, A.P.; Ji, H.; Dunn, C.C.; Black-Ziegelbein, E.A.; Casavant, T.L.; Braun, T.A.; Scheetz, T.E.; et al. Prediction of Cochlear Implant Performance by Genetic Mutation: The Spiral Ganglion Hypothesis. Hear. Res. 2012, 292, 51–58. [Google Scholar] [CrossRef]
- Meleca, J.B.; Stillitano, G.; Lee, M.Y.; Lyle, W.; Carol Liu, Y.C.; Anne, S. Outcomes of Audiometric Testing in Children with Auditory Neuropathy Spectrum Disorder. Int. J. Pediatr. Otorhinolaryngol. 2020, 129, 109757. [Google Scholar] [CrossRef]
- Haustein, M.D.; Read, D.J.; Steinert, J.R.; Pilati, N.; Dinsdale, D.; Forsythe, I.D. Acute Hyperbilirubinaemia Induces Presynaptic Neurodegeneration at a Central Glutamatergic Synapse. J. Physiol. 2010, 588, 4683–4693. [Google Scholar] [CrossRef]
- Chandan, H.S.; Prabhu, P. Audiological Changes over Time in Adolescents and Young Adults with Auditory Neuropathy Spectrum Disorder. Eur. Arch. Oto-Rhino-Laryngol. 2015, 272, 1801–1807. [Google Scholar] [CrossRef]
- Rance, G.; Beer, D.E.; Cone-Wesson, B.; Shepherd, R.K.; Dowell, R.C.; King, A.M.; Rickards, F.W.; Clark, G.M. Clinical Findings for a Group of Infants and Young Children with Auditory Neuropathy. Ear Hear. 1999, 20, 238–252. [Google Scholar] [CrossRef]
- Kraus, N.; Bradlow, A.R.; Cheatham, M.A.; Cunningham, J.; King, C.D.; Koch, D.B.; Nicol, T.G.; McGee, T.J.; Stein, L.K.; Wright, B.A. Consequences of Neural Asynchrony: A Case of Auditory Neuropathy. JARO J. Assoc. Res. Otolaryngol. 2000, 1, 33–45. [Google Scholar] [CrossRef]
- Dimitrijevic, A.; Starr, A.; Bhatt, S.; Michalewski, H.J.; Zeng, F.G.; Pratt, H. Auditory Cortical N100 in Pre- and Post-Synaptic Auditory Neuropathy to Frequency or Intensity Changes of Continuous Tones. Clin. Neurophysiol. 2011, 122, 594–604. [Google Scholar] [CrossRef]
- Kemp, D.T. Stimulated Acoustic Emissions from within the Human Auditory System. J. Acoust. Soc. Am. 1978, 64, 1386–1391. [Google Scholar] [CrossRef]
- Prieve, B.; Fitzgerald, T. Otoacoustic Emissions. In Handbook of Clinical Audiology; Katz, J., Ed.; Wolters Kluwer Health: Philadelphia, PA, USA, 2015. [Google Scholar]
- Glattke, T.J. Otoacoustic Emissions—Clinical Applications, 2nd ed.; Thieme: New York, NY, USA, 2002. [Google Scholar]
- Uus, K. Transient Auditory Neuropathy in Infants: How to Conceptualize the Recovery of Auditory Brain Stem Response in the Context of Newborn Hearing Screening? Semin. Hear. 2011, 32, 123–128. [Google Scholar] [CrossRef]
- Pappa, A.K.; Hutson, K.A.; Scott, W.C.; David Wilson, J.; Fox, K.E.; Masood, M.M.; Giardina, C.K.; Pulver, S.H.; Grana, G.D.; Askew, C.; et al. Hair Cell and Neural Contributions to the Cochlear Summating Potential. J. Neurophysiol. 2019, 121, 2163–2180. [Google Scholar] [CrossRef]
- Dallos, P.; Cheatham, M.A. Production of Cochlear Potentials by Inner and Outer Hair Cells. J. Acoust. Soc. Am. 1976, 60, 510–512. [Google Scholar] [CrossRef]
- do Amaral Soares, I.; de Lemos Menezes, P.; Carnaúba, A.T.L.; de Andrade, K.C.L.; Lins, O.G. Estudo Do Microfonismo Coclear Na Neuropatia Auditiva. Braz. J. Otorhinolaryngol. 2016, 82, 722–736. [Google Scholar] [CrossRef]
- Santarelli, R. Information from Cochlear Potentials and Genetic Mutations Helps Localize the Lesion Site in Auditory Neuropathy. Genome Med. 2010, 2, 91. [Google Scholar] [CrossRef]
- Santarelli, R.; Arslan, E. Electrocochleography in Auditory Neuropathy. Hear. Res. 2002, 170, 32–47. [Google Scholar] [CrossRef]
- Abbas, P.J.; Brown, C.J. Assessment of Responses to Cochlear Implant Stimulation at Different Levels of the Auditory Pathway. Hear. Res. 2015, 322, 67–76. [Google Scholar] [CrossRef]
- Fitzpatrick, D.C.; Campbell, A.T.; Choudhury, B.; Dillon, M.P.; Forgues, M.; Buchman, C.A.; Adunka, O.F. Round Window Electrocochleography Just before Cochlear Implantation: Relationship to Word Recognition Outcomes in Adults. Otol. Neurotol. 2014, 35, 64–71. [Google Scholar] [CrossRef]
- Formeister, E.J.; McClellan, J.H.; Merwin, W.H.; Iseli, C.E.; Calloway, N.H.; Teagle, H.F.B.; Buchman, C.A.; Adunka, O.F.; Fitzpatrick, D.C. Intraoperative Round Window Electrocochleography and Speech Perception Outcomes in Pediatric Cochlear Implant Recipients. Ear Hear. 2015, 36, 249–260. [Google Scholar] [CrossRef]
- Durrant, J.D.; Wang, J.; Ding, D.L.; Salvi, R.J. Are Inner or Outer Hair Cells the Source of Summating Potentials Recorded from the Round Window? J. Acoust. Soc. Am. 1998, 104, 370–377. [Google Scholar] [CrossRef]
- Snyder, R.L.; Schreiner, C.E. The Auditory Neurophonic: Basic Properties. Hear. Res. 1984, 15, 261–280. [Google Scholar] [CrossRef]
- Henry, K.R. Auditory Nerve Neurophonic Recorded from the Round Window of the Mongolian Gerbil. Hear. Res. 1995, 90, 176–184. [Google Scholar] [CrossRef]
- Santarelli, R.; Scimemi, P.; Dal Monte, E.; Arslan, E. Cochlear Microphonic Potential Recorded by Transtympanic Electrocochleography in Normally-Hearing and Hearing-Impaired Ears. Acta Otorhinolaryngol. Ital. 2006, 26, 78–95. [Google Scholar]
- Wichmann, C.; Moser, T. Relating Structure and Function of Inner Hair Cell Ribbon Synapses. Cell Tissue Res. 2015, 361, 95–114. [Google Scholar] [CrossRef]
- Santarelli, R.; Arslan, E. Electrocochleography. In Handbook of Clinical Audiology, 7th ed.; Katz, J., Ed.; Wolters Kluwer Health Adis (ESP): London, UK, 2014. [Google Scholar]
- Korczak, P.; Smart, J.; Delgado, R.; Strobel, T.M.; Bradford, C. Auditory Steady-State Responses. J. Am. Acad. Audiol. 2012, 23, 146–170. [Google Scholar] [CrossRef]
- Lu, P.; Huang, Y.; Chen, W.-X.; Jiang, W.; Hua, N.-Y.; Wang, Y.; Wang, B.; Xu, Z.-M. Measurement of Thresholds Using Auditory Steady-State Response and Cochlear Microphonics in Children with Auditory Neuropathy. J. Am. Acad. Audiol. 2019, 30, 672–676. [Google Scholar] [CrossRef]
- Sharma, A.; Cardon, G.; Henion, K.; Roland, P. Cortical Maturation and Behavioral Outcomes in Children with Auditory Neuropathy Spectrum Disorder. Int. J. Audiol. 2011, 50, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Michalewski, H.J.; Starr, A.; Zeng, F.G.; Dimitrijevic, A. N100 Cortical Potentials Accompanying Disrupted Auditory Nerve Activity in Auditory Neuropathy (AN): Effects of Signal Intensity and Continuous Noise. Clin. Neurophysiol. 2009, 120, 1352–1363. [Google Scholar] [CrossRef][Green Version]
- Michalewski, H.J.; Starr, A.; Nguyen, T.T.; Kong, Y.Y.; Zeng, F.G. Auditory Temporal Processes in Normal-Hearing Individuals and in Patients with Auditory Neuropathy. Clin. Neurophysiol. 2005, 116, 669–680. [Google Scholar] [CrossRef]
- Emami, S.F.; Abdoli, A. Cortical Auditory Evoked Potentials in Children with Auditory Neuropathy/Dys-Synchrony. Indian J. Otolaryngol. Head Neck Surg. 2019, 71, 238–242. [Google Scholar] [CrossRef]
- Narne, V.K.; Vanaja, C.S. Speech Identification and Cortical Potentials in Individuals with Auditory Neuropathy. Behav. Brain Funct. 2008, 4, 15. [Google Scholar] [CrossRef]
- Näätänen, R. The Mismatch Negativity: A Powerful Tool for Cognitive Neuroscience. Ear Hear. 1995, 16, 6–18. [Google Scholar] [CrossRef]
- Squires, N.K.; Squires, K.C.; Hillyard, S.A. Two Varieties of Long-Latency Positive Waves Evoked by Unpredictable Auditory Stimuli in Man. Electroencephalogr. Clin. Neurophysiol. 1975, 38, 387–401. [Google Scholar] [CrossRef]
- Kumar, A.U.; Jayaram, M. Auditory Processing in Individuals with Auditory Neuropathy. Behav. Brain Funct. 2005, 1, 21. [Google Scholar] [CrossRef]
- Rance, G.; Starr, A. Pathophysiological Mechanisms and Functional Hearing Consequences of Auditory Neuropathy. Brain 2015, 138, 3141–3158. [Google Scholar] [CrossRef]
- Walker, E.; McCreery, R.; Spratford, M.; Roush, P. Children with Auditory Neuropathy Spectrum Disorder Fitted with Hearing AIDS Applying the American Academy of Audiology Pediatric Amplification Guideline: Current Practice and Outcomes. J. Am. Acad. Audiol. 2016, 27, 204–218. [Google Scholar] [CrossRef]
- Giraudet, F.; Avan, P. Auditory Neuropathies. Curr. Opin. Neurol. 2012, 25, 50–56. [Google Scholar] [CrossRef]
- Breneman, A.I.; Gifford, R.H.; Dejong, M.D. Cochlear Implantation in Children with Auditory Neuropathy Spectrum Disorder: Long-Term Outcomes. J. Am. Acad. Audiol. 2012, 23, 5–17. [Google Scholar] [CrossRef]
- Teagle, H.F.B.; Roush, P.A.; Woodard, J.S.; Hatch, D.R.; Zdanski, C.J.; Buss, E.; Buchman, C.A. Cochlear Implantation in Children with Auditory Neuropathy Spectrum Disorder. Ear Hear. 2010, 31, 325–335. [Google Scholar] [CrossRef]
- Crispino, G.; Di Pasquale, G.; Scimemi, P.; Rodriguez, L.; Ramirez, F.G.; de Siati, R.D.; Santarelli, R.M.; Arslan, E.; Bortolozzi, M.; Chiorini, J.A.; et al. BAAV Mediated GJB2 Gene Transfer Restores Gap Junction Coupling in Cochlear Organotypic Cultures from Deaf Cx26Sox10Cre Mice. PLoS ONE 2011, 6, e23279. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Siati, R.D.; Rosenzweig, F.; Gersdorff, G.; Gregoire, A.; Rombaux, P.; Deggouj, N. Auditory Neuropathy Spectrum Disorders: From Diagnosis to Treatment: Literature Review and Case Reports. J. Clin. Med. 2020, 9, 1074. https://doi.org/10.3390/jcm9041074
De Siati RD, Rosenzweig F, Gersdorff G, Gregoire A, Rombaux P, Deggouj N. Auditory Neuropathy Spectrum Disorders: From Diagnosis to Treatment: Literature Review and Case Reports. Journal of Clinical Medicine. 2020; 9(4):1074. https://doi.org/10.3390/jcm9041074
Chicago/Turabian StyleDe Siati, Romolo Daniele, Flora Rosenzweig, Guillaume Gersdorff, Anaïs Gregoire, Philippe Rombaux, and Naïma Deggouj. 2020. "Auditory Neuropathy Spectrum Disorders: From Diagnosis to Treatment: Literature Review and Case Reports" Journal of Clinical Medicine 9, no. 4: 1074. https://doi.org/10.3390/jcm9041074
APA StyleDe Siati, R. D., Rosenzweig, F., Gersdorff, G., Gregoire, A., Rombaux, P., & Deggouj, N. (2020). Auditory Neuropathy Spectrum Disorders: From Diagnosis to Treatment: Literature Review and Case Reports. Journal of Clinical Medicine, 9(4), 1074. https://doi.org/10.3390/jcm9041074