New Horizons in the Genetic Etiology of Systemic Lupus Erythematosus and Lupus-Like Disease: Monogenic Lupus and Beyond


Abstract
1. Introduction
2. Genes and Molecular Pathways Associated with Lupus Phenotype

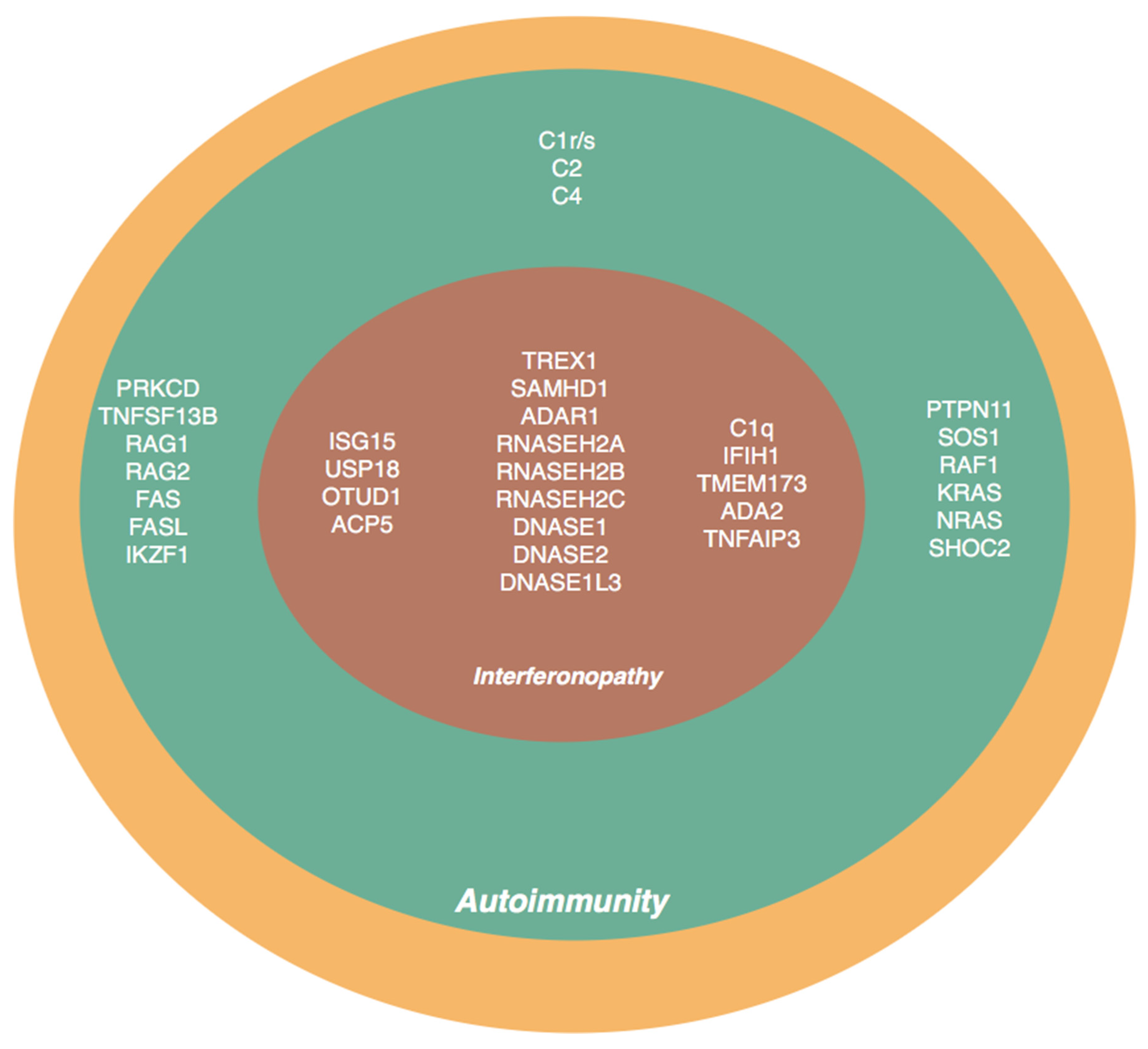{kind=link}
| Pathway | Gene | Inheritance | Disease | Reference |
|---|---|---|---|---|
| Complement pathway | C1q | AR | SLE, AGS | [18,19,20,21,22,23,24] |
| C1r/C1s | AD | [7,25,26] | ||
| C2 | AR | [27] | ||
| C4 | AR | [28,29,30,31,32] | ||
| Type I IFN pathway | DNASE1 | AD | HUV, lupus | [33,34,35] |
| DNASE2 | AR | [36,37] | ||
| DNASE1L3 | AR | [38,39] | ||
| TREX1 | AD/AR | AGS, SLE | [40,41,42,43,44,45,46,47] | |
| IFIH1 | AD | SLE, AGS, FCL, Singleton-Merton syndrome | [48,49,50,51] | |
| DDX58 | AD | SMS, glaucoma and skeletal abnormalities | [52,53] | |
| ISG15 | AR | basal ganglia calcification, AGS, pseudo-TORCH syndrome, MSMD, SLE | [54,55,56,57] | |
| USP18 | AR | |||
| SAMHD1 | AD | AGS, SLE, FCL, CLL, deforming arthropathy and recurrent oral ulcers | [58,59,60,61] | |
| OTUD1 | AR | SLE, autoimmune disease | [62] | |
| ACP5 | AD/AR | SPENCD, SLE, skeletal abnormalities | [63,64,65,66] | |
| TMEM173 | AD | SAVI, SLE, FCL, SCID | [67,68,69,70] | |
| RNASEH2A | AR | FCL, AGS, SLE | [71,72] | |
| RNASEH2B | ||||
| RNASEH2C | ||||
| ADAR1 | ||||
| Self-tolerance pathway | PRKCD | AR | SLE | [73,74,75,76,77] |
| TNFSF13B | SLE | [78,79] | ||
| RAS pathway | PTPN11 SOS1 RAF KRAS NRAS SHOC2 | AD | NS, SLE | [80,81,82,83,84,85,86,87,88,89,90,91] |
| Other pathways | RAG1 | AR | SLE, SCID | [92,93] |
| RAG2 | AD | |||
| FAS/FASL | AD | SLE, ALPS | [94,95] | |
| TNFAIP3 | AD | SLE, ALPS, BD-like | [96,97,98,99,100] | |
| ADA2 | AR | SLE, DADA2 | [101,102] | |
| IKZF1 | AD | SLE, ITP, CVID | [103,104,105] |
2.1. Complement Pathway
2.2. Type I Interferon (IFN) Pathway
2.2.1. Deoxyribonuclease Deficiencies
2.2.2. SAMHD1
2.2.3. Cytosolic RNA Nucleic Acid Degradation/Editing Pathways
2.2.4. Nucleic Acid Sensing Pathways
2.3. Negative Regulators of Type I Interferon Signaling Pathway
2.3.1. ISG15 and USP18
2.3.2. OTUD1
2.3.3. ACP5
2.4. Self-Tolerance Pathway
2.4.1. PRKCD
2.4.2. TNFSF13B/BAFF
2.5. Other Pathways Associted with Susceptibility to Lupus-like Phenotypes
2.5.1. RAG1 and RAG2
2.5.2. FAS/FASL
2.5.3. RASopathies
2.5.4. RELopathies
2.5.5. Deficiency of ADA2 (DADA2)
2.5.6. IKZF1
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bentham, J.; Morris, D.L.; Cunninghame Graham, D.S.; Pinder, C.L.; Tombleson, P.; Behrens, T.W.; Martín, J.; Fairfax, B.P.; Knight, J.C.; Chen, L.; et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat. Genet. 2015, 47, 1457–1464. [Google Scholar] [CrossRef] [PubMed]
- Julià, A.; López-Longo, F.J.; Pérez Venegas, J.J.; Bonàs-Guarch, S.; Olivé, À.; Andreu, J.L.; Aguirre-Zamorano, M.Á.; Vela, P.; Nolla, J.M.; de la Fuente, J.L.M.; et al. Genome-wide association study meta-analysis identifies five new loci for systemic lupus erythematosus. Arthritis Res. Ther. 2018, 20, 100. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Bueno, M.; Alarcón-Riquelme, M.E. Exploring Impact of Rare Variation in Systemic Lupus Erythematosus by a Genome Wide Imputation Approach. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Molineros, J.E.; Yang, W.; Zhou, X.j.; Sun, C.; Okada, Y.; Zhang, H.; Chua, K.H.; Lau, Y.L.; Kochi, Y.; Suzuki, A.; et al. Confirmation of five novel susceptibility loci for systemic lupus erythematosus (SLE) and integrated network analysis of 82 SLE susceptibility loci. Hum. Mol. Genet. 2017, 26, 1205–1216. [Google Scholar] [CrossRef] [PubMed]
- Morris, D.L.; Sheng, Y.; Zhang, Y.; Wang, Y.-F.; Zhu, Z.; Tombleson, P.; Chen, L.; Cunninghame Graham, D.S.; Bentham, J.; Roberts, A.L.; et al. Genome-wide association meta-analysis in Chinese and European individuals identifies ten new loci associated with systemic lupus erythematosus. Nat. Genet. 2016, 48, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Pullabhatla, V.; Roberts, A.L.; Lewis, M.J.; Mauro, D.; Morris, D.L.; Odhams, C.A.; Tombleson, P.; Liljedahl, U.; Vyse, S.; Simpson, M.A.; et al. De novo mutations implicate novel genes in systemic lupus erythematosus. Hum. Mol. Genet. 2018, 27, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Demirkaya, E.; Zhou, Q.; Smith, C.K.; Ombrello, M.J.; Deuitch, N.; Tsai, W.L.; Hoffmann, P.; Remmers, E.F.; Takeuchi, M.; Park, Y.H.; et al. Brief Report: Deficiency of Complement 1r Subcomponent in Early-Onset Systemic Lupus Erythematosus: The Role of Disease-Modifying Alleles in a Monogenic Disease. Arthritis Rheumatol. 2017, 69, 1832–1839. [Google Scholar] [CrossRef]
- Ellyard, J.I.; Jerjen, R.; Martin, J.L.; Lee, A.Y.S.; Field, M.A.; Jiang, S.H.; Cappello, J.; Naumann, S.K.; Andrews, T.D.; Scott, H.S.; et al. Brief Report: Identification of a Pathogenic Variant in TREX1 in Early-Onset Cerebral Systemic Lupus Erythematosus by Whole-Exome Sequencing. Arthritis Rheumatol. 2014, 66, 3382–3386. [Google Scholar] [CrossRef]
- Günther, C.; Kind, B.; Reijns, M.A.M.; Berndt, N.; Martinez-Bueno, M.; Wolf, C.; Tüngler, V.; Chara, O.; Lee, Y.A.; Hübner, N.; et al. Defective removal of ribonucleotides from DNA promotes systemic autoimmunity. J. Clin. Investig. 2015, 125, 413–424. [Google Scholar] [CrossRef]
- Hagberg, N.; Rönnblom, L. Systemic Lupus Erythematosus—A Disease with A Dysregulated Type I Interferon System. Scand. J. Immunol. 2015, 82, 199–207. [Google Scholar] [CrossRef]
- Baechler, E.C.; Batliwalla, F.M.; Karypis, G.; Gaffney, P.M.; Ortmann, W.A.; Espe, K.J.; Shark, K.B.; Grande, W.J.; Hughes, K.M.; Kapur, V.; et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. USA 2003, 100, 2610–2615. [Google Scholar] [CrossRef]
- Dale, R.C.; Ping Tang, S.; Heckmatt, J.Z.; Tatnall, M.F. Familial Systemic Lupus Erythematosus and Congenital Infection-Like Syndrome. Neuropediatrics 2000, 31, 155–158. [Google Scholar] [CrossRef]
- De Laet, C.; Goyens, P.; Christophe, C.; Ferster, A.; Mascart, F.; Dan, B. Phenotypic Overlap between Infantile Systemic Lupus Erythematosus and Aicardi-Goutières Syndrome. Neuropediatrics 2005, 36, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Weill, O.; Decramer, S.; Malcus, C.; Kassai, B.; Rouvet, I.; Ginhoux, T.; Crow, Y.J.; Rieux-Laucat, F.; Soulas-Sprauel, P.; Pagnier, A.; et al. Familial and syndromic lupus share the same phenotype as other early-onset forms of lupus. Jt. Bone Spine 2017, 84, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Deapen, D.; Escalante, A.; Weinrib, L.; Horwitz, D.; Bachman, B.; Roy-Burman, P.; Walker, A.; Mack, T.M. A revised estimate of twin concordance in systemic lupus erythematosus. Arthritis Rheum. 1992, 35, 311–318. [Google Scholar] [PubMed]
- Alarcón-Segovia, D.; Alarcón-Riquelme, M.E.; Cardiel, M.H.; Caeiro, F.; Massardo, L.; Villa, A.R.; Pons-Estel, B.A. Familial aggregation of systemic lupus erythematosus, rheumatoid arthritis, and other autoimmune diseases in 1,177 lupus patients from the GLADEL cohort. Arthritis Rheum. 2005, 52, 1138–1147. [Google Scholar] [CrossRef]
- Webb, R.; Kelly, J.A.; Somers, E.C.; Hughes, T.; Kaufman, K.M.; Sanchez, E.; Nath, S.K.; Bruner, G.; Alarcon-Riquelme, M.E.; Gilkeson, G.S.; et al. Early disease onset is predicted by a higher genetic risk for lupus and is associated with a more severe phenotype in lupus patients. Ann. Rheum. Dis. 2011, 70, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Al-Mayouf, S.M.; AlSaleem, A.; AlMutairi, N.; AlSonbul, A.; Alzaid, T.; Alazami, A.M.; Al-Mousa, H. Monogenic interferonopathies: Phenotypic and genotypic findings of CANDLE syndrome and its overlap with C1q deficient SLE. Int. J. Rheum. Dis. 2018, 21, 208–213. [Google Scholar] [CrossRef]
- Lood, C.; Gullstrand, B.; Truedsson, L.; Olin, A.I.; Alm, G.V.; Rönnblom, L.; Sturfelt, G.; Eloranta, M.-L.; Bengtsson, A.A. C1q inhibits immune complex-induced interferon-α production in plasmacytoid dendritic cells: A novel link between C1q deficiency and systemic lupus erythematosus pathogenesis. Arthritis Rheum. 2009, 60, 3081–3090. [Google Scholar] [CrossRef]
- Troedson, C.; Wong, M.; Dalby-Payne, J.; Wilson, M.; Dexter, M.; Rice, G.I.; Crow, Y.J.; Dale, R.C. Systemic lupus erythematosus due to C1q deficiency with progressive encephalopathy, intracranial calcification and acquired moyamoya cerebral vasculopathy. Lupus 2013, 22, 639–643. [Google Scholar] [CrossRef]
- Stegert, M.; Bock, M.; Trendelenburg, M. Clinical presentation of human C1q deficiency: How much of a lupus? Mol. Immunol. 2015, 67, 3–11. [Google Scholar] [CrossRef]
- Arkwright, P.D.; Riley, P.; Hughes, S.M.; Alachkar, H.; Wynn, R.F. Successful cure of C1q deficiency in human subjects treated with hematopoietic stem cell transplantation. J. Allergy Clin. Immunol. 2014, 133, 265–267. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.; Norsworthy, P.J.; Hall, A.E.; Kelly, S.J.; Walport, M.J.; Botto, M.; Pickering, M.C. SLE with C1q deficiency treated with fresh frozen plasma: A 10-year experience. Rheumatology 2010, 49, 823–824. [Google Scholar] [CrossRef]
- van Schaarenburg, R.A.; Magro-Checa, C.; Bakker, J.A.; Teng, Y.K.O.; Bajema, I.M.; Huizinga, T.W.; Steup-Beekman, G.M.; Trouw, L.A. C1q Deficiency and Neuropsychiatric Systemic Lupus Erythematosus. Front. Immunol. 2016, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bienaimé, F.; Quartier, P.; Dragon-Durey, M.-A.; Frémeaux-Bacchi, V.; Bader-Meunier, B.; Patey, N.; Salomon, R.; Noël, L.-H. Lupus nephritis associated with complete C1s deficiency efficiently treated with rituximab: A case report. Arthritis Care Res. 2010, 62, 1346–1350. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Brookshire, B.; Yu, C.-Y.; Arnett, F. Molecular basis of complement C1r deficiency in a male African American patient with systemic lupus erythematosus. Mol. Immunol. 2010, 47, 2219–2220. [Google Scholar] [CrossRef]
- Jönsson, G.; Sjöholm, A.G.; Truedsson, L.; Bengtsson, A.A.; Braconier, J.H.; Sturfelt, G. Rheumatological manifestations, organ damage and autoimmunity in hereditary C2 deficiency. Rheumatology 2007, 46, 1133–1139. [Google Scholar] [CrossRef]
- Blanchong, C.A.; Chung, E.K.; Rupert, K.L.; Yang, Y.; Yang, Z.; Zhou, B.; Moulds, J.M.; Yu, C.Y. Genetic, structural and functional diversities of human complement components C4A and C4B and their mouse homologues, Slp and C4. Int. Immunopharmacol. 2001, 1, 365–392. [Google Scholar] [CrossRef]
- Yang, Y.; Chung, E.K.; Wu, Y.L.; Savelli, S.L.; Nagaraja, H.N.; Zhou, B.; Hebert, M.; Jones, K.N.; Shu, Y.; Kitzmiller, K.; et al. Gene Copy-Number Variation and Associated Polymorphisms of Complement Component C4 in Human Systemic Lupus Erythematosus (SLE): Low Copy Number Is a Risk Factor for and High Copy Number Is a Protective Factor against SLE Susceptibility in European America. Am. J. Hum. Genet. 2007, 80, 1037–1054. [Google Scholar] [CrossRef]
- Yih Chen, J.; Ling Wu, Y.; Yin Mok, M.; Jan Wu, Y.-J.; Lintner, K.E.; Wang, C.-M.; Chung, E.K.; Yang, Y.; Zhou, B.; Wang, H.; et al. Effects of Complement C4 Gene Copy Number Variations, Size Dichotomy, and C4A Deficiency on Genetic Risk and Clinical Presentation of Systemic Lupus Erythematosus in East Asian Populations. Arthritis Rheumatol. 2016, 68, 1442–1453. [Google Scholar] [CrossRef]
- Jüptner, M.; Flachsbart, F.; Caliebe, A. Low copy numbers of complement C4 and homozygous deficiency of C4A may predispose to severe disease and earlier disease onset in patients with systemic lupus erythematosus. Lupus 2017, 096120331773518. [Google Scholar] [CrossRef] [PubMed]
- Pereira, K.M.C.; Faria, A.G.A.; Liphaus, B.L.; Jesus, A.A.; Silva, C.A.; Carneiro-Sampaio, M.; Andrade, L.E.C. LowC4,C4AandC4Bgene copy numbers are stronger risk factors for juvenile-onset than for adult-onset systemic lupus erythematosus. Rheumatology 2016, 55, 869–873. [Google Scholar] [CrossRef] [PubMed]
- Bodaño, A.; Amarelo, J.; González, A.; Gómez-Reino, J.J.; Conde, C. Novel DNASE I mutations related to systemic lupus erythematosus. Arthritis Rheum. 2004, 50, 4070–4071. [Google Scholar] [CrossRef] [PubMed]
- Yasutomo, K.; Horiuchi, T.; Kagami, S.; Tsukamoto, H.; Hashimura, C.; Urushihara, M.; Kuroda, Y. Mutation of DNASE1 in people with systemic lupus erythematosus. Nat. Genet. 2001, 28, 313–314. [Google Scholar] [CrossRef]
- Bodaño, A.; González, A.; Ferreiros-Vidal, I.; Balada, E.; Ordi, J.; Carreira, P.; Goómez-Reino, J.J.; Conde, C. Association of a non-synonymous single-nucleotide polymorphism of DNASEI with SLE susceptibility. Rheumatology 2006, 45, 819–823. [Google Scholar] [CrossRef]
- Rodero, M.P.; Tesser, A.; Bartok, E.; Rice, G.I.; Della Mina, E.; Depp, M.; Beitz, B.; Bondet, V.; Cagnard, N.; Duffy, D.; et al. Type I interferon-mediated autoinflammation due to DNase II deficiency. Nat. Commun. 2017, 8, 2176. [Google Scholar] [CrossRef]
- Kawane, K. Requirement of DNase II for Definitive Erythropoiesis in the Mouse Fetal Liver. Science 2001, 292, 1546–1549. [Google Scholar] [CrossRef]
- Al-Mayouf, S.M.; Sunker, A.; Abdwani, R.; Abrawi, S.A.; Almurshedi, F.; Alhashmi, N.; Al Sonbul, A.; Sewairi, W.; Qari, A.; Abdallah, E.; et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat. Genet. 2011, 43, 1186–1188. [Google Scholar] [CrossRef]
- Özçakar, Z.B.; Foster, J.; Diaz-Horta, O.; Kasapcopur, O.; Fan, Y.-S.; Yalçınkaya, F.; Tekin, M. DNASE1L3 Mutations in Hypocomplementemic Urticarial Vasculitis Syndrome. Arthritis Rheum. 2013, 65, 2183–2189. [Google Scholar] [CrossRef]
- Ablasser, A.; Hemmerling, I.; Schmid-Burgk, J.L.; Behrendt, R.; Roers, A.; Hornung, V. TREX1 Deficiency Triggers Cell-Autonomous Immunity in a cGAS-Dependent Manner. J. Immunol. 2014, 192, 5993–5997. [Google Scholar] [CrossRef]
- Lehtinen, D.A.; Harvey, S.; Mulcahy, M.J.; Hollis, T.; Perrino, F.W. The TREX1 Double-stranded DNA Degradation Activity Is Defective in Dominant Mutations Associated with Autoimmune Disease. J. Biol. Chem. 2008, 283, 31649–31656. [Google Scholar] [CrossRef] [PubMed]
- Grimbacher, B.; Warnatz, K.; Yong, P.F.K.; Korganow, A.-S.; Peter, H.-H. The crossroads of autoimmunity and immunodeficiency: Lessons from polygenic traits and monogenic defects. J. Allergy Clin. Immunol. 2016, 137, 3–17. [Google Scholar] [CrossRef] [PubMed]
- de Vries, B.; Steup-Beekman, G.M.; Haan, J.; Bollen, E.L.; Luyendijk, J.; Frants, R.R.; Terwindt, G.M.; van Buchem, M.A.; Huizinga, T.W.J.; van den Maagdenberg, A.M.J.M.; et al. TREX1 gene variant in neuropsychiatric systemic lupus erythematosus. Ann. Rheum. Dis. 2010, 69, 1886–1887. [Google Scholar] [CrossRef] [PubMed]
- Lee-Kirsch, M.A.; Gong, M.; Chowdhury, D.; Senenko, L.; Engel, K.; Lee, Y.-A.; de Silva, U.; Bailey, S.L.; Witte, T.; Vyse, T.J.; et al. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat. Genet. 2007, 39, 1065–1067. [Google Scholar] [CrossRef] [PubMed]
- Namjou, B.; Kothari, P.H.; Kelly, J.A.; Glenn, S.B.; Ojwang, J.O.; Adler, A.; Alarcón-Riquelme, M.E.; Gallant, C.J.; Boackle, S.A.; Criswell, L.A.; et al. Evaluation of the TREX1 gene in a large multi-ancestral lupus cohort. Genes Immun. 2011, 12, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Barizzone, N.; Monti, S.; Mellone, S.; Godi, M.; Marchini, M.; Scorza, R.; Danieli, M.G.; D’Alfonso, S. Rare Variants in the TREX1 Gene and Susceptibility to Autoimmune Diseases. Biomed Res. Int. 2013, 2013, 1–6. [Google Scholar] [CrossRef]
- Abe, J.; Izawa, K.; Nishikomori, R.; Awaya, T.; Kawai, T.; Yasumi, T.; Hiragi, N.; Hiragi, T.; Ohshima, Y.; Heike, T. Heterozygous TREX1 p.Asp18Asn mutation can cause variable neurological symptoms in a family with Aicardi-Goutieres syndrome/familial chilblain lupus. Rheumatology 2013, 52, 406–408. [Google Scholar] [CrossRef]
- Bursztejn, A.C.; Briggs, T.A.; del Toro Duany, Y.; Anderson, B.H.; O’Sullivan, J.; Williams, S.G.; Bodemer, C.; Fraitag, S.; Gebhard, F.; Leheup, B.; et al. Unusual cutaneous features associated with a heterozygous gain-of-function mutation in IFIH1: Overlap between Aicardi-Goutières and Singleton-Merten syndromes. Br. J. Dermatol. 2015, 173, 1505–1513. [Google Scholar] [CrossRef]
- Rice, G.I.; Del Toro Duany, Y.; Jenkinson, E.M.; Forte, G.M.A.; Anderson, B.H.; Ariaudo, G.; Bader-Meunier, B.; Baildam, E.M.; Battini, R.; Beresford, M.W.; et al. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type i interferon signaling. Nat. Genet. 2014, 46, 503–509. [Google Scholar] [CrossRef]
- Van Eyck, L.; De Somer, L.; Pombal, D.; Bornschein, S.; Frans, G.; Humblet-Baron, S.; Moens, L.; De Zegher, F.; Bossuyt, X.; Wouters, C.; et al. Brief report: IFIH1 mutation causes systemic lupus erythematosus with selective IgA deficiency. Arthritis Rheumatol. 2015, 67, 1592–1597. [Google Scholar] [CrossRef]
- Rutsch, F.; MacDougall, M.; Lu, C.; Buers, I.; Mamaeva, O.; Nitschke, Y.; Rice, G.I.; Erlandsen, H.; Kehl, H.G.; Thiele, H.; et al. A specific IFIH1 gain-of-function mutation causes Singleton-Merten syndrome. Am. J. Hum. Genet. 2015, 96, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Funabiki, M.; Kato, H.; Miyachi, Y.; Toki, H.; Motegi, H.; Inoue, M.; Minowa, O.; Yoshida, A.; Deguchi, K.; Sato, H.; et al. Autoimmune Disorders Associated with Gain of Function of the Intracellular Sensor MDA5. Immunity 2014, 40, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.A.; Kim, E.K.; Now, H.; Nguyen, N.T.; Kim, W.J.; Yoo, J.Y.; Lee, J.; Jeong, Y.M.; Kim, C.H.; Kim, O.H.; et al. Mutations in DDX58, which encodes RIG-I, cause atypical Singleton-Merten syndrome. Am. J. Hum. Genet. 2015, 96, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Meuwissen, M.E.C.; Schot, R.; Buta, S.; Oudesluijs, G.; Tinschert, S.; Speer, S.D.; Li, Z.; van Unen, L.; Heijsman, D.; Goldmann, T.; et al. Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome. J. Exp. Med. 2016, 213, 1163–1174. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Bogunovic, D.; Payelle-Brogard, B.; Francois-Newton, V.; Speer, S.D.; Yuan, C.; Volpi, S.; Li, Z.; Sanal, O.; Mansouri, D.; et al. Human intracellular ISG15 prevents interferon-α/β over-amplification and auto-inflammation. Nature 2015, 517, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Huang, J.; Liu, Y.; Xiao, L.; Wang, D.; Hua, B.; Tsao, B.P.; Sun, L. Identification of interferon-inducible genes as diagnostic biomarker for systemic lupus erythematosus. Clin. Rheumatol. 2015, 34, 71–79. [Google Scholar] [CrossRef]
- Yuan, Y.; Ma, H.; Ye, Z.; Jing, W.; Jiang, Z. Interferon-stimulated gene 15 expression in systemic lupus erythematosus. Z. Für Rheumatol. 2018, 77, 256–262. [Google Scholar] [CrossRef]
- Kretschmer, S.; Wolf, C.; König, N.; Staroske, W.; Guck, J.; Häusler, M.; Luksch, H.; Nguyen, L.A.; Kim, B.; Alexopoulou, D.; et al. SAMHD1 prevents autoimmunity by maintaining genome stability. Ann. Rheum. Dis. 2015, 74, e17. [Google Scholar] [CrossRef]
- Clifford, R.; Louis, T.; Robbe, P.; Ackroyd, S.; Burns, A.; Timbs, A.T.; Wright Colopy, G.; Dreau, H.; Sigaux, F.; Judde, J.G.; et al. SAMHD1 is mutated recurrently in chronic lymphocytic leukemia and is involved in response to DNA damage. Blood 2014, 123, 1021–1031. [Google Scholar] [CrossRef]
- Ravenscroft, J.C.; Suri, M.; Rice, G.I.; Szynkiewicz, M.; Crow, Y.J. Autosomal dominant inheritance of a heterozygous mutation in SAMHD1 causing familial chilblain lupus. Am. J. Med Genet. Part A 2011, 155, 235–237. [Google Scholar] [CrossRef]
- Dale, R.C.; Gornall, H.; Singh-Grewal, D.; Alcausin, M.; Rice, G.I.; Crow, Y.J. Familial Aicardi-Goutières syndrome due to SAMHD1 mutations is associated with chronic arthropathy and contractures. Am. J. Med Genet. Part A 2010, 152A, 938–942. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Song, J.; Sun, Y.; Qi, F.; Liu, L.; Jin, Y.; McNutt, M.A.; Yin, Y. Mutations of deubiquitinase OTUD1 are associated with autoimmune disorders. J. Autoimmun. 2018, 94, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Bilginer, Y.; Düzova, A.; Topaloğlu, R.; Batu, E.D.; Boduroğlu, K.; Güçer, Ş.; Bodur, İ.; Alanay, Y. Three cases of spondyloenchondrodysplasia (SPENCD) with systemic lupus erythematosus: A case series and review of the literature. Lupus 2016, 25, 760–765. [Google Scholar] [CrossRef] [PubMed]
- Briggs, T.A.; Rice, G.I.; Daly, S.; Urquhart, J.; Gornall, H.; Bader-Meunier, B.; Baskar, K.; Baskar, S.; Baudouin, V.; Beresford, M.W.; et al. Tartrate-resistant acid phosphatase deficiency causes a bone dysplasia with autoimmunity and a type I interferon expression signature. Nat. Genet. 2011, 43, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Lausch, E.; Janecke, A.; Bros, M.; Trojandt, S.; Alanay, Y.; De Laet, C.; Hübner, C.A.; Meinecke, P.; Nishimura, G.; Matsuo, M.; et al. Genetic deficiency of tartrate-resistant acid phosphatase associated with skeletal dysplasia, cerebral calcifications and autoimmunity. Nat. Genet. 2011, 43, 132–137. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Briggs, T.A.; Dumax-Vorzet, A.; Alarcón-Riquelme, M.E.; Belot, A.; Beresford, M.; Bruce, I.N.; Carvalho, C.; Chaperot, L.; Frostegård, J.; et al. Tartrate-Resistant Acid Phosphatase Deficiency in the Predisposition to Systemic Lupus Erythematosus. Arthritis Rheumatol. 2017, 69, 131–142. [Google Scholar] [CrossRef]
- Jeremiah, N.; Neven, B.; Gentili, M.; Callebaut, I.; Maschalidi, S.; Stolzenberg, M.-C.; Goudin, N.; Frémond, M.-L.; Nitschke, P.; Molina, T.J.; et al. Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. J. Clin. Investig. 2014, 124, 5516–5520. [Google Scholar] [CrossRef]
- Bennion, B.G.; Ingle, H.; Ai, T.L.; Miner, C.A.; Platt, D.J.; Smith, A.M.; Baldridge, M.T.; Miner, J.J. A Human Gain-of-Function STING Mutation Causes Immunodeficiency and Gammaherpesvirus-Induced Pulmonary Fibrosis in Mice. J. Virol. 2019, 93, e01806-18. [Google Scholar] [CrossRef]
- König, N.; Fiehn, C.; Wolf, C.; Schuster, M.; Cura Costa, E.; Tüngler, V.; Alvarez, H.A.; Chara, O.; Engel, K.; Goldbach-Mansky, R.; et al. Familial chilblain lupus due to a gain-of-function mutation in STING. Ann. Rheum. Dis. 2017, 76, 468–472. [Google Scholar] [CrossRef]
- Bouis, D.; Kirstetter, P.; Arbogast, F.; Lamon, D.; Delgado, V.; Jung, S.; Ebel, C.; Jacobs, H.; Knapp, A.M.; Jeremiah, N.; et al. Severe combined immunodeficiency in stimulator of interferon genes (STING) V154M/wild-type mice. J. Allergy Clin. Immunol. 2019, 143, 712–725.e715. [Google Scholar] [CrossRef]
- Crow, Y.J.; Chase, D.S.; Lowenstein Schmidt, J.; Szynkiewicz, M.; Forte, G.M.A.; Gornall, H.L.; Oojageer, A.; Anderson, B.; Pizzino, A.; Helman, G.; et al. Characterization of human disease phenotypes associated with mutations in TREX1, 61, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am. J. Med Genet. Part A 2015, 167, 296–312. [Google Scholar] [CrossRef] [PubMed]
- Rice, G.I.; Kasher, P.R.; Forte, G.M.A.; Mannion, N.M.; Greenwood, S.M.; Szynkiewicz, M.; Dickerson, J.E.; Bhaskar, S.S.; Zampini, M.; Briggs, T.A.; et al. Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat. Genet. 2012, 44, 1243–1248. [Google Scholar] [CrossRef] [PubMed]
- Belot, A.; Kasher, P.R.; Trotter, E.W.; Foray, A.-P.; Debaud, A.-L.; Rice, G.I.; Szynkiewicz, M.; Zabot, M.-T.; Rouvet, I.; Bhaskar, S.S.; et al. Protein Kinase Cδ Deficiency Causes Mendelian Systemic Lupus Erythematosus With B Cell-Defective Apoptosis and Hyperproliferation. Arthritis Rheum. 2013, 65, 2161–2171. [Google Scholar] [CrossRef] [PubMed]
- Kiykim, A.; Ogulur, I.; Baris, S.; Salzer, E.; Karakoc-Aydiner, E.; Ozen, A.O.; Garncarz, W.; Hirschmugl, T.; Krolo, A.; Yucelten, A.D.; et al. Potentially Beneficial Effect of Hydroxychloroquine in a Patient with a Novel Mutation in Protein Kinase Cδ Deficiency. J. Clin. Immunol. 2015, 35, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Kuehn, H.S.; Niemela, J.E.; Rangel-Santos, A.; Zhang, M.; Pittaluga, S.; Stoddard, J.L.; Hussey, A.A.; Evbuomwan, M.O.; Priel, D.A.L.; Kuhns, D.B.; et al. Loss-of-function of the protein kinase C (PKC ) causes a B-cell lymphoproliferative syndrome in humans. Blood 2013, 121, 3117–3125. [Google Scholar] [CrossRef] [PubMed]
- Nanthapisal, S.; Omoyinmi, E.; Murphy, C.; Standing, A.; Eisenhut, M.; Eleftheriou, D.; Brogan, P.A. Early-Onset Juvenile SLE Associated With a Novel Mutation in Protein Kinase C δ. Pediatrics 2017, 139, e20160781. [Google Scholar] [CrossRef]
- Salzer, E.; Santos-Valente, E.; Klaver, S.; Ban, S.A.; Emminger, W.; Prengemann, N.K.; Garncarz, W.; Mullauer, L.; Kain, R.; Boztug, H.; et al. B-cell deficiency and severe autoimmunity caused by deficiency of protein kinase C. Blood 2013, 121, 3112–3116. [Google Scholar] [CrossRef]
- Theodorou, E.; Nezos, A.; Antypa, E.; Ioakeimidis, D.; Koutsilieris, M.; Tektonidou, M.; Moutsopoulos, H.M.; Mavragani, C.P. B-cell activating factor and related genetic variants in lupus related atherosclerosis. J. Autoimmun. 2018, 92, 87–92. [Google Scholar] [CrossRef]
- Gonzalez-Serna, D.; Ortiz-Fernandez, L.; Vargas, S.; Garcia, A.; Raya, E.; Fernandez-Gutierrez, B.; Lopez-Longo, F.J.; Balsa, A.; Gonzalez-Alvaro, I.; Narvaez, J.; et al. Association of a rare variant of the TNFSF13B gene with susceptibility to Rheumatoid Arthritis and Systemic Lupus Erythematosus. Sci. Rep. 2018, 8, 8195. [Google Scholar] [CrossRef]
- Quaio, C.R.D.C.; Carvalho, J.F.; da Silva, C.A.; Bueno, C.; Brasil, A.S.; Pereira, A.C.; Jorge, A.A.L.; Malaquias, A.C.; Kim, C.A.; Bertola, D.R. Autoimmune disease and multiple autoantibodies in 42 patients with RASopathies. Am. J. Med Genet. Part A 2012, 158A, 1077–1082. [Google Scholar] [CrossRef]
- Şimşek-Kiper, P.; Alanay, Y.; Gülhan, B.; Lissewski, C.; Türkyılmaz, D.; Alehan, D.; Çetin, M.; Utine, G.E.; Zenker, M.; Boduroğlu, K. Clinical and molecular analysis of RASopathies in a group of Turkish patients. Clin. Genet. 2013, 83, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Alanay, Y.; Balci, S.; Ozen, S. Noonan syndrome and systemic lupus erythematosus: Presentation in childhood. Clin. Dysmorphol. 2004, 13, 161–163. [Google Scholar] [CrossRef] [PubMed]
- Amoroso, A.; Garzia, P.; Vadacca, M.; Galluzzo, S.; Delporto, F.; Mitterhofer, A.; Afeltra, A. The unusual association of three autoimmune diseases in a patient with Noonan syndrome. J. Adolesc. Health 2003, 32, 94–97. [Google Scholar] [CrossRef]
- Bader-Meunier, B.; Cavé, H.; Jeremiah, N.; Magerus, A.; Lanzarotti, N.; Rieux-Laucat, F.; Cormier-Daire, V. Are RASopathies new monogenic predisposing conditions to the development of systemic lupus erythematosus? Case report and systematic review of the literature. Semin. Arthritis Rheum. 2013, 43, 217–219. [Google Scholar] [CrossRef] [PubMed]
- Hanaya, A.; Miyamae, T.; Kishi, T.; Sahara, M.; Tani, Y.; Yamanaka, H.; Nagata, S. Systemic lupus erythematosus associated with RASopathy. Mod. Rheumatol. Case Rep. 2017, 1, 94–98. [Google Scholar] [CrossRef]
- Leventopoulos, G.; Denayer, E.; Makrythanasis, P.; Papapolychroniou, C.; Fryssira, H. Noonan syndrome and systemic lupus erythematosus in a patient with a novel KRAS mutation. Clin. Exp. Rheumatol. 2010, 28, 556–557. [Google Scholar]
- Lisbona, M.P.; Moreno, M.; Orellana, C.; Gratacos, J.; Larrosa, M. Noonan syndrome associated with systemic lupus erythematosus. Lupus 2009, 18, 267–269. [Google Scholar] [CrossRef]
- Lopez-Rangel, E.; Malleson, P.N.; Lirenman, D.S.; Roa, B.; Wiszniewska, J.; Suzanne Lewis, M.E. Systemic lupus erythematosus and other autoimmune disorders in children with Noonan syndrome. Am. J. Med. Genet. Part A 2005, 139A, 239–242. [Google Scholar] [CrossRef]
- Martin, D.M.; Gencyuz, C.F.; Petty, E.M. Systemic lupus erythematosus in a man with Noonan syndrome. Am. J. Med. Genet. 2001, 102, 59–62. [Google Scholar] [CrossRef][Green Version]
- Ragotte, R.J.; Dhanrajani, A.; Pleydell-Pearce, J.; Del Bel, K.L.; Tarailo-Graovac, M.; van Karnebeek, C.; Terry, J.; Senger, C.; McKinnon, M.L.; Seear, M.; et al. The importance of considering monogenic causes of autoimmunity: A somatic mutation in KRAS causing pediatric Rosai-Dorfman syndrome and systemic lupus erythematosus. Clin. Immunol. 2017, 175, 143–146. [Google Scholar] [CrossRef]
- Uehara, T.; Hosogaya, N.; Matsuo, N.; Kosaki, K. Systemic lupus erythematosus in a patient with Noonan syndrome-like disorder with loose anagen hair 1: More than a chance association. Am. J. Med. Genet. Part A 2018, 176, 1662–1666. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Wu, W.; Mathew, D.; Zhang, Y.; Browne, S.K.; Rosen, L.B.; McManus, M.P.; Pulsipher, M.A.; Yandell, M.; Bohnsack, J.F.; et al. Autoimmunity due to RAG deficiency and estimated disease incidence in RAG1/2 mutations. J. Allergy Clin. Immunol. 2014, 133, 880–882.e810. [Google Scholar] [CrossRef] [PubMed]
- Walter, J.E.; Lo, M.S.; Kis-Toth, K.; Tirosh, I.; Frugoni, F.; Lee, Y.N.; Csomos, K.; Chen, K.; Pillai, S.; Dunham, J.; et al. Impaired receptor editing and heterozygous RAG2 mutation in a patient with systemic lupus erythematosus and erosive arthritis. J. Allergy Clin. Immunol. 2015, 135, 272–273. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wilson, J.; He, J.; Xiang, L.; Schur, P.H.; Mountz, J.D. Fas ligand mutation in a patient with systemic lupus erythematosus and lymphoproliferative disease. J. Clin. Investig. 1996, 98, 1107–1113. [Google Scholar] [CrossRef] [PubMed]
- Xiang, N.; Li, X.-m.; Wang, G.-s.; Tao, J.-h.; Li, X.-p. Association of Fas gene polymorphisms with systemic lupus erythematosus: A meta-analysis. Mol. Biol. Rep. 2013, 40, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Aeschlimann, F.A.; Batu, E.D.; Canna, S.W.; Go, E.; Gül, A.; Hoffmann, P.; Leavis, H.L.; Ozen, S.; Schwartz, D.M.; Stone, D.L.; et al. A20 haploinsufficiency (HA20): Clinical phenotypes and disease course of patients with a newly recognised NF-kB-mediated autoinflammatory disease. Ann. Rheum. Dis. 2018, 77, 728–735. [Google Scholar] [CrossRef]
- Zhou, Q.; Wang, H.; Schwartz, D.M.; Stoffels, M.; Park, Y.H.; Zhang, Y.; Yang, D.; Demirkaya, E.; Takeuchi, M.; Tsai, W.L.; et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat. Genet. 2016, 48, 67–73. [Google Scholar] [CrossRef]
- Kadowaki, T.; Ohnishi, H.; Kawamoto, N.; Hori, T.; Nishimura, K.; Kobayashi, C.; Shigemura, T.; Ogata, S.; Inoue, Y.; Kawai, T.; et al. Haploinsufficiency of A20 causes autoinflammatory and autoimmune disorders. J. Allergy Clin. Immunol. 2018, 141, 1485–1488.e1411. [Google Scholar] [CrossRef]
- Schwartz, D.M.; Blackstone, S.A.; Sampaio-Moura, N.; Rosenzweig, S.; Burma, A.M.; Stone, D.; Hoffmann, P.; Jones, A.; Romeo, T.; Barron, K.S.; et al. Type I interferon signature predicts response to JAK inhibition in haploinsufficiency of A20. Ann. Rheum. Dis. 2020, 79, 429–431. [Google Scholar] [CrossRef]
- Takagi, M.; Ogata, S.; Ueno, H.; Yoshida, K.; Yeh, T.; Hoshino, A.; Piao, J.; Yamashita, M.; Nanya, M.; Okano, T.; et al. Haploinsufficiency of TNFAIP3 ( A20 ) by germline mutation is involved in autoimmune lymphoproliferative syndrome. J. Allergy Clin. Immunol. 2017, 139, 1914–1922. [Google Scholar] [CrossRef]
- Schepp, J.; Bulashevska, A.; Mannhardt-Laakmann, W.; Cao, H.; Yang, F.; Seidl, M.; Kelly, S.; Hershfield, M.; Grimbacher, B. Deficiency of Adenosine Deaminase 2 Causes Antibody Deficiency. J. Clin. Immunol. 2016, 36, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Skrabl-Baumgartner, A.; Plecko, B.; Schmidt, W.M.; Konig, N.; Hershfield, M.; Gruber-Sedlmayr, U.; Lee-Kirsch, M.A. Autoimmune phenotype with type I interferon signature in two brothers with ADA2 deficiency carrying a novel CECR1 mutation. Pediatric Rheumatol. Online J. 2017, 15, 67. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Okada, S.; Yoshida, K.; Nishida, N.; Okuno, Y.; Ueno, H.; Yamashita, M.; Okano, T.; Tsumura, M.; Nishimura, S.; et al. Abnormal hematopoiesis and autoimmunity in human subjects with germline IKZF1 mutations. J. Allergy Clin. Immunol. 2017, 140, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Kuehn, H.S.; Boisson, B.; Cunningham-Rundles, C.; Reichenbach, J.; Stray-Pedersen, A.; Gelfand, E.W.; Maffucci, P.; Pierce, K.R.; Abbott, J.K.; Voelkerding, K.V.; et al. Loss of B Cells in Patients with Heterozygous Mutations in IKAROS. N. Engl. J. Med. 2016, 374, 1032–1043. [Google Scholar] [CrossRef]
- Zhang, Y.M.; Zhou, X.J.; Cheng, F.J.; Qi, Y.Y.; Hou, P.; Zhao, M.H.; Zhang, H. Association of the IKZF1 5’ UTR variant rs1456896 with lupus nephritis in a northern Han Chinese population. Scand. J. Rheumatol. 2017, 46, 210–214. [Google Scholar] [CrossRef]
- Lintner, K.E.; Wu, Y.L.; Yang, Y.; Spencer, C.H.; Hauptmann, G.; Hebert, L.A.; Atkinson, J.P.; Yu, C.Y. Early Components of the Complement Classical Activation Pathway in Human Systemic Autoimmune Diseases. Front. Immunol. 2016, 7, 1–22. [Google Scholar] [CrossRef]
- Roozendaal, R.; Carroll, M.C. Complement Receptors CD21 and CD35 in Humoral Immunity. Immunol. Rev. 2007, 219, 157–166. [Google Scholar] [CrossRef]
- Macedo, A.C.L.; Isaac, L. Systemic Lupus Erythematosus and Deficiencies of Early Components of the Complement Classical Pathway. Front. Immunol. 2016, 7, 1–7. [Google Scholar] [CrossRef]
- Lewis, M.J.; Botto, M. Complement deficiencies in humans and animals: Links to autoimmunity. Autoimmunity 2006, 39, 367–378. [Google Scholar] [CrossRef]
- Alperin, J.M.; Ortiz-Fernández, L.; Sawalha, A.H. Monogenic Lupus: A Developing Paradigm of Disease. Front. Immunol. 2018, 9, 2496. [Google Scholar] [CrossRef]
- Batu, E.D. Monogenic systemic lupus erythematosus: Insights in pathophysiology. Rheumatol. Int. 2018, 38, 1763–1775. [Google Scholar] [CrossRef] [PubMed]
- Costa-Reis, P.; Sullivan, K.E. Monogenic lupus: it’s all new! Curr. Opin. Immunol. 2017, 49, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Hiraki, L.T.; Silverman, E.D. Genomics of Systemic Lupus Erythematosus. Rheum. Dis. Clin. N. Am. 2017, 43, 415–434. [Google Scholar] [CrossRef] [PubMed]
- Lipsker, D.; Hauptmann, G. Cutaneous manifestations of complement deficiencies. Lupus 2010, 19, 1096–1106. [Google Scholar] [CrossRef] [PubMed]
- Bhattad, S.; Rawat, A.; Gupta, A.; Suri, D.; Garg, R.; de Boer, M.; Kuijpers, T.W.; Singh, S. Early Complement Component Deficiency in a Single-Centre Cohort of Pediatric Onset Lupus. J. Clin. Immunol. 2015, 35, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Prodeus, A.P.; Goerg, S.; Shen, L.-M.; Pozdnyakova, O.O.; Chu, L.; Alicot, E.M.; Goodnow, C.C.; Carroll, M.C. A Critical Role for Complement in Maintenance of Self-Tolerance. Immunity 1998, 9, 721–731. [Google Scholar] [CrossRef]
- Furie, R.; Khamashta, M.; Merrill, J.T.; Werth, V.P.; Kalunian, K.; Brohawn, P.; Illei, G.G.; Drappa, J.; Wang, L.; Yoo, S. Anifrolumab, an Anti-Interferon-alpha Receptor Monoclonal Antibody, in Moderate-to-Severe Systemic Lupus Erythematosus. Arthritis Rheumatol. 2017, 69, 376–386. [Google Scholar] [CrossRef]
- Takeuchi, T.; Tanaka, Y.; Matsumura, R.; Saito, K.; Yoshimura, M.; Amano, K.; Atsumi, T.; Suematsu, E.; Hayashi, N.; Wang, L.; et al. Safety and tolerability of sifalimumab, an anti-interferon-alpha monoclonal antibody, in Japanese patients with systemic lupus erythematosus: A multicenter, phase 2, open-label study. Mod. Rheumatol./Jpn. Rheum. Assoc. 2020, 30, 93–100. [Google Scholar] [CrossRef]
- Khamashta, M.; Merrill, J.T.; Werth, V.P.; Furie, R.; Kalunian, K.; Illei, G.G.; Drappa, J.; Wang, L.; Greth, W. Sifalimumab, an anti-interferon-alpha monoclonal antibody, in moderate to severe systemic lupus erythematosus: A randomised, double-blind, placebo-controlled study. Ann. Rheum. Dis. 2016, 75, 1909–1916. [Google Scholar] [CrossRef]
- van Vollenhoven, R.F.; Hahn, B.H.; Tsokos, G.C.; Wagner, C.L.; Lipsky, P.; Touma, Z.; Werth, V.P.; Gordon, R.M.; Zhou, B.; Hsu, B.; et al. Efficacy and safety of ustekinumab, an IL-12 and IL-23 inhibitor, in patients with active systemic lupus erythematosus: Results of a multicentre, double-blind, phase 2, randomised, controlled study. Lancet 2018, 392, 1330–1339. [Google Scholar] [CrossRef]
- You, H.; Zhang, G.; Wang, Q.; Zhang, S.; Zhao, J.; Tian, X.; Li, H.; Li, M.; Zeng, X. Successful treatment of arthritis and rash with tofacitinib in systemic lupus erythematosus: The experience from a single centre. Ann. Rheum. Dis. 2019, 78, 1441–1443. [Google Scholar] [CrossRef] [PubMed]
- Bruschi, M.; Bonanni, A.; Petretto, A.; Vaglio, A.; Pratesi, F.; Santucci, L.; Migliorini, P.; Bertelli, R.; Galetti, M.; Belletti, S.; et al. Neutrophil Extracellular Traps Profiles in Patients with Incident Systemic Lupus Erythematosus and Lupus Nephritis. J. Rheumatol. 2019, 47. [Google Scholar] [CrossRef] [PubMed]
- Napirei, M.; Karsunky, H.; Zevnik, B.; Stephan, H.; Mannherz, H.G.; Möröy, T. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat. Genet. 2000, 25, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Sisirak, V.; Sally, B.; D’Agati, V.; Martinez-Ortiz, W.; Ozcakar, Z.B.; David, J.; Rashidfarrokhi, A.; Yeste, A.; Panea, C.; Chida, A.S.; et al. Digestion of Chromatin in Apoptotic Cell Microparticles Prevents Autoimmunity. Cell 2016, 166, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.D.; Park, B.L.; Kim, L.H.; Lee, H.-S.; Kim, T.-Y.; Bae, S.-C. Common DNase I polymorphism associated with autoantibody production among systemic lupus erythematosus patients. Hum. Mol. Genet. 2004, 13, 2343–2350. [Google Scholar] [CrossRef][Green Version]
- Stetson, D.B.; Ko, J.S.; Heidmann, T.; Medzhitov, R. Trex1 Prevents Cell-Intrinsic Initiation of Autoimmunity. Cell 2008, 134, 587–598. [Google Scholar] [CrossRef]
- Rice, G.I.; Rodero, M.P.; Crow, Y.J. Human Disease Phenotypes Associated With Mutations in TREX1. J. Clin. Immunol. 2015, 35, 235–243. [Google Scholar] [CrossRef]
- Rodero, M.P.; Crow, Y.J. Type I interferon–mediated monogenic autoinflammation: The type I interferonopathies, a conceptual overview. J. Exp. Med. 2016, 213, 2527–2538. [Google Scholar] [CrossRef]
- Liu, Y.; Jesus, A.A.; Marrero, B.; Yang, D.; Ramsey, S.E.; Sanchez, G.A.M.; Tenbrock, K.; Wittkowski, H.; Jones, O.Y.; Kuehn, H.S.; et al. Activated STING in a vascular and pulmonary syndrome. N. Engl. J. Med. 2014, 371, 507–518. [Google Scholar] [CrossRef]
- Gray, E.E.; Treuting, P.M.; Woodward, J.J.; Stetson, D.B. Cutting Edge: cGAS Is Required for Lethal Autoimmune Disease in the Trex1-Deficient Mouse Model of Aicardi–Goutières Syndrome. J. Immunol. 2015, 195, 1939–1943. [Google Scholar] [CrossRef]
- Abdel-Salam, G.M.H.; El-Kamah, G.Y.; Rice, G.I.; El-Darouti, M.; Gornall, H.; Szynkiewicz, M.; Aymard, F.; Zaki, M.S.; Abdel-Aleem, A.K.; Lebon, P.; et al. Chilblains as a Diagnostic Sign of Aicardi-Goutières Syndrome. Neuropediatrics 2010, 41, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Ramantani, G.; Kohlhase, J.; Hertzberg, C.; Innes, A.M.; Engel, K.; Hunger, S.; Borozdin, W.; Mah, J.K.; Ungerath, K.; Walkenhorst, H.; et al. Expanding the phenotypic spectrum of lupus erythematosus in Aicardi-Goutières syndrome. Arthritis Rheum. 2010, 62, 1469–1477. [Google Scholar] [CrossRef] [PubMed]
- Cattalini, M.; Galli, J.; Andreoli, L.; Olivieri, I.; Ariaudo, G.; Fredi, M.; Orcesi, S.; Tincani, A.; Fazzi, E. Exploring Autoimmunity in a Cohort of Children with Genetically Confirmed Aicardi–Goutières Syndrome. J. Clin. Immunol. 2016, 36, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, E.; Vanderver, A.; Brown, K.J.; Sandza, A.; Takanohashi, A.; Jansen, M.H.; Anink, J.; Herron, B.; Orcesi, S.; Olivieri, I.; et al. Aicardi–Goutières syndrome harbours abundant systemic and brain-reactive autoantibodies. Ann. Rheum. Dis. 2015, 74, 1931–1939. [Google Scholar] [CrossRef]
- Dostert, C.; Meylan, E.; Tschopp, J. Intracellular pattern recognition receptors in the host response. Nature 2006, 442, 39–44. [Google Scholar]
- Robinson, T.; Kariuki, S.N.; Franek, B.S.; Kumabe, M.; Kumar, A.A.; Badaracco, M.; Mikolaitis, R.A.; Guerrero, G.; Utset, T.O.; Drevlow, B.E.; et al. Autoimmune Disease Risk Variant of IFIH1 Is Associated with Increased Sensitivity to IFN- and Serologic Autoimmunity in Lupus Patients. J. Immunol. 2011, 187, 1298–1303. [Google Scholar] [CrossRef]
- Cen, H.; Wang, W.; Leng, R.-X.; Wang, T.-Y.; Pan, H.-F.; Fan, Y.-G.; Wang, B.; Ye, D.-Q. Association of IFIH1 rs1990760 polymorphism with susceptibility to autoimmune diseases: A meta-analysis. Autoimmunity 2013, 46, 455–462. [Google Scholar] [CrossRef]
- Wang, Y. Rig-I-/- mice develop colitis associated with downregulation of G alpha i2. Cell Res. 2007, 17, 858–868. [Google Scholar] [CrossRef]
- Burdette, D.L.; Monroe, K.M.; Sotelo-Troha, K.; Iwig, J.S.; Eckert, B.; Hyodo, M.; Hayakawa, Y.; Vance, R.E. STING is a direct innate immune sensor of cyclic di-GMP. Nature 2011, 478, 515–518. [Google Scholar] [CrossRef]
- Bouis, D.; Kirstetter, P.; Arbogast, F.; Lamon, D.; Delgado, V.; Jung, S.; Ebel, C.; Jacobs, H.; Knapp, A.-M.; Jeremiah, N.; et al. Severe combined immunodeficiency in Sting V154M/WT mice. J. Allergy Clin. Immunol. 2019, 143, 712–725.e5. [Google Scholar] [CrossRef]
- Hermann, M.; Bogunovic, D. ISG15: In Sickness and in Health. Trends Immunol. 2017, 38, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Salzer, E.; Santos-Valente, E.; Keller, B.; Warnatz, K.; Boztug, K. Protein Kinase C δ: A Gatekeeper of Immune Homeostasis. J. Clin. Immunol. 2016, 36, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, A.; Nakayama, K.; Imaki, H.; Hirose, S.; Jiang, Y.; Abe, M.; Tsukiyama, T.; Nagahama, H.; Ohno, S.; Hatakeyama, S.; et al. Increased proliferation of B cells and auto-immunity in mice lacking protein kinase Cδ. Nature 2002, 416, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Steri, M.; Orru, V.; Idda, M.L.; Pitzalis, M.; Pala, M.; Zara, I.; Sidore, C.; Faa, V.; Floris, M.; Deiana, M.; et al. Overexpression of the Cytokine BAFF and Autoimmunity Risk. N. Engl. J. Med. 2017, 376, 1615–1626. [Google Scholar] [CrossRef]
- Marin-Rosales, M.; Cruz, A.; Salazar-Camarena, D.C.; Santillan-Lopez, E.; Espinoza-Garcia, N.; Munoz-Valle, J.F.; Ramirez-Duenas, M.G.; Oregon-Romero, E.; Orozco-Barocio, G.; Palafox-Sanchez, C.A. High BAFF expression associated with active disease in systemic lupus erythematosus and relationship with rs9514828C>T polymorphism in TNFSF13B gene. Clin. Exp. Med. 2019, 19, 183–190. [Google Scholar] [CrossRef]
- St Jean, P.L.; Roth, D.A.; McCarthy, L.C.; Hughes, A.R. Pharmacogenetic analysis of belimumab fails to identify robust genetic predictors of efficacy in lupus. Pharm. Genom. 2019, 29, 132–135. [Google Scholar] [CrossRef]
- Glesse, N.; Vianna, P.; Paim, L.M.G.; Matte, M.C.C.; Aguiar, A.K.K.; Palhano, P.L.; Monticielo, O.A.; Brenol, C.V.; Xavier, R.M.; Chies, J.A.B. Evaluation of polymorphic variants in apoptotic genes and their role in susceptibility and clinical progression to systemic lupus erythematosus. Lupus 2017, 26, 746–755. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Demirkaya, E.; Sahin, S.; Romano, M.; Zhou, Q.; Aksentijevich, I. New Horizons in the Genetic Etiology of Systemic Lupus Erythematosus and Lupus-Like Disease: Monogenic Lupus and Beyond. J. Clin. Med. 2020, 9, 712. https://doi.org/10.3390/jcm9030712
Demirkaya E, Sahin S, Romano M, Zhou Q, Aksentijevich I. New Horizons in the Genetic Etiology of Systemic Lupus Erythematosus and Lupus-Like Disease: Monogenic Lupus and Beyond. Journal of Clinical Medicine. 2020; 9(3):712. https://doi.org/10.3390/jcm9030712
Chicago/Turabian StyleDemirkaya, Erkan, Sezgin Sahin, Micol Romano, Qing Zhou, and Ivona Aksentijevich. 2020. "New Horizons in the Genetic Etiology of Systemic Lupus Erythematosus and Lupus-Like Disease: Monogenic Lupus and Beyond" Journal of Clinical Medicine 9, no. 3: 712. https://doi.org/10.3390/jcm9030712
APA StyleDemirkaya, E., Sahin, S., Romano, M., Zhou, Q., & Aksentijevich, I. (2020). New Horizons in the Genetic Etiology of Systemic Lupus Erythematosus and Lupus-Like Disease: Monogenic Lupus and Beyond. Journal of Clinical Medicine, 9(3), 712. https://doi.org/10.3390/jcm9030712