Molecular Genetics of Niemann–Pick Type C Disease in Italy: An Update on 105 Patients and Description of 18 NPC1 Novel Variants

 ,
, 
 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Analysis of NPC1 and NPC2 Genes
2.3. Filipin Staining
2.4. mRNA Analysis
2.5. MLPA Assay
2.6. Western Blot Analysis
2.7. Nomenclature
3. Results
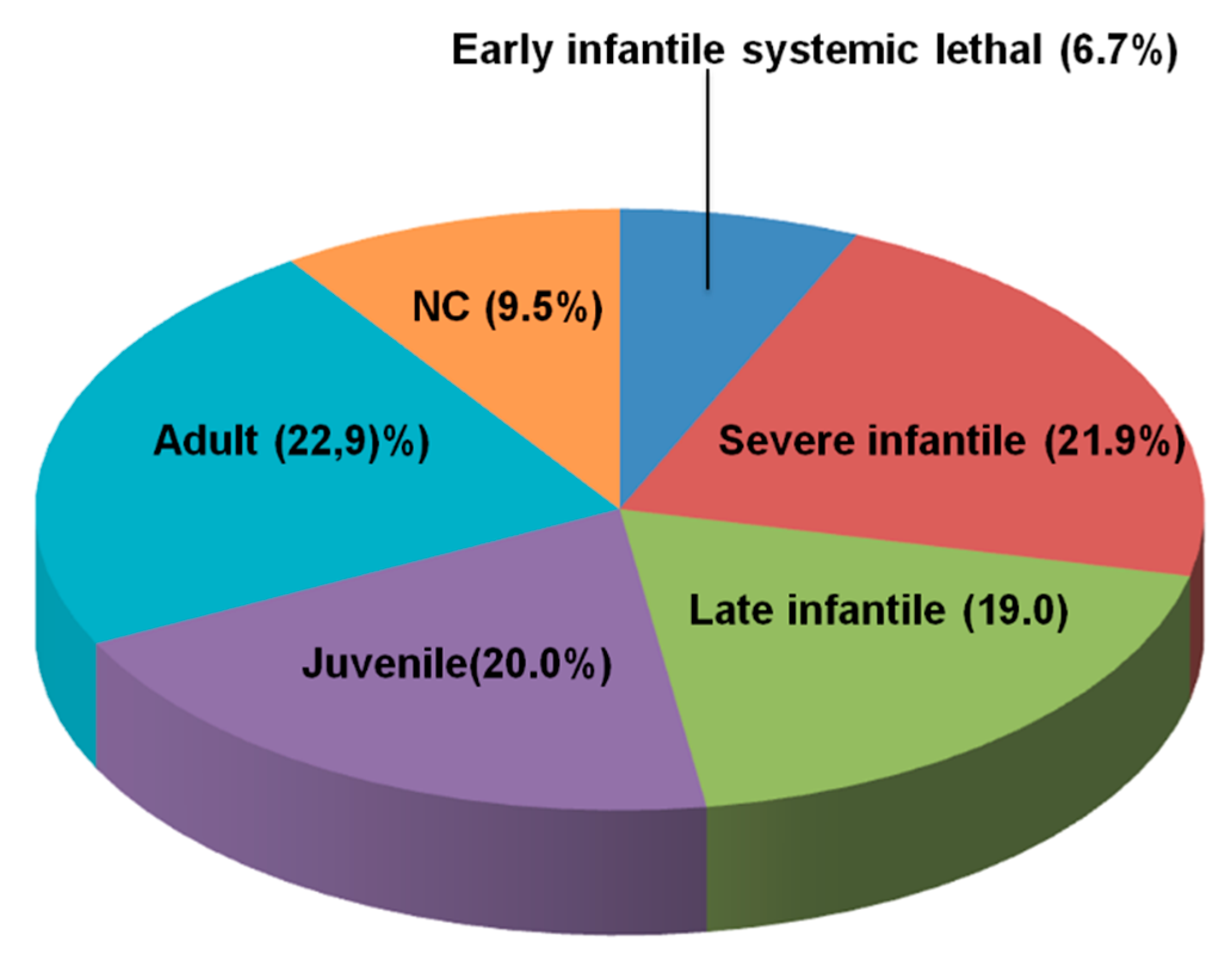
3.1. Distribution of NPC Clinical Phenotypes
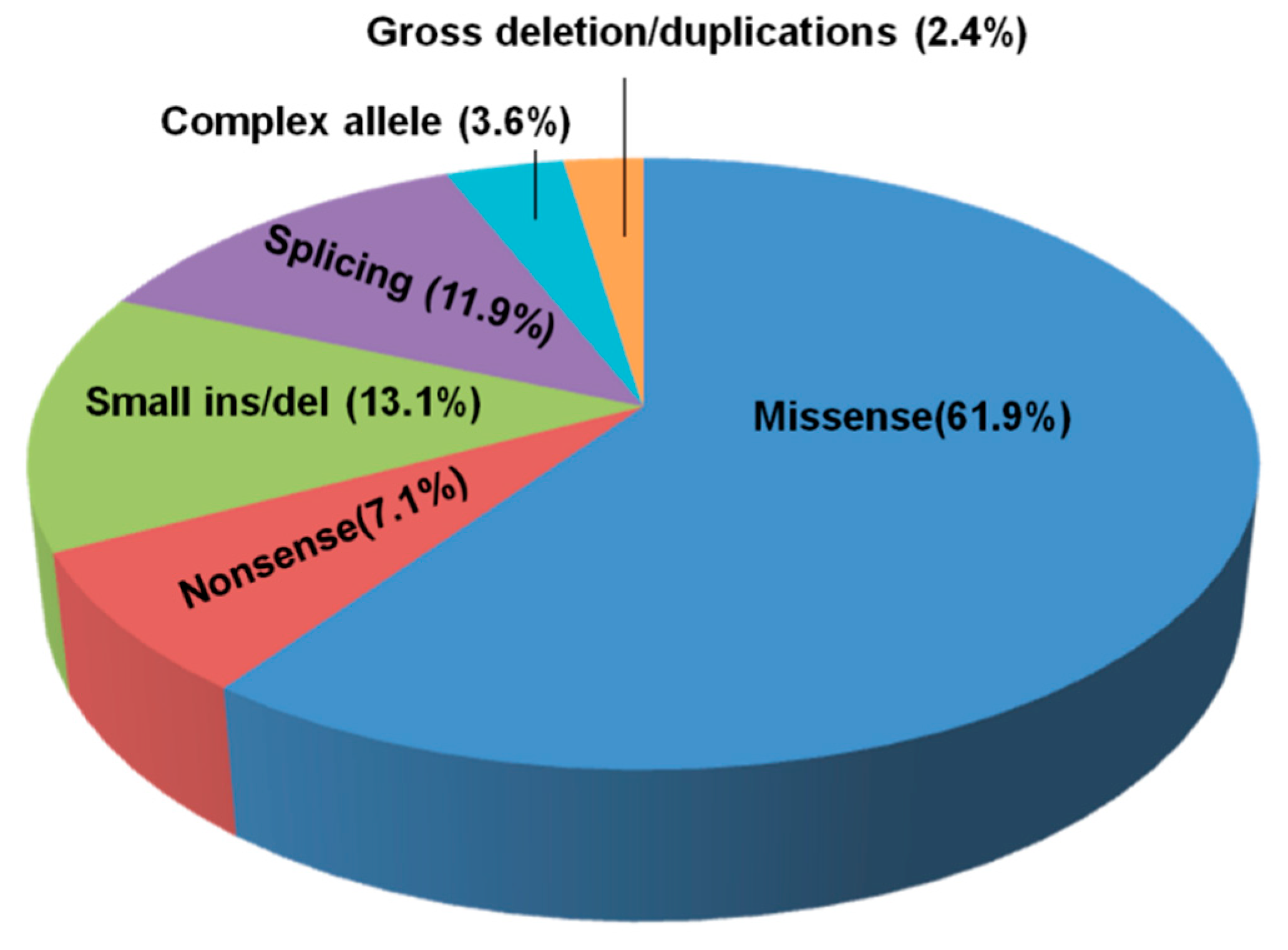
3.2. NPC1 and NPC2 Mutational Profile
3.3. Filipin Staining
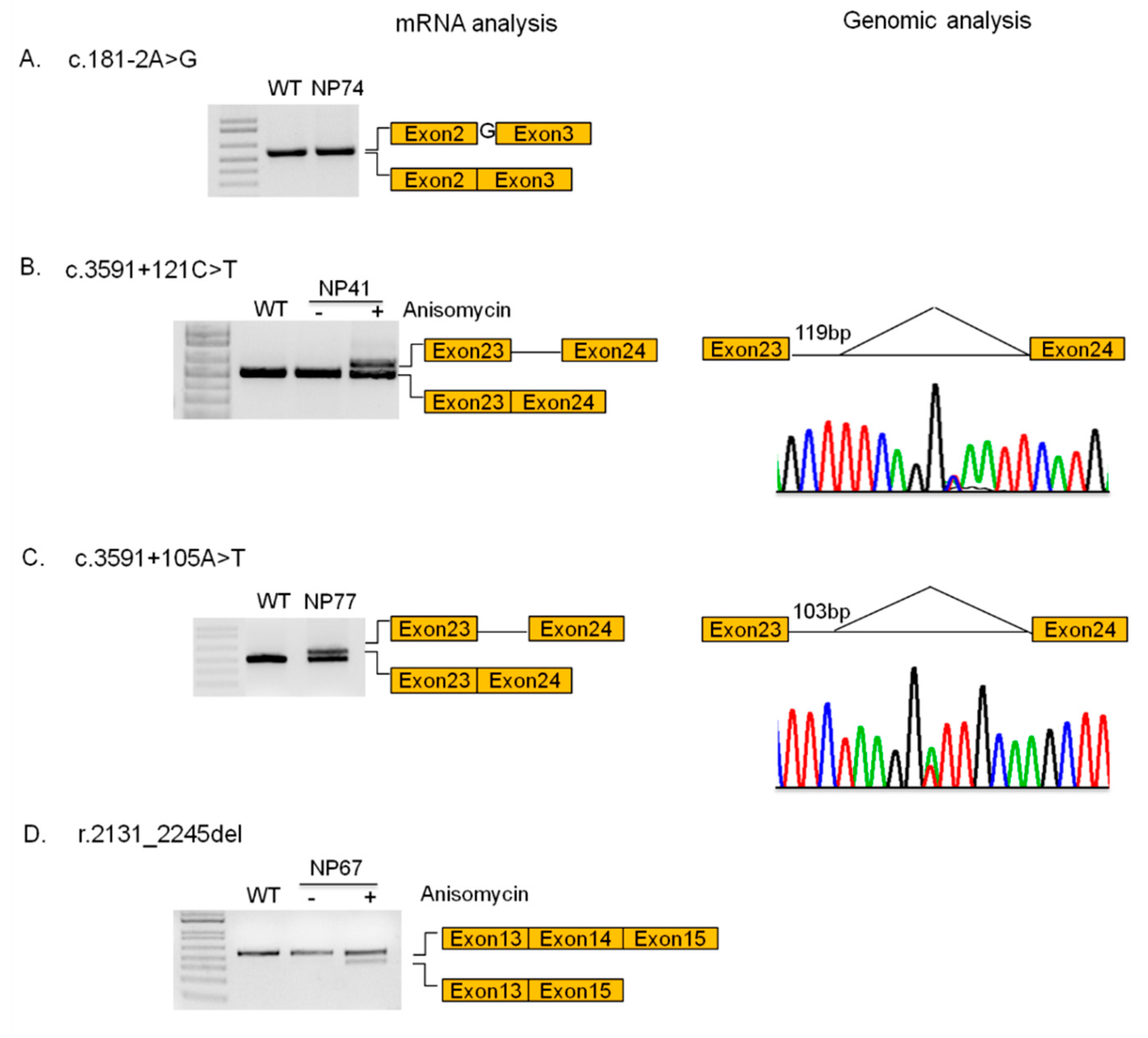
3.4. Novel Mutations
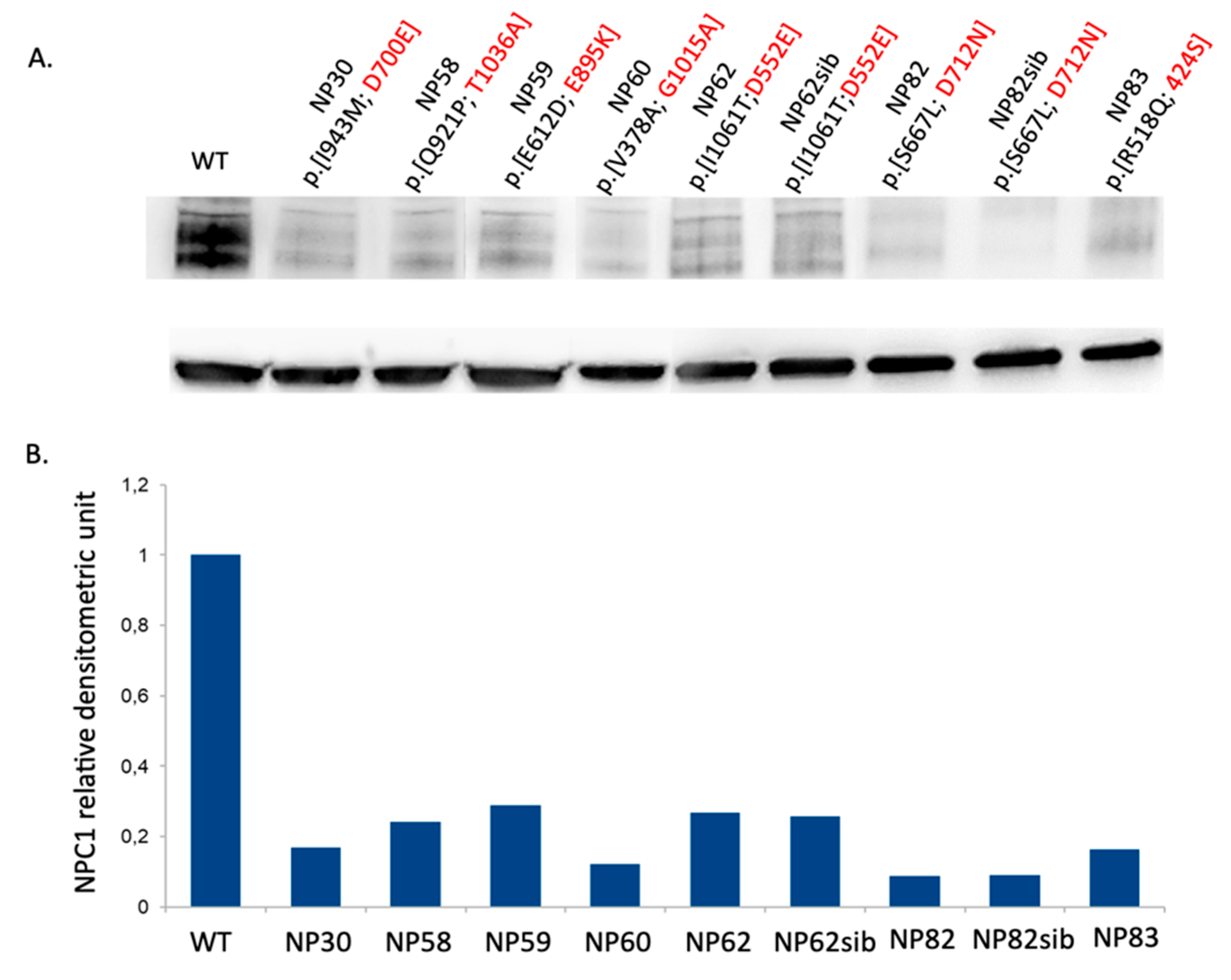
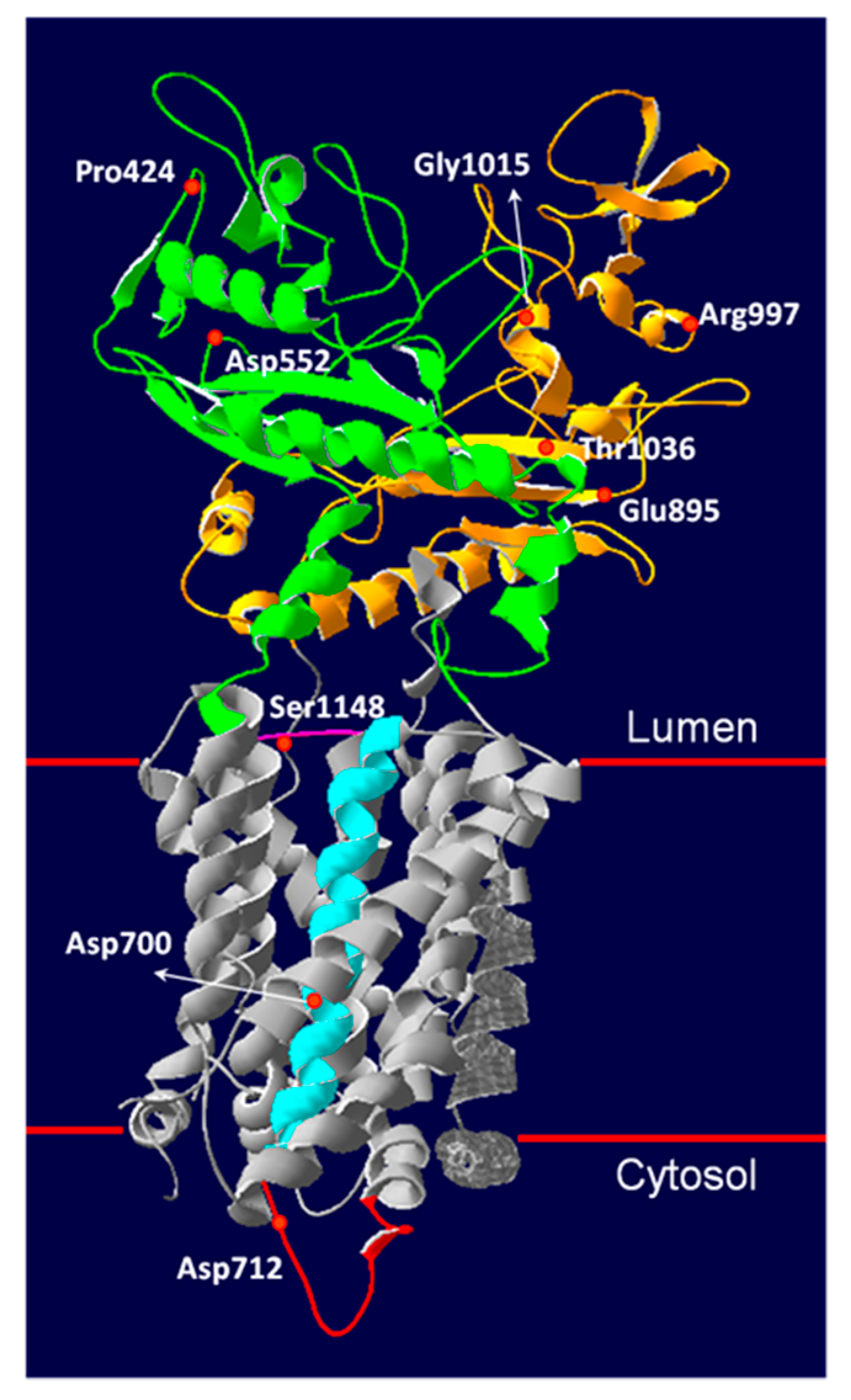
3.5. Novel NPC1 Mutations and Protein Structure
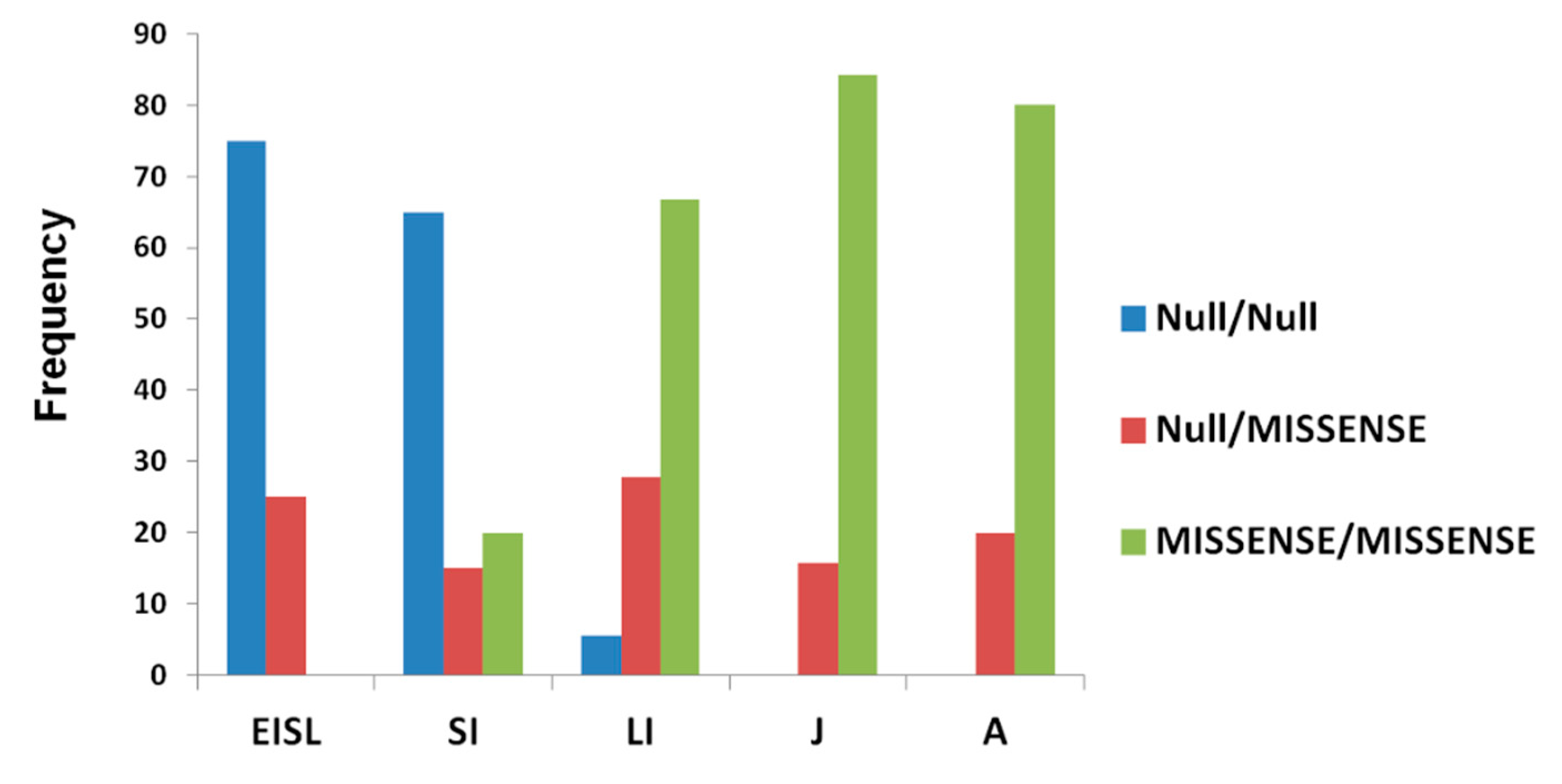
3.6. Genotype–Phenotype Correlation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Vanier, M.T. Niemann-Pick disease type C. Orphanet J. Rare Dis. 2010, 5, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Wassif, C.A.; Cross, J.L.; Iben, J.; Sanchez-Pulido, L.; Cougnoux, A.; Platt, F.M.; Ory, D.S.; Ponting, C.P.; Bailey-Wilson, J.E.; Biesecker, L.G.; et al. High incidence of unrecognized visceral/neurological late-onset Niemann-Pick disease, type C1, predicted by analysis of massively parallel sequencing data sets. Genet. Med. 2016, 18, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.; Ioannou, Y.A. The NPC1 protein: Structure implies function. Biochim. Biophys. Acta 2004, 1685, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Hua, X.; Nohturfft, A.; Goldstein, J.L.; Brown, M.S. Sterol resistance in CHO cells traced to point mutation in SREBP cleavage-activating protein. Cell 1996, 87, 415–426. [Google Scholar] [CrossRef]
- Kuwabara, P.E.; Labouesse, M. The sterol-sensing domain: Multiple families, a unique role? Trends Genet. 2002, 18, 193–201. [Google Scholar] [CrossRef]
- Li, X.; Saha, P.; Li, J.; Blobel, G.; Pfeffer, S.R. Clues to the mechanism of cholesterol transfer from the structure of NPC1 middle lumenal domain bound to NPC2. Proc. Natl. Acad. Sci. USA 2016, 113, 10079–10084. [Google Scholar] [CrossRef]
- Millat, G.; Marçais, C.; Rafi, M.A.; Yamamoto, T.; Morris, J.A.; Pentchev, P.G.; Ohno, K.; Wenger, D.A.; Vanier, M.T. Niemann-Pick C1 disease: The I1061T substitution is a frequent mutant allele in patients of Western European descent and correlates with a classic juvenile phenotype. Am. J. Hum. Genet. 1999, 65, 1321–1329. [Google Scholar] [CrossRef]
- Imrie, J.; Heptinstall, L.; Knight, S.; Strong, K. Observational cohort study of the natural history of Niemann-Pick disease type C in the UK: A 5-year update from the UK clinical database. BMC Neurol. 2015, 15, 257. [Google Scholar]
- Macías-Vidal, J.; Rodríguez-Pascau, L.; Sánchez-Ollé, G.; Lluch, M.; Vilageliu, L.; Grinberg, D.; Coll, M.J.; Spanish NPC Working Group. Molecular analysis of 30 Niemann-Pick type C patients from Spain. Clin. Genet. 2011, 80, 39–49. [Google Scholar] [CrossRef]
- Mavridou, I.; Dimitriou, E.; Vanier, M.T.; Vilageliu, L.; Grinberg, D.; Latour, P.; Xaidara, A.; Lycopoulou, L.; Bostantjopoulou, S.; Zafeiriou, D.; et al. The Spectrum of Niemann-Pick Type C Disease in Greece. JIMD Rep. 2017, 36, 41–48. [Google Scholar]
- Greer, W.L.; Dobson, M.J.; Girouard, G.S.; Byers, D.M.; Riddell, D.C.; Neumann, P.E. Mutations in NPC1 highlight a conserved NPC1-specific cysteine-rich domain. Am. J. Hum. Genet. 1999, 65, 1252–1260. [Google Scholar] [CrossRef] [PubMed]
- Millat, G.; Baïlo, N.; Molinero, S.; Rodriguez, C.; Chikh, K.; Vanier, M.T. Niemann-Pick C disease: Use of denaturing high performance liquid chromatography for the detection of NPC1 and NPC2 genetic variations and impact on management of patients and families. Mol. Genet. Metab. 2005, 86, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Fancello, T.; Dardis, A.; Rosano, C.; Tarugi, P.; Tappino, B.; Zampieri, S.; Pinotti, E.; Corsolini, F.; Fecarotta, S.; D’Amico, A.; et al. Molecular analysis of NPC1 and NPC2 gene in 34 Niemann-Pick C Italian patients: Identification and structural modeling of novel mutations. Neurogenetics 2009, 10, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Geberhiwot, T.; Moro, A.; Dardis, A.; Ramaswami, U.; Sirrs, S.; Marfa, M.P.; Vanier, M.T.; Walterfang, M.; Bolton, S.; Dawson, C.; et al. International Niemann-Pick Disease Registry (INPDR). Consensus clinical management guidelines for Niemann-Pick disease type C. Orphanet J. Rare Dis. 2018, 13, 50. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramen- sky, V.E.; Gerasimova, A.; Bork, P.; Kon-drashov, A.S.; Sunyaev, S.R. A method and server for pre- dicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Adebali, O.; Reznik, A.O.; Ory, D.S.; Zhulin, I.B. Establishing the precise evolutionary history of a gene improves prediction of disease-causing missense mutations. Genet. Med. 2016, 18, 1029–1036. [Google Scholar] [CrossRef]
- Li, X.; Lu, F.; Trinh, M.N.; Schmiege, P.; Seemann, J.; Wang, J.; Blobel, G. 3.3 Å structure of Niemann-Pick C1 protein reveals insights into the function of the C-terminal luminal domain in cholesterol transport. Proc. Natl. Acad. Sci. USA 2017, 114, 9116–9121. [Google Scholar] [CrossRef]
- Blanchette-Mackie, E.J.; Dwyer, N.K.; Amende, L.M.; Kruth, H.S.; Butler, J.D.; Sokol, J.; Comly, M.E.; Vanier, M.T.; August, J.T.; Brady, R.O. Type-C Niemann-Pick disease: Low density lipoprotein uptake is associated with premature cholesterol accumulation in the Golgi complex and excessive cholesterol storage in lysosomes. Proc. Natl. Acad. Sci. USA 1988, 85, 8022–8026. [Google Scholar] [CrossRef]
- Tarugi, P.; Ballarini, G.; Bembi, B.; Battisti, C.; Palmeri, S.; Panzani, F.; Di Leo, E.; Martini, C.; Federico, A.; Calandra, S. Niemann-Pick type C disease: Mutations of NPC1 gene and evidence of abnormal expression of some mutant alleles in fibroblasts. J. Lipid Res. 2002, 43, 1908–1919. [Google Scholar] [CrossRef]
- Den Dunnen, J.T.; Antonarakis, S.E. Mutation nomenclature extensions and suggestions to describe complex mutations: A discussion. Hum. Mutat. 2000, 15, 7–12. [Google Scholar] [CrossRef]
- Den Dunnen, J.T.; Paalman, M.H. Standardizing mutation nomenclature: Why bother? Hum. Mutat. 2003, 22, 181–182. [Google Scholar] [CrossRef] [PubMed]
- Romanello, M.; Zampieri, S.; Bortolotti, N.; Deroma, L.; Sechi, A.; Fiumara, A.; Parini, R.; Borroni, B.; Brancati, F.; Bruni, A.; et al. Comprehensive Evaluation of Plasma 7-Ketocholesterol and Cholestan-3β,5α,6β-Triol in an Italian Cohort of Patients Affected by Niemann-Pick Disease due to NPC1 and SMPD1 Mutations. Clin. Chim. Acta 2016, 455, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Boenzi, S.; Deodato, F.; Taurisano, R.; Martinelli, D.; Verrigni, D.; Carrozzo, R.; Bertini, E.; Pastore, A.; Dionisi-Vici, C.; Johnson, D.W. A new simple and rapid LC-ESI-MS/MS method for quantification of plasma oxysterols as dimethylaminobutyrate esters. Its successful use for the diagnosis of Niemann-Pick type C disease. Clin. Chim. Acta 2014, 437, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Fecarotta, S.; Romano, A.; Della Casa, R.; Del Giudice, E.; Bruschini, D.; Mansi, G.; Bembi, B.; Dardis, A.; Fiumara, A.; Di Rocco, M.; et al. Long term follow-up to evaluate the efficacy of miglustat treatment in Italian patients with Niemann-Pick disease type C. Orphanet J. Rare Dis. 2015, 10, 22. [Google Scholar] [CrossRef]
- Ginocchio, V.M.; D’Amico, A.; Bertini, E.; Ceravolo, F.; Dardis, A.; Verrigni, D.; Bembi, B.; Dionisi-Vici, C.; Deodato, F. Efficacy of miglustat in Niemann-Pick C disease: A single centre experience. Mol. Genet. Metab. 2013, 110, 229–235. [Google Scholar] [CrossRef]
- Zampieri, S.; Bembi, B.; Rosso, N.; Filocamo, M.; Dardis, A. Treatment of Human Fibroblasts Carrying NPC1 Missense Mutations with MG132 Leads to an Improvement of Intracellular Cholesterol Trafficking. JIMD Rep. 2012, 2, 59–69. [Google Scholar]
- Di Leo, E.; Panico, F.; Tarugi, P.; Battisti, C.; Federico, A.; Calandra, S. A point mutation in the lariat branch point of intron 6 of NPC1 as the cause of abnormal pre-mRNA splicing in Niemann-Pick type C disease. Hum. Mutat. 2004, 24, 440. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.; Minikel, E.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef]
- Jahnova, H.; Dvorakova, L.; Vlaskova, H.; Hulkova, H.; Poupetova, H.; Hrebicek, M.; Jesina, P. Observational, retrospective study of a large cohort of patients with Niemann-Pick disease type C in the Czech Republic: A surprisingly stable diagnostic rate spanning almost 40 years. Orphanet J. Rare Dis. 2014, 9, 140. [Google Scholar] [CrossRef]
- Millat, G.; Marçais, C.; Tomasetto, C.; Chikh, K.; Fensom, A.H.; Harzer, K.; Wenger, D.A.; Ohno, K.; Vanier, M.T. Niemann-Pick C1 Disease: Correlations between NPC1 Mutations, Levels of NPC1 Protein, and Phenotypes Emphasize the Functional Significance of the Putative Sterol-Sensing Domain and of the Cysteine-Rich Luminal Loop. Am. J. Hum. Genet. 2001, 68, 1373–1385. [Google Scholar] [CrossRef] [PubMed]
- Tängemo, C.; Weber, D.; Theiss, S.; Mengel, E.; Runz, H. Niemann-Pick Type C disease: Characterizing lipid levels in patients with variant lysosomal cholesterol storage. J. Lipid. Res. 2011, 52, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.C.; Su, Y.N.; Chiou, P.C.; Fietz, M.J.; Yu, C.L.; Hwu, W.L.; Lee, M.J. Six novel NPC1 mutations in Chinese patients with Niemann-Pick disease type C. J. Neurol. Neurosurg. Psychiatry 2005, 76, 592–595. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Valero, E.M.; Ballart, A.; Iturriaga, C.; Lluch, M.; Macias, J.; Vanier, M.T.; Pineda, M.; Coll, M.J. Identification of 25 new mutations in 40 unrelated Spanish Niemann-Pick type C patients: Genotype-phenotype correlations. Clin. Genet. 2005, 68, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Millard, E.E.; Gale, S.E.; Dudley, N.; Zhang, J.; Schaffer, J.E.; Ory, D.S. The sterol-sensing domain of the Niemann-Pick C1 (NPC1) protein regulates trafficking of low density lipoprotein cholesterol. J. Biol. Chem. 2005, 280, 28581–28590. [Google Scholar] [CrossRef]
- Park, W.D.; O’Brien, J.F.; Lundquist, P.A.; Kraft, D.L.; Vockley, C.W.; Karnes, P.S.; Patterson, M.C.; Snow, K. Identification of 58 novel mutations in Niemann-Pick disease type C: Correlation with biochemical phenotype and importance of PTC1-like domains in NPC1. Hum. Mutat. 2003, 22, 313–325. [Google Scholar] [CrossRef]
- Nadjar, Y.; Hütter-Moncada, A.L.; Latour, P.; Ayrignac, X.; Kaphan, E.; Tranchant, C.; Cintas, P.; Degardin, A.; Goizet, C.; Laurencin, C.; et al. Adult Niemann-Pick disease type C in France: Clinical phenotypes and long-term miglustat treatment effect. Orphanet J. Rare Dis. 2018, 13, 175. [Google Scholar] [CrossRef]
- Greenberg, C.R.; Barnes, J.G.; Kogan, S.; Seargeant, L.E. A rare case of Niemann-Pick disease type C without neurological involvement in a 66-year-old patient. Mol. Genet. Metab. Rep. 2015, 3, 18–20. [Google Scholar] [CrossRef]
- Palmeri, S.; Tarugi, P.; Sicurelli, F.; Buccoliero, R.; Malandrini, A.; De Santi, M.M.; Marcianò, G.; Battisti, C.; Dotti, M.T.; Calandra, S.; et al. Lung involvement in Niemann-Pick disease type C1: Improvement with bronchoalveolar lavage. Neurol. Sci. 2005, 26, 71–73. [Google Scholar] [CrossRef]
- Kawazoe, T.; Yamamoto, T.; Narita, A.; Ohno, K.; Adachi, K.; Nanba, E.; Noguchi, A.; Takahashi, T.; Maekawa, M.; Eto, Y.; et al. Phenotypic variability of Niemann-Pick disease type C including a case with clinically pure schizophrenia: A case report. BMC Neurol. 2018, 18, 117. [Google Scholar] [CrossRef]
- Walterfang, M.; Fietz, M.; Abel, L.; Bowman, E.; Mocellin, R.; Velakoulis, D. Gender dimorphism in siblings with schizophrenia-like psychosis due to Niemann-Pick disease type C. J. Inherit. Metab. Dis. 2009, 32, S221–S226. [Google Scholar] [CrossRef] [PubMed]
- Benussi, A.; Alberici, A.; Premi, E.; Bertasi, V.; Cotelli, M.S.; Turla, M.; Dardis, A.; Zampieri, S.; Marchina, E.; Paghera, B.; et al. Phenotypic heterogeneity of Niemann-Pick disease type C in monozygotic twins. J. Neurol. 2015, 262, 642–647. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Erickson, R.P. A modifier of Niemann Pick C 1 maps to mouse chromosome 19. Mamm. Genome 2000, 11, 69–71. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Li, H.; Repa, J.J.; Turley, S.D.; Dietschy, J.M. Genetic variations and treatments that affect the lifespan of the NPC1 mouse. J. Lipid. Res. 2008, 49, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.A.; Watkins-Chow, D.E.; Palladino, G.; Deutsch, G.; Chandran, K.; Pavan, W.J.; Erickson, R.P. In Niemann-Pick C1 mouse models, glial-only expression of the normal gene extends survival much further than do changes in genetic background or treatment with hydroxypropyl-beta-cyclodextrin. Gene 2018, 643, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Parra, J.; Klein, A.D.; Castro, J.; Morales, M.G.; Mosqueira, M.; Valencia, I.; Cortés, V.; Rigotti, A.; Zanlungo, S. Npc1 deficiency in the C57BL/6J genetic background enhances Niemann-Pick disease type C spleen pathology. Biochem. Biophys. Res. Commun. 2011, 413, 400–406. [Google Scholar] [CrossRef]
- Rodriguez-Gil, J.L.; Watkins-Chow, D.E.; Baxter, L.L.; Elliot, G.; Harper, U.L.; Wincovitch, S.M.; Wedel, J.C.; Incao, A.A.; Huebecker, M.; Boehm, F.J.; et al. Genetic background modifies phenotypic severity and longevity in a mouse model of Niemann-Pick disease type C1. Dis. Model. Mech. 2020. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suñer, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J.; et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Nat. Acad. Sci. 2005, 102, 10604–10609. [Google Scholar] [CrossRef]
- Ballestar, E. Epigenetic alterations in autoimmune rheumatic diseases. Nat. Rev. Rheumatol. 2011, 7, 263–271. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Code | Allele1 | Allele 2 | Clinical Phenotype | Filipin | Gene | Reference |
|---|---|---|---|---|---|---|
| NP3 sib | c.852delT (p.F284Lfs*26) | c.852delT (p.F284Lfs*26) | EISL | NA | NPC1 | [13] |
| NP10 sib | c.1819C>T (p.R607*) | c.3614C>A (p.T1205K) | EISL | NA | NPC1 | [13] |
| NP18 sib | c.2800C>T (p.R934*) | c.2872C>T (R958*) | EISL | NA | NPC1 | [13] |
| NP35 | c.1029dupG (p.S344Vfs*36) | c.1029dupG (p.S344Vfs*36) | EISL | NA | NPC1 | [23] |
| NP20 | c.1A>T (p.M1L) | c.1A>T (p.M1L) | EISL | CLASSIC | NPC2 | [13] |
| NP78 | c.133C>T (p.Q45*) | c.133C>T (p.Q45*) | EISL | CLASSIC | NPC2 | [24] |
| NP36 | c.436C>T (P.Q146*) | c.436C>T (Q146*) | EISL | CLASSIC | NPC2 | [12] |
| NP3 | c.852delT (p.F284Lfs*26) | c.852delT (p.F284Lfs*26) | EI | NA | NPC1 | [13] |
| NP7 | c.2972_2973delAG (p.Q991Rfs*15) | c.2972_2973delAG (p.Q991Rfs*15) | EI | CLASSIC | NPC1 | [13] |
| NP9 | c.1901A>G (p.Y634C) | c.3562delG (p.E1188Kfs*54) | EI | CLASSIC | NPC1 | [13] |
| NP10 | c.1819C>T (p.R607*) | c.3614C>A (p.T1205K) | EI | CLASSIC | NPC1 | [13] |
| NP12 | c.93_94delTG (p.C31Wfs*26) | c.93_94delTG (p.C31Wfs*26) | EI | NA | NPC1 | [13] |
| NP13 | r.0 | r.0 | EI | NA | NPC1 | [13] |
| NP13sib | r.0 | r.0 | EI | NA | NPC1 | [13] |
| NP14 | c.3467A>G (p.N1156S) | c.3467A>G (p.N1156S) | EI | NA | NPC1 | [13] |
| NP16 | c. 3613-3614insA (p.T1205Nfs*53) | c. 3613-3614insA (p.T1205Nfs*53) | EI | CLASSIC | NPC1 | [13] |
| NP18 | c.2800C>T (p.R934*) | c.2872C>T (p.R958*) | EI | CLASSIC | NPC1 | [13] |
| NP22 | c.2762A>C (p.Q921P) | c.2339T>G (p.V780G) | EI | CLASSIC | NPC1 | [13] |
| NP37 | c.852delT (p.F284Lfs*26) | c.852delT (p.F284Lfs*26) | EI | CLASSIC | NPC1 | [25] |
| NP38 | c.2800C>T (p.R934*) | c.3235T>C (p.F1079L) | EI | CLASSIC | NPC1 | [23] |
| NP39 | c.2761C>T (p.Q921*) | c.2761C>T (p.Q921*) | EI | NA | NPC1 | [23] |
| NP40 | c.852delT (p.F284Lfs*26) | c.852delT (p.F284Lfs*26) | EI | CLASSIC | NPC1 | [26] |
| NP71 | c.665A>G (p.N222S) | c.2861C>T (p.S954L) | EI | VARIANT | NPC1 | This study |
| NP74 | c.181-2A>G (p.E61Gfs*24) | c.1553+1G>A (p.R518Gfs*7) | EI | CLASSIC | NPC1 | This study |
| NP75 | c.3322dupG (p.A1108Gfs*13) | c.3322dupG (p.A1108Gfs*13) | EI | CLASSIC | NPC1 | [24] |
| NP76 | c.2819C>T (p.S940L) | c.2819C>T (p.S940L) | EI | NA | NPC1 | This study |
| NP82 | c.1554_1757dup (p.A519_E586dup) | c.1554_1757dup (p.A519_E586dup) | EI | CLASSIC | NPC1 | This study |
| NP34 | c.58G>T (p.E20*) | c.58G>T (p.E20*) | EI | NA | NPC2 | [13] |
| NP34Sib | c.58G>T (p.E20*) | c.58G>T (p.E20*) | EI | NA | NPC2 | [13] |
| NP80 | c.1A>T (p.M1L) | c.1A>T (p.M1L) | EI | CLASSIC | NPC2 | This study |
| NP1 | c.2776G>A (p.A926T) | c.3731 T>C (p.L1244P) | LI | CLASSIC | NPC1 | [13] |
| NP2 | c.3571C>T (p.L1191F) | c.3571C>T (p.L1191F) | LI | CLASSIC | NPC1 | [13] |
| NP4 | c.3068T>G (p.V1023G) | c.3068T>G (p.V1023G) | LI | NA | NPC1 | [13] |
| NP5 | c.1535A>G (p.H512R) | c.3056A>G (p.Y1019C) | LI | CLASSIC | NPC1 | [13] |
| NP6 | c.852delT (p.F284Lfs*26) | c.3056A>G (p.Y1019C) | LI | NA | NPC1 | [13] |
| NP11 | c.2972_2973delAG (p.Q991Rfs*15) | ? | LI | VARIANT | NPC1 | [13] |
| NP15 | c.852delT (p.F284Lfs*26) | c.852delT (p.F284Lfs*26) | LI | CLASSIC | NPC1 | [13] |
| NP17 | c.710C>T (p.P237L) | c.3304C>T (p.L1102F) | LI | NA | NPC1 | [13] |
| NP19 | c.3019C>G (p.P1007A) | c.3614C>A (p.T1205K) | LI | NA | NPC1 | [13] |
| NP25 | c. 3182 T>C (p.I1061T) | c. 3182 T>C (p.I1061T) | LI | CLASSIC | NPC1 | [13] |
| NP26 | c. 298T>A (p.C100S) | c.[3424T>C ; 1943T>A] (p.[L648H; M1142T]) | LI | CLASSIC | NPC1 | [13] |
| NP27 | c.2662C>T (p.P888S) | c.2761C>T (p.Q921*) | LI | NA | NPC1 | [13] |
| NP28 | c.2762A>C (p.Q921P) | c. 3182 T>C (p.I1061T) | LI | CLASSIC | NPC1 | [13] |
| NP30 | c.2829C>G (p.I943M) | c.2100C>G (p.D700E) | LI | CLASSIC | NPC1 | [25] |
| NP41 | c.3493G>A (p.V1165M) | c.3591+121C>T (p.0) | LI | CLASSIC | NPC1 | [26] |
| NP42 | c.3019 C>G (p.P1007A) | c.3734_3735delCT (p.P1245Rfs12*) | LI | CLASSIC | NPC1 | [26] |
| NP43 | c.3493G>A (p.V1165M) | c.58-3T>G (p.?) | LI | CLASSIC | NPC1 | [26] |
| NP61 | c.3614C>A (p.T1205K) | c.3614C>A (p.T1205K) | LI | NA | NPC1 | This study |
| NP81 | c.681T>A (p.C227*) | c.2861C>T (p.S954L) | LI | NA | NPC1 | This study |
| NP58 | c.3106A>G (p.T1036A) | c.2762A>G (p.Q921P) | J | CLASSIC | NPC1 | This study |
| NP19sib | c.3019 C>G (p.P1007A) | c.3614C>A (p.T1205K) | J | NA | NPC1 | [13] |
| NP24 | c.2800C>T (p.R934*) | c.2292G>A (p.A750_G765del) | J | CLASSIC | NPC1 | [13] |
| NP24sib | c.2800C>T (p.R934*) | c.2292G>A (p.A750_G765del) | J | CLASSIC | NPC1 | [13] |
| NP25Sib | c. 3182 T>C (p.I1061T) | c.3182 T>C (p.I1061T) | J | CLASSIC | NPC1 | [13] |
| NP29 | c.3056A>G (p.Y1019C) | c.3056A>G (p.Y1019C) | J | CLASSIC | NPC1 | [13] |
| NP32Sib | c.2762A>C (p.Q921P) | c.2903A>G (p.N968S) | J | NA | NPC1 | [13] |
| NP32Sib1 | c.2762A>C (p.Q921P) | c.2903A>G (p.N968S) | J | NA | NPC1 | [13] |
| NP43sib | c.3493G>A (p.V1165M) | c.58-3T>G (p.?) | J | NA | NPC1 | [26] |
| NP44 | c.2662C>T (p.P888S) | c.2662C>T (p.P888S) | J | CLASSIC | NPC1 | [23] |
| NP44sib | c.2662C>T (p.P888S) | c.2662C>T (p.P888S) | J | NA | NPC1 | [23] |
| NP45 | c.3467A>G (p.N1156S) | c.3467A>G (p.N1156S) | J | NA | NPC1 | [23] |
| NP46 | c.1501G>T (p.D501Y) | c.1421 C>T (p.P474L) | J | CLASSIC | NPC1 | [23] |
| NP47 | c.2819C>T (p.S940L) | c.2291C>T (p.A764V) | J | CLASSIC | NPC1 | [23] |
| NP56sib1 | c.2974G>T (p.G992W) | c.1351G>A (p.E451K) | J | VARIANT | NPC1 | [23] |
| NP57 | c.3493G>A (p.V1165M) | c.2795+1G>C (p.0) | J | CLASSIC | NPC1 | [27] |
| NP63 | c.1421C>T (p.P474L) | c.1421C>T (p.P474L) | J | CLASSIC | NPC1 | [25] |
| NP59 | c.2683G>A (p.E895K) | c.1836A>C (p.E612D) | J | CLASSIC | NPC1 | This study |
| NP70 | c.352_353delAG (p.Q119FS*8) | c.2861C>T (p.S954L) | J | NA | NPC1 | This study |
| NP77 | c.3591+105A>T (p.F1199Sfs*21) | c.3467A>G (p.N1156S) | J | CLASSIC | NPC1 | This study |
| NP79 | c.3443G>T (p.S1148I) | c.3019C>G (p.P1007A) | J | NA | NPC1 | This study |
| NP84 | c.1270C>T (p.P424S) | c.1553G>A (p.R518Q) | A | CLASSIC | NPC1 | This study |
| NP21 | c.1415T>A (p.L472H) | c.3230G>A (p.R1077Q) | A | CLASSIC | NPC1 | [13] |
| NP23 | c.1907C>T (p.S636F) | c.(?) | A | CLASSIC | NPC1 | [13] |
| NP31 | c.665A>G (p.N222S) | c.3560C>T (p.A1187V) | A | VARIANT | NPC1 | [13] |
| NP33 | c.1166G>T (p.R389L) | c.1166G>T (p.R389L) | A | NA | NPC1 | [13] |
| NP48 | c.882-28A>G (p.0) | c.2932C>T (p.R978C) | A | VARIANT | NPC1 | [28] |
| NP48sib | c.882-28A>G (p.0) | c.2932C>T (p.R978C) | A | NA | NPC1 | [28] |
| NP50 | c.3493G>A (p.V1165M) | c.3019C>G (p.P1007A) | A | VARIANT | NPC1 | [25] |
| NP51 | c.2974G>C (p.G992R) | c. 2130dupG (p.R711Efs*3) | A | CLASSIC | NPC1 | [23] |
| NP51Sib | c.2974G>C (p.G992R) | c. 2130dupG (p.R711Efs*3) | A | NA | NPC1 | [23] |
| NP52 | c.3019C>G (p.P1007A) | c.3019C>G (p.P1007A) | A | NA | NPC1 | [23] |
| NP56sib2 | c.2974G>T (p.G992W) | c.1351G>A (p.E451K) | A | VARIANT | NPC1 | [23] |
| NP56sib3 | c.2974G>T (p.G992W) | c.1351G>A (p.E451K) | A | VARIANT | NPC1 | [23] |
| NP60 | c.1133T>C (p.V378A) | c.3044G>C (p.G1015A) | A | CLASSIC | NPC1 | This study |
| NP60sib | c.1133T>C (p.V378A) | c.3044G>C (p.G1015A) | A | NA | NPC1 | This study |
| NP62 | c.1656T>A (p.D552E) | c.3182T>C (p.I1061T) | A | CLASSIC | NPC1 | This study |
| NP62sib | c.1656T>A (p.D552E) | c.3182T>C (p.I1061T) | A | VARIANT | NPC1 | This study |
| NP64 | c.632_1553del (p.D211Gfs*6) | c.1654+6delTA (p.?) | A | NA | NPC1 | This study |
| NP65 | c.3493G>A (p.V1165M) | c.3493G>A (p.V1165M) | A | NA | NPC1 | This study |
| NP66 | c.3019C>G (p.P1007A) | c.3019C>G (p.P1007A) | A | NA | NPC1 | This study |
| NP67 | c.2728G>A (p.G910S) | r.2131_2245del (p.0; p.=) | A | VARIANT | NPC1 | This study |
| NP68 | c.1907C>T (p.S636F)* c.2932C>T (p.R978C) c.3467A>G (p.N1156S) | A | NA | NPC1 | This study | |
| NP69 | c.2991A>T (p.R997S) | c.3493G>A (p.V1165M) | A | NA | NPC1 | This study |
| NP49 | c.26T>C (p.L9P) | c.26T>C (p.L9P) | A | CLASSIC | NPC2 | [25] |
| NP49sib | c.26T>C (p.L9P) | c.26T>C (p.L9P) | A | NA | NPC2 | This study |
| NP8 | c.1339C>T (p.Q447*) | c.1339C>T (p.Q447*) | NC | CLASSIC | NPC1 | [13] |
| NP29 | c.3056A>G (p.Y1019C) | c.3056A>G (p.Y1019C) | NC | CLASSIC | NPC1 | [13] |
| NP53 | c.2972_2973delAG (p.Q991Rfs*15) | c. 3182 T>C (p.I1061T) | NC | CLASSIC | NPC1 | [23] |
| NP54 | c.2689C>A (p.H897N) | c.665A>G (p.N222S) | NC | VARIANT | NPC1 | [23] |
| NP56 | c.2974G>T (p.G992W) | c.1351G>A (p.E451K) | NC | VARIANT | NPC1 | [23] |
| NP56sib | c.2974G>T (p.G992W) | c.1351G>A (p.E451K) | NC | VARIANT | NPC1 | [23] |
| NP72 | c. 3613-3614insA (p.T1205Nfs*53) | c. 3182 T>C (p.I1061T) | NC | NA | NPC1 | This study |
| NP73 | c.[478T>C; c.665A>G] (p.[C160R; N222S]) | c.3106A>G (p.T1036A) | NC | NA | NPC1 | This study |
| NP83 | c.2134G>A (p.D712N) | c.2000C>T (p.S667L) | NC | VARIANT | NPC1 | This study |
| NP83 sib | c.2134G>A (p.D712N) | c.2000C>T (p.S667L) | NC | VARIANT | NPC1 | This study |
| NPC1 Variant | n of Alleles | Frequency (%) |
|---|---|---|
| p.F284Lfs*26 | 9 | 5.8 |
| p.P1007A | 8 | 5.2 |
| p.V1165M | 7 | 4.5 |
| p.N1156S | 6 | 3.9 |
| p.I1061T | 6 | 3.9 |
| p.Q991Rfs*15 | 4 | 2.6 |
| p.T1205K | 4 | 2.6 |
| p.Y1019C | 4 | 2.6 |
| p.Q921P | 4 | 2.6 |
| p.S954L | 3 | 1.95 |
| p.R934* | 3 | 1.95 |
| p.Q921* | 3 | 1.95 |
| p.P888S | 3 | 1.95 |
| p.P474L | 3 | 1.95 |
| p.S940L | 3 | 1.95 |
| p.N222S | 3 | 1.95 |
| p.S334Vfs*36 | 2 | 1.3 |
| p.C31Wfs*26 | 2 | 1.3 |
| p.T1205Nfs*53 | 2 | 1.3 |
| p.L1191F | 2 | 1.3 |
| p.V1023G | 2 | 1.3 |
| p.S636F | 2 | 1.3 |
| p.R389L | 2 | 1.3 |
| p.R978C | 2 | 1.3 |
| p.Q447* | 2 | 1.3 |
| p.A1108Gfs*13 | 2 | 1.3 |
| p.T1036A | 2 | 1.3 |
| Patient Code | Nucleotide Substitution | Predicted Aminoacid Change | Filipin Staining | SIFT | Polyphen | Classification Following Adebali et al. [17] |
|---|---|---|---|---|---|---|
| NP74 | c.181-2A>G | p.E61Gfs*24 | CLASSIC | NA | NA | NA |
| NP73 | c. [478T>C; c.665A>G] | p [C160R; N222S] | NA | NA | ||
| NP64 | c.632_1553del | p.D211Gfs*6 | NA | NA | NA | NA |
| NP81 | c.681T>A | p.C227* | NA | NA | NA | NA |
| NP84 | c.1270C>T | p.P424S | CLASSIC | Benign | Benign | Damaging |
| NP82 | c.1554_1757dup | p.A519_E586dup | CLASSIC | NA | NA | NA |
| NP64 | c.1654+6delTA | p.? | NA | NA | NA | NA |
| NP62 NP62sib | c.1656T>A | p.D552E | CLASSIC VARIANT | Benign | Benign | Benign |
| NP30 | c.2100C>G | p.D700E | CLASSIC | Damaging | Damaging | Damaging |
| NP83 NP83sib | c.2134G>A | p.D712N | VARIANT | Damaging | Damaging | Damaging |
| NP59 | c.2683G>A | p.E895K | CLASSIC | Benign | Benign | Benign |
| NP69 | c.2991A>T | p.R997S | NA | Tolerated | Benign | Benign |
| NP60 NP60sib | c.3044G>C | p.G1015A | CLASSIC | Damaging | Damaging | Damaging |
| NP58 # | c.3106A>G | p.T1036A | CLASSIC | Damaging | Damaging | Damaging |
| NP79 | c.3443G>T | p.S1148I | NA | Damaging | Probably damaging | Damaging |
| NP77 | c.3591+105A>T | p.F1199Sfs*21 | CLASSIC | NA | NA | NA |
| NP41 | c.3591+121C>T | p = 0 | CLASSIC | NA | NA | NA |
| NP67 | r.2131_2245del | (p.0; p.=) | VARIANT | NA | NA | NA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dardis, A.; Zampieri, S.; Gellera, C.; Carrozzo, R.; Cattarossi, S.; Peruzzo, P.; Dariol, R.; Sechi, A.; Deodato, F.; Caccia, C.; et al. Molecular Genetics of Niemann–Pick Type C Disease in Italy: An Update on 105 Patients and Description of 18 NPC1 Novel Variants. J. Clin. Med. 2020, 9, 679. https://doi.org/10.3390/jcm9030679
Dardis A, Zampieri S, Gellera C, Carrozzo R, Cattarossi S, Peruzzo P, Dariol R, Sechi A, Deodato F, Caccia C, et al. Molecular Genetics of Niemann–Pick Type C Disease in Italy: An Update on 105 Patients and Description of 18 NPC1 Novel Variants. Journal of Clinical Medicine. 2020; 9(3):679. https://doi.org/10.3390/jcm9030679
Chicago/Turabian StyleDardis, Andrea, Stefania Zampieri, Cinzia Gellera, Rosalba Carrozzo, Silvia Cattarossi, Paolo Peruzzo, Rosalia Dariol, Annalisa Sechi, Federica Deodato, Claudio Caccia, and et al. 2020. "Molecular Genetics of Niemann–Pick Type C Disease in Italy: An Update on 105 Patients and Description of 18 NPC1 Novel Variants" Journal of Clinical Medicine 9, no. 3: 679. https://doi.org/10.3390/jcm9030679
APA StyleDardis, A., Zampieri, S., Gellera, C., Carrozzo, R., Cattarossi, S., Peruzzo, P., Dariol, R., Sechi, A., Deodato, F., Caccia, C., Verrigni, D., Gasperini, S., Fiumara, A., Fecarotta, S., Carecchio, M., Filosto, M., Santoro, L., Borroni, B., Bordugo, A., ... Bembi, B. (2020). Molecular Genetics of Niemann–Pick Type C Disease in Italy: An Update on 105 Patients and Description of 18 NPC1 Novel Variants. Journal of Clinical Medicine, 9(3), 679. https://doi.org/10.3390/jcm9030679