rAAV-Mediated Cochlear Gene Therapy: Prospects and Challenges for Clinical Application

 , , and
, , and 
Abstract
1. Introduction
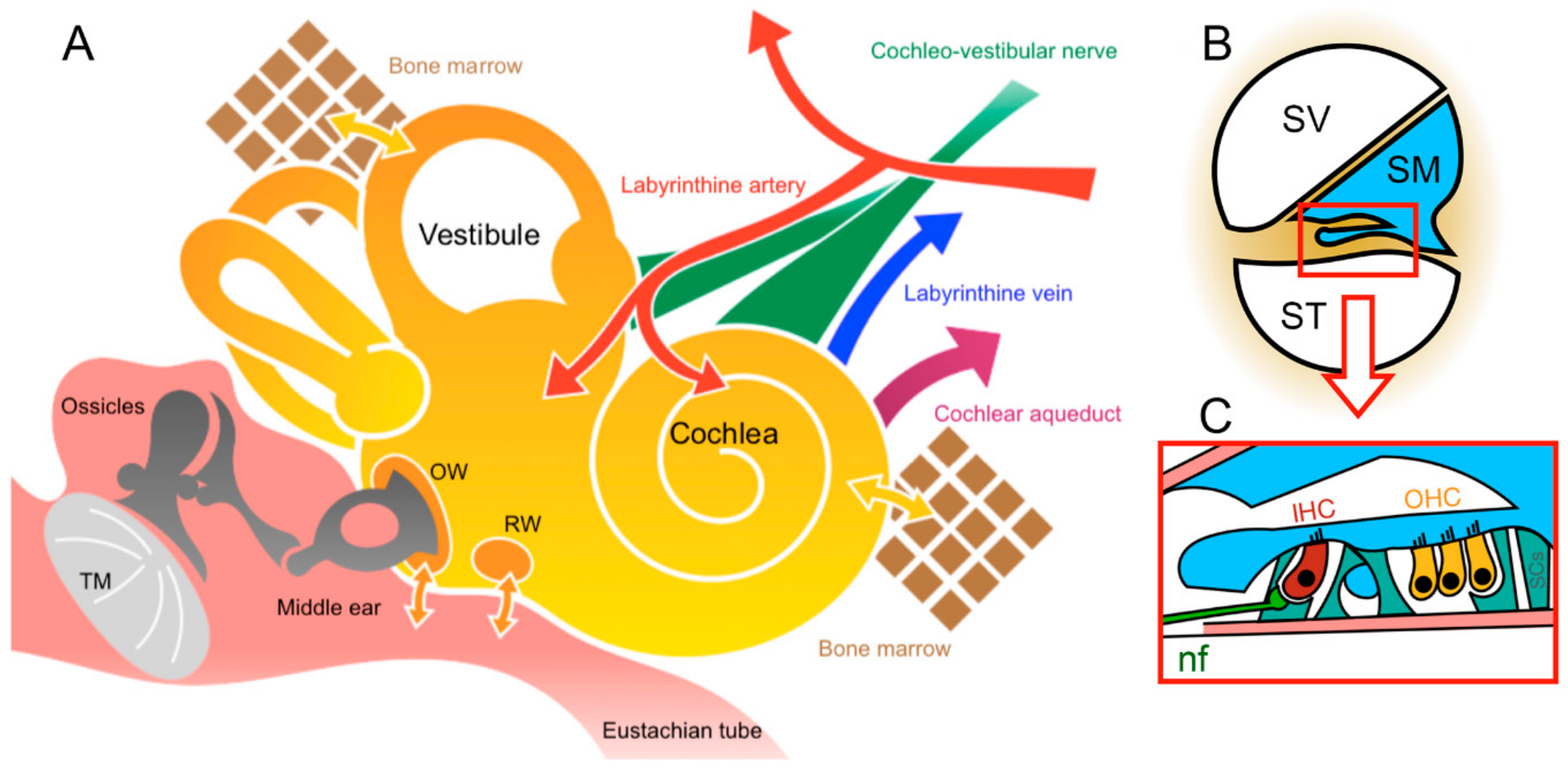
2. Challenges and Advantages of Inner-Ear Anatomy for Cochlear Gene Therapy
3. Recombinant Adeno-Associated Virus Vectors
3.1. rAAV Trafficking and Transduction
3.2. Approaches Used for Selective Tissue—Or Organ Targeting
3.3. AAV Immunogenicity and Strategies to Avoid
4. rAAV-Mediated Cochlear Gene Therapy
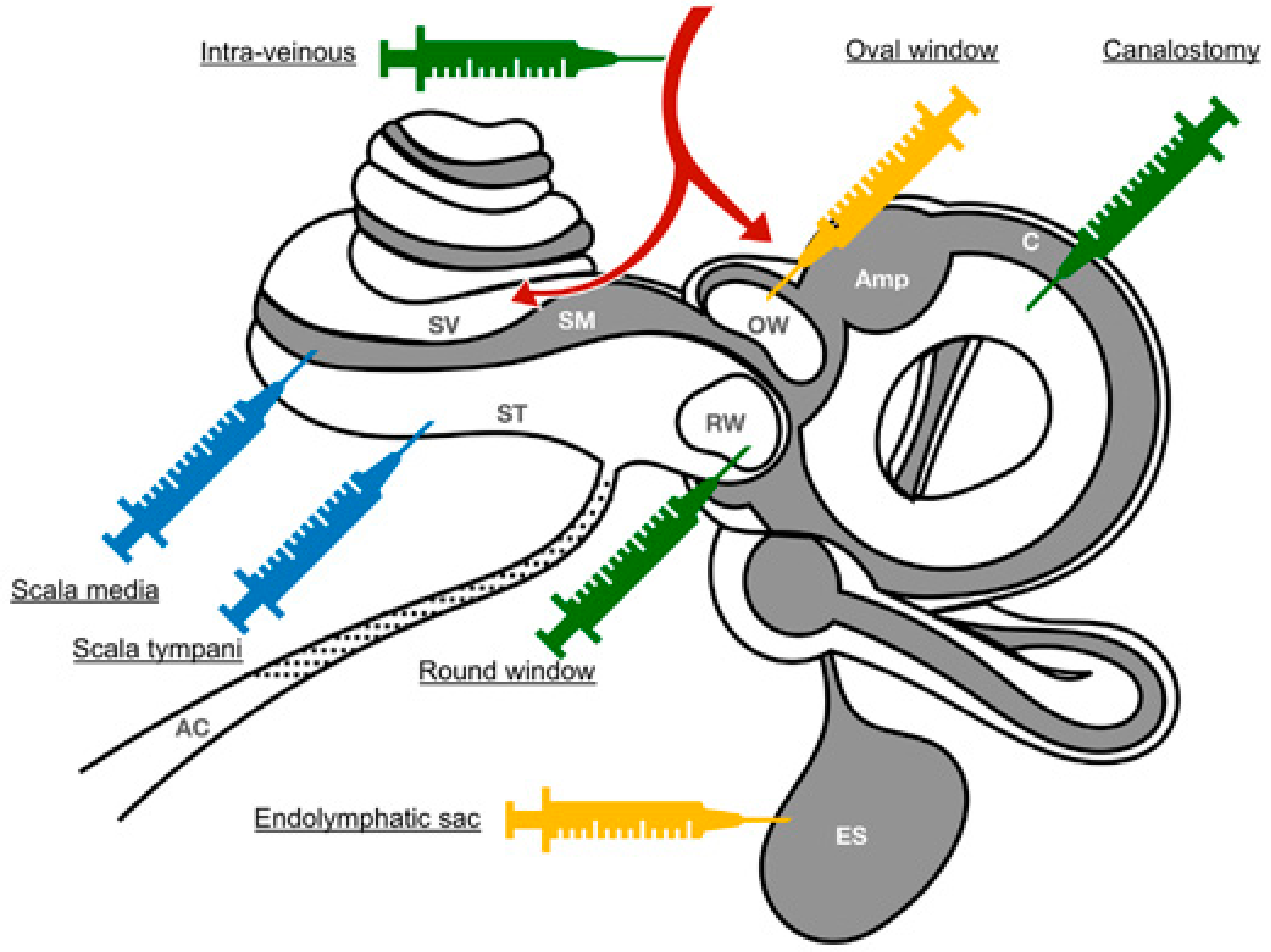
4.1. Routes of Application
4.1.1. Systemic Route of Administration
4.1.2. Local Routes of Administration
Round Window Injection
Round-Window Membrane Diffusion
Delivery via a Cochleostomy
Delivery via a Canalostomy
Delivery via a Combined Approach
Oval-Window/Trans-Stapedial Injection
Endolymphatic sac Injection
4.2. Preclinical Targets
4.2.1. Gene Addition
4.2.2. RNAi
4.2.3. Antisense Oligonucleotide
4.2.4. Gene Editing
4.3. Risk of Getting Side-Effects
5. Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Deafness and Hearing Loss. Available online: https://www.who.int/news-room/fact-sheets/detail/deafness-and-hearing-loss (accessed on 5 September 2019).
- Liberman, M.C.; Kujawa, S.G. Cochlear synaptopathy in acquired sensorineural hearing loss: Manifestations and mechanisms. Hear. Res. 2017, 349, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Angeli, S.; Lin, X.; Liu, X.Z. Genetics of hearing and deafness. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 2012, 295, 1812–1829. [Google Scholar] [CrossRef]
- Yan, D.; Liu, X.-Z. Cochlear molecules and hereditary deafness. Front. Biosci. J. Virtual Libr. 2008, 13, 4972–4983. [Google Scholar] [CrossRef]
- Wesarg, T.; Richter, N.; Hessel, H.; Günther, S.; Arndt, S.; Aschendorff, A.; Laszig, R.; Hassepass, F. Binaural integration of periodically alternating speech following cochlear implantation in subjects with profound sensorineural unilateral hearing loss. Audiol. Neurootol. 2015, 20 (Suppl. 1), 73–78. [Google Scholar] [CrossRef] [PubMed]
- Jiam, N.T.; Caldwell, M.T.; Limb, C.J. What Does Music Sound Like for a Cochlear Implant User? Otol. Neurotol. 2017, 38, e240–e247. [Google Scholar] [CrossRef]
- Mulligan, R.C. The basic science of gene therapy. Science 1993, 260, 926–932. [Google Scholar] [CrossRef]
- Al-Moyed, H.; Cepeda, A.P.; Jung, S.; Moser, T.; Kügler, S.; Reisinger, E. A dual-AAV approach restores fast exocytosis and partially rescues auditory function in deaf otoferlin knock-out mice. EMBO Mol. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Akil, O.; Dyka, F.; Calvet, C.; Emptoz, A.; Lahlou, G.; Nouaille, S.; Boutet de Monvel, J.; Hardelin, J.-P.; Hauswirth, W.W.; Avan, P.; et al. Dual AAV-mediated gene therapy restores hearing in a DFNB9 mouse model. Proc. Natl. Acad. Sci. USA 2019. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.; Askew, C.; Galvin, A.; Heman-Ackah, S.; Asai, Y.; Indzhykulian, A.A.; Jodelka, F.M.; Hastings, M.L.; Lentz, J.J.; Vandenberghe, L.H.; et al. Gene therapy restores auditory and vestibular function in a mouse model of Usher syndrome type 1c. Nat. Biotechnol. 2017, 35, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Salt, A.N.; Hirose, K. Communication pathways to and from the inner ear and their contributions to drug delivery. Hear. Res. 2018, 362, 25–37. [Google Scholar] [CrossRef]
- Ishiyama, G.; Lopez, I.A.; Ishiyama, P.; Vinters, H.V.; Ishiyama, A. The blood labyrinthine barrier in the human normal and Meniere’s disease macula utricle. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Le, T.N.; Blakley, B.W. Mannitol and the blood-labyrinth barrier. J. Otolaryngol. Head Neck Surg. 2017, 46, 66. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Puel, J.-L. Toward Cochlear Therapies. Physiol. Rev. 2018, 98, 2477–2522. [Google Scholar] [CrossRef] [PubMed]
- Sacheli, R.; Delacroix, L.; Vandenackerveken, P.; Nguyen, L.; Malgrange, B. Gene transfer in inner ear cells: A challenging race. Gene. Ther. 2013, 20, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.; Nakai, H.; Xiao, W. Characterization of Genome Integrity for Oversized Recombinant AAV Vector. Mol. Ther. 2010, 18, 87–92. [Google Scholar] [CrossRef]
- Naso, M.F.; Tomkowicz, B.; Perry, W.L.; Strohl, W.R. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs Clin. Immunother. Biopharm. Gene Ther. 2017, 31, 317–334. [Google Scholar] [CrossRef]
- Le Meur, G.; Lebranchu, P.; Billaud, F.; Adjali, O.; Schmitt, S.; Bézieau, S.; Péréon, Y.; Valabregue, R.; Ivan, C.; Darmon, C.; et al. Safety and Long-Term Efficacy of AAV4 Gene Therapy in Patients with RPE65 Leber Congenital Amaurosis. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 256–268. [Google Scholar] [CrossRef]
- Bennett, J.; Wellman, J.; Marshall, K.A.; McCague, S.; Ashtari, M.; DiStefano-Pappas, J.; Elci, O.U.; Chung, D.C.; Sun, J.; Wright, J.F.; et al. Safety and durability of effect of contralateral-eye administration of AAV2 gene therapy in patients with childhood-onset blindness caused by RPE65 mutations: A follow-on phase 1 trial. Lancet Lond. Engl. 2016, 388, 661–672. [Google Scholar] [CrossRef]
- Hastie, E.; Samulski, R.J. Adeno-associated virus at 50: A golden anniversary of discovery, research, and gene therapy success—A personal perspective. Hum. Gene Ther. 2015, 26, 257–265. [Google Scholar] [CrossRef]
- Powell, S.K.; Rivera-Soto, R.; Gray, S.J. Viral expression cassette elements to enhance transgene target specificity and expression in gene therapy. Discov. Med. 2015, 19, 49–57. [Google Scholar]
- Chamberlain, K.; Riyad, J.M.; Weber, T. Expressing Transgenes That Exceed the Packaging Capacity of Adeno-Associated Virus Capsids. Hum. Gene Ther. Methods 2016, 27, 1–12. [Google Scholar] [CrossRef]
- Daya, S.; Berns, K.I. Gene Therapy Using Adeno-Associated Virus Vectors. Clin. Microbiol. Rev. 2008, 21, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Asokan, A.; Samulski, R.J. Adeno-associated virus serotypes: Vector toolkit for human gene therapy. Mol. Ther. J. Am. Soc. Gene Ther. 2006, 14, 316–327. [Google Scholar] [CrossRef]
- Asokan, A.; Schaffer, D.V.; Jude Samulski, R. The AAV Vector Toolkit: Poised at the Clinical Crossroads. Mol. Ther. 2012, 20, 699–708. [Google Scholar] [CrossRef] [PubMed]
- DiMattia, M.A.; Nam, H.-J.; Van Vliet, K.; Mitchell, M.; Bennett, A.; Gurda, B.L.; McKenna, R.; Olson, N.H.; Sinkovits, R.S.; Potter, M.; et al. Structural Insight into the Unique Properties of Adeno-Associated Virus Serotype 9. J. Virol. 2012, 86, 6947–6958. [Google Scholar] [CrossRef] [PubMed]
- Halder, S.; Van Vliet, K.; Smith, J.K.; Duong, T.T.P.; McKenna, R.; Wilson, J.M.; Agbandje-McKenna, M. Structure of neurotropic adeno-associated virus AAVrh.8. J. Struct. Biol. 2015, 192, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Pillay, S.; Meyer, N.L.; Puschnik, A.S.; Davulcu, O.; Diep, J.; Ishikawa, Y.; Jae, L.T.; Wosen, J.E.; Nagamine, C.M.; Chapman, M.S.; et al. An essential receptor for adeno-associated virus infection. Nature 2016, 530, 108–112. [Google Scholar] [CrossRef]
- Summerford, C.; Bartlett, J.S.; Samulski, R.J. αVβ5 integrin: A co-receptor for adeno-associated virus type 2 infection. Nat. Med. 1999, 5, 78–82. [Google Scholar] [CrossRef]
- Qing, K.; Mah, C.; Hansen, J.; Zhou, S.; Dwarki, V.; Srivastava, A. Human fibroblast growth factor receptor 1 is a co-receptor for infection by adeno-associated virus 2. Nat. Med. 1999, 5, 71–77. [Google Scholar] [CrossRef]
- Kashiwakura, Y.; Tamayose, K.; Iwabuchi, K.; Hirai, Y.; Shimada, T.; Matsumoto, K.; Nakamura, T.; Watanabe, M.; Oshimi, K.; Daida, H. Hepatocyte Growth Factor Receptor Is a Coreceptor for Adeno-Associated Virus Type 2 Infection. J. Virol. 2005, 79, 609–614. [Google Scholar] [CrossRef]
- Asokan, A.; Hamra, J.B.; Govindasamy, L.; Agbandje-McKenna, M.; Samulski, R.J. Adeno-Associated Virus Type 2 Contains an Integrin 5 1 Binding Domain Essential for Viral Cell Entry. J. Virol. 2006, 80, 8961–8969. [Google Scholar] [CrossRef]
- Akache, B.; Grimm, D.; Pandey, K.; Yant, S.R.; Xu, H.; Kay, M.A. The 37/67-Kilodalton Laminin Receptor Is a Receptor for Adeno-Associated Virus Serotypes 8, 2, 3, and 9. J. Virol. 2006, 80, 9831–9836. [Google Scholar] [CrossRef]
- Sonntag, F.; Bleker, S.; Leuchs, B.; Fischer, R.; Kleinschmidt, J.A. Adeno-Associated Virus Type 2 Capsids with Externalized VP1/VP2 Trafficking Domains Are Generated prior to Passage through the Cytoplasm and Are Maintained until Uncoating Occurs in the Nucleus. J. Virol. 2006, 80, 11040–11054. [Google Scholar] [CrossRef]
- Xiao, P.-J.; Samulski, R.J. Cytoplasmic Trafficking, Endosomal Escape, and Perinuclear Accumulation of Adeno-Associated Virus Type 2 Particles Are Facilitated by Microtubule Network. J. Virol. 2012, 86, 10462–10473. [Google Scholar] [CrossRef] [PubMed]
- Nicolson, S.C.; Samulski, R.J. Recombinant Adeno-Associated Virus Utilizes Host Cell Nuclear Import Machinery To Enter the Nucleus. J. Virol. 2014, 88, 4132–4144. [Google Scholar] [CrossRef] [PubMed]
- Kelich, J.M.; Ma, J.; Dong, B.; Wang, Q.; Chin, M.; Magura, C.M.; Xiao, W.; Yang, W. Super-resolution imaging of nuclear import of adeno-associated virus in live cells. Mol. Ther. Methods Clin. Dev. 2015, 2, 15047. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef]
- Zinn, E.; Pacouret, S.; Khaychuk, V.; Turunen, H.T.; Carvalho, L.S.; Andres-Mateos, E.; Shah, S.; Shelke, R.; Maurer, A.C.; Plovie, E.; et al. In Silico Reconstruction of the Viral Evolutionary Lineage Yields a Potent Gene Therapy Vector. Cell Rep. 2015, 12, 1056–1068. [Google Scholar] [CrossRef]
- Suzuki, J.; Hashimoto, K.; Xiao, R.; Vandenberghe, L.H.; Liberman, M.C. Cochlear gene therapy with ancestral AAV in adult mice: Complete transduction of inner hair cells without cochlear dysfunction. Sci. Rep. 2017, 7, 45524. [Google Scholar] [CrossRef]
- Dashkoff, J.; Lerner, E.P.; Truong, N.; Klickstein, J.A.; Fan, Z.; Mu, D.; Maguire, C.A.; Hyman, B.T.; Hudry, E. Tailored transgene expression to specific cell types in the central nervous system after peripheral injection with AAV9. Mol. Ther. Methods Clin. Dev. 2016, 3, 16081. [Google Scholar] [CrossRef]
- Xu, R.; Janson, C.; Mastakov, M.; Lawlor, P.; Young, D.; Mouravlev, A.; Fitzsimons, H.; Choi, K.-L.; Ma, H.; Dragunow, M.; et al. Quantitative comparison of expression with adeno-associated virus (AAV-2) brain-specific gene cassettes. Gene Ther. 2001, 8, 1323–1332. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Chai, R.; Guo, L.; Dong, B.; Li, W.; Shu, Y.; Huang, X.; Li, H. Transduction of Adeno-Associated Virus Vectors Targeting Hair Cells and Supporting Cells in the Neonatal Mouse Cochlea. Front. Cell Neurosci. 2019, 13. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, J.S.; Kleinschmidt, J.; Boucher, R.C.; Samulski, R.J. Targeted adeno-associated virus vector transduction of nonpermissive cells mediated by a bispecific F(ab’γ)2 antibody. Nat. Biotechnol. 1999, 17, 181–186. [Google Scholar] [CrossRef]
- Arnold, G.S.; Sasser, A.K.; Stachler, M.D.; Bartlett, J.S. Metabolic Biotinylation Provides a Unique Platform for the Purification and Targeting of Multiple AAV Vector Serotypes. Mol. Ther. 2006, 14, 97–106. [Google Scholar] [CrossRef]
- Ponnazhagan, S.; Mahendra, G.; Curiel, D.T.; Shaw, D.R. Adeno-Associated Virus Type 2-Mediated Transduction of Human Monocyte-Derived Dendritic Cells: Implications for Ex Vivo Immunotherapy. J. Virol. 2001, 75, 9493–9501. [Google Scholar] [CrossRef] [PubMed]
- Okano, T.; Nakagawa, T.; Kita, T.; Kada, S.; Yoshimoto, M.; Nakahata, T.; Ito, J. Bone marrow-derived cells expressing Iba1 are constitutively present as resident tissue macrophages in the mouse cochlea. J. Neurosci. Res. 2008, 86, 1758–1767. [Google Scholar] [CrossRef] [PubMed]
- Fredelius, L.; Rask-Andersen, H. The role of macrophages in the disposal of degeneration products within the organ of corti after acoustic overstimulation. Acta Otolaryngol. 1990, 109, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.J.T.; Thorne, P.R.; Vlajkovic, S.M. Characterisation of cochlear inflammation in mice following acute and chronic noise exposure. Histochem. Cell Biol. 2016, 146, 219–230. [Google Scholar] [CrossRef]
- So, H.; Kim, H.; Lee, J.-H.; Park, C.; Kim, Y.; Kim, E.; Kim, J.-K.; Yun, K.-J.; Lee, K.-M.; Lee, H.-Y.; et al. Cisplatin Cytotoxicity of Auditory Cells Requires Secretions of Proinflammatory Cytokines via Activation of ERK and NF-κB. J. Assoc. Res. Otolaryngol. 2007, 8, 338–355. [Google Scholar] [CrossRef]
- Zaiss, A.-K.; Liu, Q.; Bowen, G.P.; Wong, N.C.W.; Bartlett, J.S.; Muruve, D.A. Differential activation of innate immune responses by adenovirus and adeno-associated virus vectors. J. Virol. 2002, 76, 4580–4590. [Google Scholar] [CrossRef]
- Li, H.; Lasaro, M.O.; Jia, B.; Lin, S.W.; Haut, L.H.; High, K.A.; Ertl, H.C.J. Capsid-specific T-cell responses to natural infections with adeno-associated viruses in humans differ from those of nonhuman primates. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 2021–2030. [Google Scholar] [CrossRef] [PubMed]
- Boutin, S.; Monteilhet, V.; Veron, P.; Leborgne, C.; Benveniste, O.; Montus, M.F.; Masurier, C. Prevalence of Serum IgG and Neutralizing Factors Against Adeno-Associated Virus (AAV) Types 1, 2, 5, 6, 8, and 9 in the Healthy Population: Implications for Gene Therapy Using AAV Vectors. Hum. Gene Ther. 2010, 21, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Calcedo, R.; Bell, P.; Lin, J.; Grant, R.L.; Siegel, D.L.; Wilson, J.M. Impact of pre-existing immunity on gene transfer to nonhuman primate liver with adeno-associated virus 8 vectors. Hum. Gene Ther. 2011, 22, 1389–1401. [Google Scholar] [CrossRef]
- Landegger, L.D.; Pan, B.; Askew, C.; Wassmer, S.J.; Gluck, S.D.; Galvin, A.; Taylor, R.; Forge, A.; Stankovic, K.M.; Holt, J.R.; et al. A synthetic AAV vector enables safe and efficient gene transfer to the mammalian inner ear. Nat. Biotechnol. 2017, 35, 280–284. [Google Scholar] [CrossRef]
- Cao, O.; Hoffman, B.E.; Moghimi, B.; Nayak, S.; Cooper, M.; Zhou, S.; Ertl, H.C.J.; High, K.A.; Herzog, R.W. Impact of the underlying mutation and the route of vector administration on immune responses to factor IX in gene therapy for hemophilia B. Mol. Ther. J. Am. Soc. Gene Ther. 2009, 17, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Nathwani, A.C.; Tuddenham, E.G.D.; Rangarajan, S.; Rosales, C.; McIntosh, J.; Linch, D.C.; Chowdary, P.; Riddell, A.; Pie, A.J.; Harrington, C.; et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N. Engl. J. Med. 2011, 365, 2357–2365. [Google Scholar] [CrossRef] [PubMed]
- Mingozzi, F.; Meulenberg, J.J.; Hui, D.J.; Basner-Tschakarjan, E.; Hasbrouck, N.C.; Edmonson, S.A.; Hutnick, N.A.; Betts, M.R.; Kastelein, J.J.; Stroes, E.S.; et al. AAV-1-mediated gene transfer to skeletal muscle in humans results in dose-dependent activation of capsid-specific T cells. Blood 2009, 114, 2077–2086. [Google Scholar] [CrossRef] [PubMed]
- Kilpatrick, L.A.; Li, Q.; Yang, J.; Goddard, J.C.; Fekete, D.M.; Lang, H. Adeno-associated virus-mediated gene delivery into the scala media of the normal and deafened adult mouse ear. Gene Ther. 2011, 18, 569–578. [Google Scholar] [CrossRef]
- Konishi, M.; Kawamoto, K.; Izumikawa, M.; Kuriyama, H.; Yamashita, T. Gene transfer into guinea pig cochlea using adeno-associated virus vectors. J. Gene Med. 2008, 10, 610–618. [Google Scholar] [CrossRef]
- Hudry, E.; Andres-Mateos, E.; Lerner, E.P.; Volak, A.; Cohen, O.; Hyman, B.T.; Maguire, C.A.; Vandenberghe, L.H. Efficient Gene Transfer to the Central Nervous System by Single-Stranded Anc80L65. Mol. Ther. Methods Clin. Dev. 2018, 10, 197–209. [Google Scholar] [CrossRef]
- Shibata, S.B.; Yoshimura, H.; Ranum, P.T.; Goodwin, A.T.; Smith, R.J.H. Intravenous rAAV2/9 injection for murine cochlear gene delivery. Sci. Rep. 2017, 7, 9609. [Google Scholar] [CrossRef] [PubMed]
- Jero, J.; Mhatre, A.N.; Tseng, C.J.; Stern, R.E.; Coling, D.E.; Goldstein, J.A.; Hong, K.; Zheng, W.W.; Hoque, A.T.; Lalwani, A.K. Cochlear gene delivery through an intact round window membrane in mouse. Hum. Gene Ther. 2001, 12, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Thorne, M.; Salt, A.N.; DeMott, J.E.; Henson, M.M.; Henson, O.W.; Gewalt, S.L. Cochlear Fluid Space Dimensions for Six Species Derived From Reconstructions of Three-Dimensional Magnetic Resonance Images. Laryngoscope 1999, 109, 1661–1668. [Google Scholar] [CrossRef] [PubMed]
- Chien, W.W.; McDougald, D.S.; Roy, S.; Fitzgerald, T.S.; Cunningham, L.L. Cochlear gene transfer mediated by adeno-associated virus: Comparison of two surgical approaches. Laryngoscope 2015, 125, 2557–2564. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.Z.; Saleh, J.; Isgrig, K.T.; Cunningham, L.L.; Chien, W.W. Hearing Loss after Round Window Surgery in Mice Is due to Middle Ear Effusion. Audiol. Neurootol. 2016, 21, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Yin, S.; Wang, J. Inner ear gene transfection in neonatal mice using adeno-associated viral vector: A comparison of two approaches. PLoS ONE 2012, 7, e43218. [Google Scholar] [CrossRef]
- Shi, X.; Wu, N.; Zhang, Y.; Guo, W.; Lin, C.; Yang, S. Adeno-associated virus transformation into the normal miniature pig and the normal guinea pigs cochlea via scala tympani. Acta Otolaryngol. 2017, 137(9), 910–916. [Google Scholar] [CrossRef]
- Liu, Y.; Okada, T.; Sheykholeslami, K.; Shimazaki, K.; Nomoto, T.; Muramatsu, S.-I.; Kanazawa, T.; Takeuchi, K.; Ajalli, R.; Mizukami, H.; et al. Specific and efficient transduction of Cochlear inner hair cells with recombinant adeno-associated virus type 3 vector. Mol. Ther. J. Am. Soc. Gene Ther. 2005, 12, 725–733. [Google Scholar] [CrossRef]
- Shibata, S.B.; Cortez, S.R.; Wiler, J.A.; Swiderski, D.L.; Raphael, Y. Hyaluronic acid enhances gene delivery into the cochlea. Hum. Gene Ther. 2012, 23, 302–310. [Google Scholar] [CrossRef]
- Yoshimura, H.; Shibata, S.B.; Ranum, P.T.; Smith, R.J.H. Enhanced viral-mediated cochlear gene delivery in adult mice by combining canal fenestration with round window membrane inoculation. Sci. Rep. 2018, 8, 2980. [Google Scholar] [CrossRef]
- Dai, C.; Lehar, M.; Sun, D.Q.; Rvt, L.S.; Carey, J.P.; MacLachlan, T.; Brough, D.; Staecker, H.; Della Santina, A.M.; Hullar, T.E.; et al. Rhesus Cochlear and Vestibular Functions Are Preserved After Inner Ear Injection of Saline Volume Sufficient for Gene Therapy Delivery. J. Assoc. Res. Otolaryngol. JARO 2017, 18, 601–617. [Google Scholar] [CrossRef] [PubMed]
- György, B.; Meijer, E.J.; Ivanchenko, M.V.; Tenneson, K.; Emond, F.; Hanlon, K.S.; Indzhykulian, A.A.; Volak, A.; Karavitaki, K.D.; Tamvakologos, P.I.; et al. Gene Transfer with AAV9-PHP.B Rescues Hearing in a Mouse Model of Usher Syndrome 3A and Transduces Hair Cells in a Non-human Primate. Mol. Ther. Methods Clin. Dev. 2019, 13, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hendricks, J.L.; Chikar, J.A.; Crumling, M.A.; Raphael, Y.; Martin, D.C. Localized cell and drug delivery for auditory prostheses. Hear. Res. 2008, 242, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Murphy, R.; Taaffe, D.; Yin, S.; Xia, L.; Hauswirth, W.W.; Bance, M.; Robertson, G.S.; Wang, J. Efficient cochlear gene transfection in guinea-pigs with adeno-associated viral vectors by partial digestion of round window membrane. Gene Ther. 2012, 19, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Tarabichi, M.; Kapadia, M. Principles of endoscopic ear surgery. Curr. Opin. Otolaryngol. Head Neck Surg. 2016, 24, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Shin, J.E.; Cunnane, M.; Fujita, K.; Henein, S.; Psaltis, D.; Stankovic, K.M. Surgical Anatomy of the Human Round Window Region: Implication for Cochlear Endoscopy Through the External Auditory Canal. Otol. Neurotol. 2016, 37, 1189–1194. [Google Scholar] [CrossRef]
- Kelso, C.M.; Watanabe, H.; Wazen, J.M.; Bucher, T.; Qian, Z.J.; Olson, E.S.; Kysar, J.W.; Lalwani, A.K. Microperforations significantly enhance diffusion across round window membrane. Otol. Neurotol. 2015, 36, 694–700. [Google Scholar] [CrossRef]
- Kawamoto, K.; Oh, S.H.; Kanzaki, S.; Brown, N.; Raphael, Y. The functional and structural outcome of inner ear gene transfer via the vestibular and cochlear fluids in mice. Mol. Ther. J. Am. Soc. Gene Ther. 2001, 4, 575–585. [Google Scholar] [CrossRef]
- Guo, J.-Y.; He, L.; Qu, T.-F.; Liu, Y.-Y.; Liu, K.; Wang, G.-P.; Gong, S.-S. Canalostomy As a Surgical Approach to Local Drug Delivery into the Inner Ears of Adult and Neonatal Mice. J. Vis. Exp. JoVE 2018. [Google Scholar] [CrossRef]
- Wang, G.-P.; Guo, J.-Y.; Peng, Z.; Liu, Y.-Y.; Xie, J.; Gong, S.-S. Adeno-associated virus-mediated gene transfer targeting normal and traumatized mouse utricle. Gene Ther. 2014, 21, 958–966. [Google Scholar] [CrossRef]
- Isgrig, K.; McDougald, D.S.; Zhu, J.; Wang, H.J.; Bennett, J.; Chien, W.W. AAV2.7m8 is a powerful viral vector for inner ear gene therapy. Nat. Commun. 2019, 10, 427. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Huang, M.; Shu, Y.; Ruprecht, A.; Wang, H.; Tang, Y.; Vandenberghe, L.H.; Wang, Q.; Gao, G.; Kong, W.-J.; et al. Delivery of Adeno-Associated Virus Vectors in Adult Mammalian Inner-Ear Cell Subtypes Without Auditory Dysfunction. Hum. Gene Ther. 2018, 29, 492–506. [Google Scholar] [CrossRef] [PubMed]
- De Morgon, A.; Aran, J.M.; Collet, L.; Dauman, R.; Fraysse, B.; Freyss, G.; Pujol, R.; Sens, A.; Serkers, O.; Tran Ba Huy, P.; et al. Données Actuelles sur la Pathologie et la Physiologie de l’oreille Interne; Rapport de la société française d’ORL; Edition Arnette; Arnette: Paris, France, 1990. [Google Scholar]
- Ekdale, E.G. Comparative Anatomy of the Bony Labyrinth (Inner Ear) of Placental Mammals. PLoS ONE 2013, 8, e66624. [Google Scholar] [CrossRef]
- Pinyon, J.L.; Tadros, S.F.; Froud, K.E.Y.; Wong, A.C.; Tompson, I.T.; Crawford, E.N.; Ko, M.; Morris, R.; Klugmann, M.; Housley, G.D. Close-Field Electroporation Gene Delivery Using the Cochlear Implant Electrode Array Enhances the Bionic Ear. Sci. Transl. Med. 2014, 6, 233ra54. [Google Scholar] [CrossRef] [PubMed]
- Rejali, D.; Lee, V.A.; Abrashkin, K.A.; Humayun, N.; Swiderski, D.L.; Raphael, Y. Cochlear implants and ex vivo BDNF gene therapy protect spiral ganglion neurons. Hear. Res. 2007, 228, 180–187. [Google Scholar] [CrossRef]
- Corrales, C.E.; Mudry, A. History of the Endolymphatic Sac: From Anatomy to Surgery. Otol. Neurotol. 2017, 38, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Wick, C.C.; Manzoor, N.F.; McKenna, C.; Semaan, M.T.; Megerian, C.A. Long-term outcomes of endolymphatic sac shunting with local steroids for Meniere’s disease. Am. J. Otolaryngol. 2017, 38, 285–290. [Google Scholar] [CrossRef]
- Salt, A.N.; Rask-Andersen, H. Responses of the endolymphatic sac to perilymphatic injections and withdrawals: Evidence for the presence of a one-way valve. Hear. Res. 2004, 191, 90–100. [Google Scholar] [CrossRef]
- Hildebrand, M.S.; Kahrizi, K.; Bromhead, C.J.; Shearer, A.E.; Webster, J.A.; Khodaei, H.; Abtahi, R.; Bazazzadegan, N.; Babanejad, M.; Nikzat, N.; et al. Mutations in TMC1 are a Common Cause of DFNB7/11 Hearing Loss in the Iranian Population. Ann. Otol. Rhinol. Laryngol. 2010, 119, 830–835. [Google Scholar] [CrossRef]
- Askew, C.; Rochat, C.; Pan, B.; Asai, Y.; Ahmed, H.; Child, E.; Schneider, B.L.; Aebischer, P.; Holt, J.R. Tmc gene therapy restores auditory function in deaf mice. Sci. Transl. Med. 2015, 7, 295ra108. [Google Scholar] [CrossRef]
- Lentz, J.J.; Gordon, W.C.; Farris, H.E.; MacDonald, G.H.; Cunningham, D.E.; Robbins, C.A.; Tempel, B.L.; Bazan, N.G.; Rubel, E.W.; Oesterle, E.C.; et al. Deafness and retinal degeneration in a novel USH1C knock-in mouse model. Dev. Neurobiol. 2010, 70, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Lentz, J.; Savas, S.; Ng, S.-S.; Athas, G.; Deininger, P.; Keats, B. The USH1C 216G?A splice-site mutation results in a 35-base-pair deletion. Hum. Genet. 2005, 116, 225–227. [Google Scholar] [CrossRef] [PubMed]
- Isgrig, K.; Shteamer, J.W.; Belyantseva, I.A.; Drummond, M.C.; Fitzgerald, T.S.; Vijayakumar, S.; Jones, S.M.; Griffith, A.J.; Friedman, T.B.; Cunningham, L.L.; et al. Gene Therapy Restores Balance and Auditory Functions in a Mouse Model of Usher Syndrome. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Emptoz, A.; Michel, V.; Lelli, A.; Akil, O.; Boutet de Monvel, J.; Lahlou, G.; Meyer, A.; Dupont, T.; Nouaille, S.; Ey, E.; et al. Local gene therapy durably restores vestibular function in a mouse model of Usher syndrome type 1G. Proc. Natl. Acad. Sci. USA 2017, 114, 9695–9700. [Google Scholar] [CrossRef] [PubMed]
- Yasunaga, S.; Grati, M.; Cohen-Salmon, M.; El-Amraoui, A.; Mustapha, M.; Salem, N.; El-Zir, E.; Loiselet, J.; Petit, C. A mutation in OTOF, encoding otoferlin, a FER-1-like protein, causes DFNB9, a nonsyndromic form of deafness. Nat. Genet. 1999, 21, 363–369. [Google Scholar] [CrossRef]
- Ruel, J.; Emery, S.; Nouvian, R.; Bersot, T.; Amilhon, B.; Van Rybroek, J.M.; Rebillard, G.; Lenoir, M.; Eybalin, M.; Delprat, B.; et al. Impairment of SLC17A8 Encoding Vesicular Glutamate Transporter-3, VGLUT3, Underlies Nonsyndromic Deafness DFNA25 and Inner Hair Cell Dysfunction in Null Mice. Am. J. Hum. Genet. 2008, 83, 278–292. [Google Scholar] [CrossRef]
- Akil, O.; Seal, R.P.; Burke, K.; Wang, C.; Alemi, A.; During, M.; Edwards, R.H.; Lustig, L.R. Restoration of Hearing in the VGLUT3 Knockout Mouse Using Virally Mediated Gene Therapy. Neuron 2012, 75, 283–293. [Google Scholar] [CrossRef]
- Maeda, Y.; Fukushima, K.; Nishizaki, K.; Smith, R.J.H. In vitro and in vivo suppression of GJB2 expression by RNA interference. Hum. Mol. Genet. 2005, 14, 1641–1650. [Google Scholar] [CrossRef]
- Lentz, J.J.; Jodelka, F.M.; Hinrich, A.J.; McCaffrey, K.E.; Farris, H.E.; Spalitta, M.J.; Bazan, N.G.; Duelli, D.M.; Rigo, F.; Hastings, M.L. Rescue of hearing and vestibular function by antisense oligonucleotides in a mouse model of human deafness. Nat. Med. 2013, 19, 345–350. [Google Scholar] [CrossRef]
- Gao, X.; Tao, Y.; Lamas, V.; Huang, M.; Yeh, W.-H.; Pan, B.; Hu, Y.-J.; Hu, J.H.; Thompson, D.B.; Shu, Y.; et al. Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature 2018, 553, 217–221. [Google Scholar] [CrossRef]
- György, B.; Nist-Lund, C.; Pan, B.; Asai, Y.; Karavitaki, K.D.; Kleinstiver, B.P.; Garcia, S.P.; Zaborowski, M.P.; Solanes, P.; Spataro, S.; et al. Allele-specific gene editing prevents deafness in a model of dominant progressive hearing loss. Nat. Med. 2019, 25, 1123–1130. [Google Scholar] [CrossRef]
- Boettcher, M.; McManus, M.T. Choosing the Right Tool for the Job: RNAi, TALEN, or CRISPR. Mol. Cell 2015, 58, 575–585. [Google Scholar] [CrossRef]
- Borel, F.; Kay, M.A.; Mueller, C. Recombinant AAV as a platform for translating the therapeutic potential of RNA interference. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 692–701. [Google Scholar] [CrossRef]
- Naito, Y.; Ui-Tei, K. Designing functional siRNA with reduced off-target effects. Methods Mol. Biol. 2013, 942, 57–68. [Google Scholar]
- Jagger, D.J.; Forge, A. Connexins and gap junctions in the inner ear--it’s not just about K+ recycling. Cell Tissue Res. 2015, 360, 633–644. [Google Scholar] [CrossRef]
- Winkler, J.; Stessl, M.; Amartey, J.; Noe, C.R. Off-target effects related to the phosphorothioate modification of nucleic acids. ChemMedChem 2010, 5, 1344–1352. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Huang, X. Methods and applications of CRISPR/Cas system for genome editing in stem cells. Cell Regen. 2019, 8, 33–41. [Google Scholar] [CrossRef]
- Campa, C.C.; Weisbach, N.R.; Santinha, A.J.; Incarnato, D.; Platt, R.J. Multiplexed genome engineering by Cas12a and CRISPR arrays encoded on single transcripts. Nat. Methods 2019, 16, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.L.; Ruan, M.Z.C.; Mahajan, V.B.; Tsang, S.H. Viral Delivery Systems for CRISPR. Viruses 2019, 11. [Google Scholar] [CrossRef]
- Chen, J.-R.; Tang, Z.-H.; Zheng, J.; Shi, H.-S.; Ding, J.; Qian, X.-D.; Zhang, C.; Chen, J.-L.; Wang, C.-C.; Li, L.; et al. Effects of genetic correction on the differentiation of hair cell-like cells from iPSCs with MYO15A mutation. Cell Death Differ. 2016, 23, 1347–1357. [Google Scholar] [CrossRef] [PubMed]
- Akil, O.; Blits, B.; Lustig, L.R.; Leake, P.A. Virally Mediated Overexpression of Glial-Derived Neurotrophic Factor Elicits Age- and Dose-Dependent Neuronal Toxicity and Hearing Loss. Hum. Gene Ther. 2019, 30, 88–105. [Google Scholar] [CrossRef] [PubMed]
- Lalwani, A.K.; Walsh, B.J.; Reilly, P.G.; Muzyczka, N.; Mhatre, A.N. Development of in vivo gene therapy for hearing disorders: Introduction of adeno-associated virus into the cochlea of the guinea pig. Gene Ther. 1996, 3, 588–592. [Google Scholar] [PubMed]
- Thalen, E.O.; Wit, H.P.; Segenhout, J.M.; Albers, F.W. Dynamics of inner ear pressure change caused by intracranial pressure manipulation in the guinea pig. Acta Otolaryngol. 2001, 121, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Tinling, S.P.; Chole, R.A. Apical cochlear nerve exposed to perilymph in the gerbil and rat. Hear. Res. 1994, 73, 203–208. [Google Scholar] [CrossRef]
- Hareendran, S.; Balakrishnan, B.; Sen, D.; Kumar, S.; Srivastava, A.; Jayandharan, G.R. Adeno-associated virus (AAV) vectors in gene therapy: Immune challenges and strategies to circumvent them. Rev. Med. Virol. 2013, 23, 399–413. [Google Scholar] [CrossRef]
- Gopen, Q.; Rosowski, J.J.; Merchant, S.N. Anatomy of the normal human cochlear aqueduct with functional implications. Hear. Res. 1997, 107, 9–22. [Google Scholar] [CrossRef]
- Jackler, R.K.; Hwang, P.H. Enlargement of the cochlear aqueduct: Fact or fiction? Otolaryngol. Head Neck Surg. 1993, 109, 14–25. [Google Scholar] [CrossRef]
- Nakashima, T.; Sone, M.; Teranishi, M.; Yoshida, T.; Terasaki, H.; Kondo, M.; Yasuma, T.; Wakabayashi, T.; Nagatani, T.; Naganawa, S. A perspective from magnetic resonance imaging findings of the inner ear: Relationships among cerebrospinal, ocular and inner ear fluids. Auris Nasus Larynx 2012, 39, 345–355. [Google Scholar] [CrossRef]
- Holden, H.B.; Schuknecht, H.F. Distribution pattern of blood in the inner ear following spontaneous subarachnoid haemorrhage. J. Laryngol. Otol. 1968, 82, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Marsot-Dupuch, K.; Djouhri, H.; Meyer, B.; Pharaboz, C.; Tran Ba Huy, P. Inner ear and subarachnoid spaces: Relations and diseases. Ann. Oto Laryngol. Chir. Cervico Faciale Bull. Soc. Oto Hopitaux Paris 2001, 118, 171–180. [Google Scholar]
- Palva, T. Cochlear aqueduct in infants. Acta Otolaryngol. 1970, 70, 83–94. [Google Scholar] [CrossRef]
- Dalbert, A.; Pfiffner, F.; Hoesli, M.; Koka, K.; Veraguth, D.; Roosli, C.; Huber, A. Assessment of Cochlear Function during Cochlear Implantation by Extra- and Intracochlear Electrocochleography. Front. Neurosci. 2018, 12, 18. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Deafness | Mouse Models | Therapeutic Strategies | Vectors and Promotors | Routes | Outcomes | Reference |
|---|---|---|---|---|---|---|
| DFNB7/11 and DFNA36 Usher 1C syndrome Usher IG syndrome DFNB9 DFNA25 | Tmc1−/−, Tmc2−/−, Tmc1/2−/−, Tmc1-Bth Ush1c c.216G > A Ush1g−/− Otof−/− Slc17a8 −/− | Tmc1 or Tmc2 gene addition harmonin-a1 or harmonin-b1 gene addition cDNA SENS addition Otoferlin cDNA addition VGLU3 cDNA addition | rAAV2/1-CBA rAAV2/Anc80L65-CMV rAAV2/8-CBA Dual rAAV-CBA Dual rAAV2/6-CBA/CMV AAV1-CBA | RWM P0-P2 RWM P1 RWM P2 RWM P10-P30 RWM P6-P7 RWM P10 or cochleostomy | Partially restored sensory transduction, ABR, and acoustic startle reflexes. Restoration of hearing and balance. Rescue of balance and low frequency hearing. Complete hearing restoration Partial restoration of IHC exocytosis and hearing. Complete restoration of ABR thresholds, partial rescue of the startle response | [92] [10] [95,96] [9] [8] [99] |
| Cx26 deafness Usher 1C syndrome | GJB2 R75W USH1C 216 Knock-In | siRNA against disease allele (R75W) ASO to block 216A cryptic splicing | Liposome complex | RWM in adult Intraperitoneal Injection P3-P16 or adult | Partial restoration of hearing Partially rescued vestibular function and hearing | [100] [101] |
| DFNA36 | Tmc1-Bth | CRISPR-Cas9 Gene Editing | Cationic lipid or rAAV2/Anc80L65-CMV | Scala media injection P1 | Effective prevention of deafness | [102,103] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blanc, F.; Mondain, M.; Bemelmans, A.-P.; Affortit, C.; Puel, J.-L.; Wang, J. rAAV-Mediated Cochlear Gene Therapy: Prospects and Challenges for Clinical Application. J. Clin. Med. 2020, 9, 589. https://doi.org/10.3390/jcm9020589
Blanc F, Mondain M, Bemelmans A-P, Affortit C, Puel J-L, Wang J. rAAV-Mediated Cochlear Gene Therapy: Prospects and Challenges for Clinical Application. Journal of Clinical Medicine. 2020; 9(2):589. https://doi.org/10.3390/jcm9020589
Chicago/Turabian StyleBlanc, Fabian, Michel Mondain, Alexis-Pierre Bemelmans, Corentin Affortit, Jean-Luc Puel, and Jing Wang. 2020. "rAAV-Mediated Cochlear Gene Therapy: Prospects and Challenges for Clinical Application" Journal of Clinical Medicine 9, no. 2: 589. https://doi.org/10.3390/jcm9020589
APA StyleBlanc, F., Mondain, M., Bemelmans, A.-P., Affortit, C., Puel, J.-L., & Wang, J. (2020). rAAV-Mediated Cochlear Gene Therapy: Prospects and Challenges for Clinical Application. Journal of Clinical Medicine, 9(2), 589. https://doi.org/10.3390/jcm9020589