Inhibition of Anaplerosis Attenuated Vascular Proliferation in Pulmonary Arterial Hypertension


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Experimental Section
2.1. Human Subjects
2.2. Rat Model of PH
2.3. Hemodynamic Measurement
2.4. Histopathological Analysis
2.5. Western Blot Analysis
2.6. Pyruvate Dehydrogenase and Oxaloacetate Assay
2.7. Statistical Analysis
3. Results
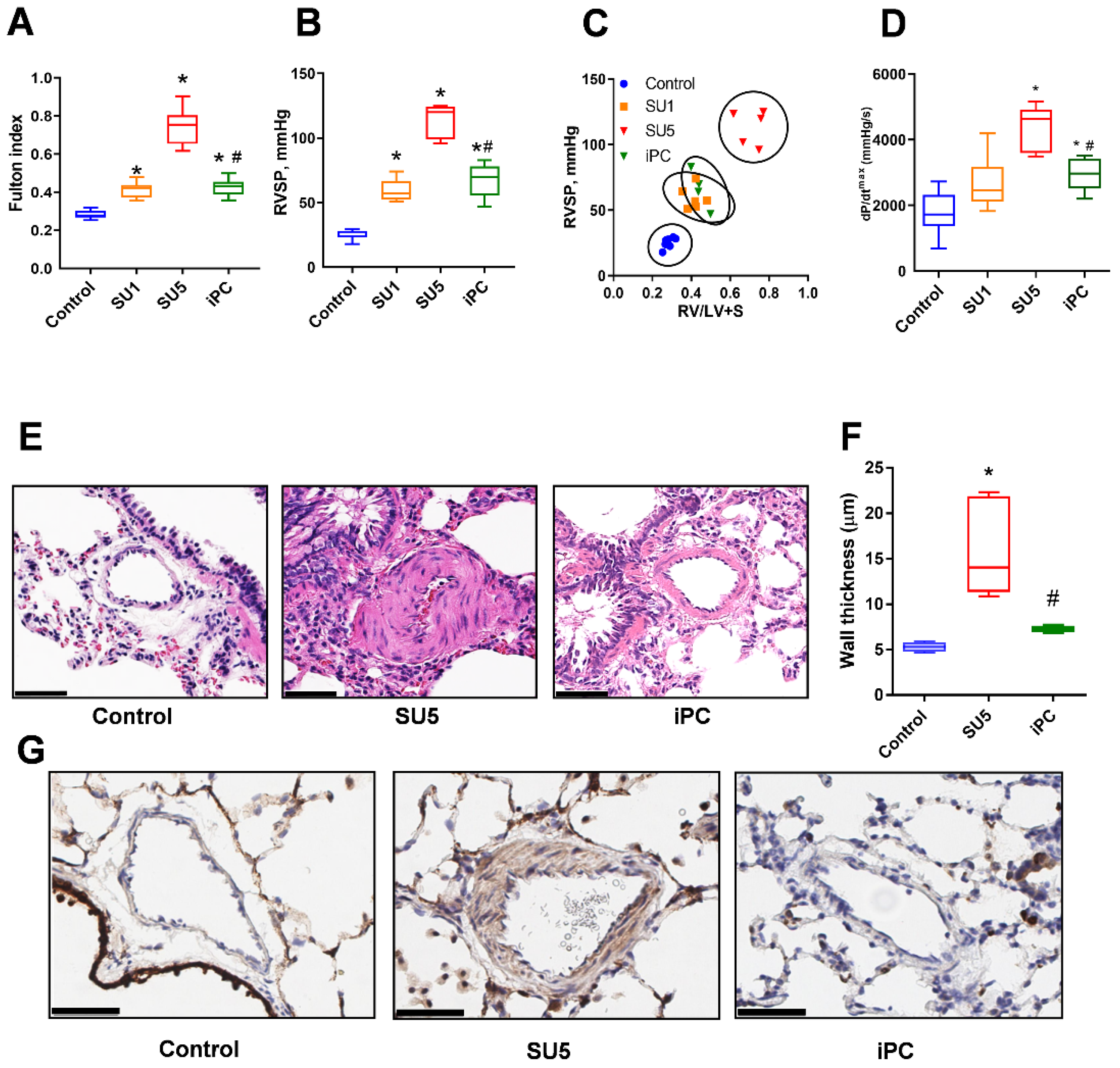
3.1. PC Inhibitor Attenuated Ventricular Pressure and Histological Changes
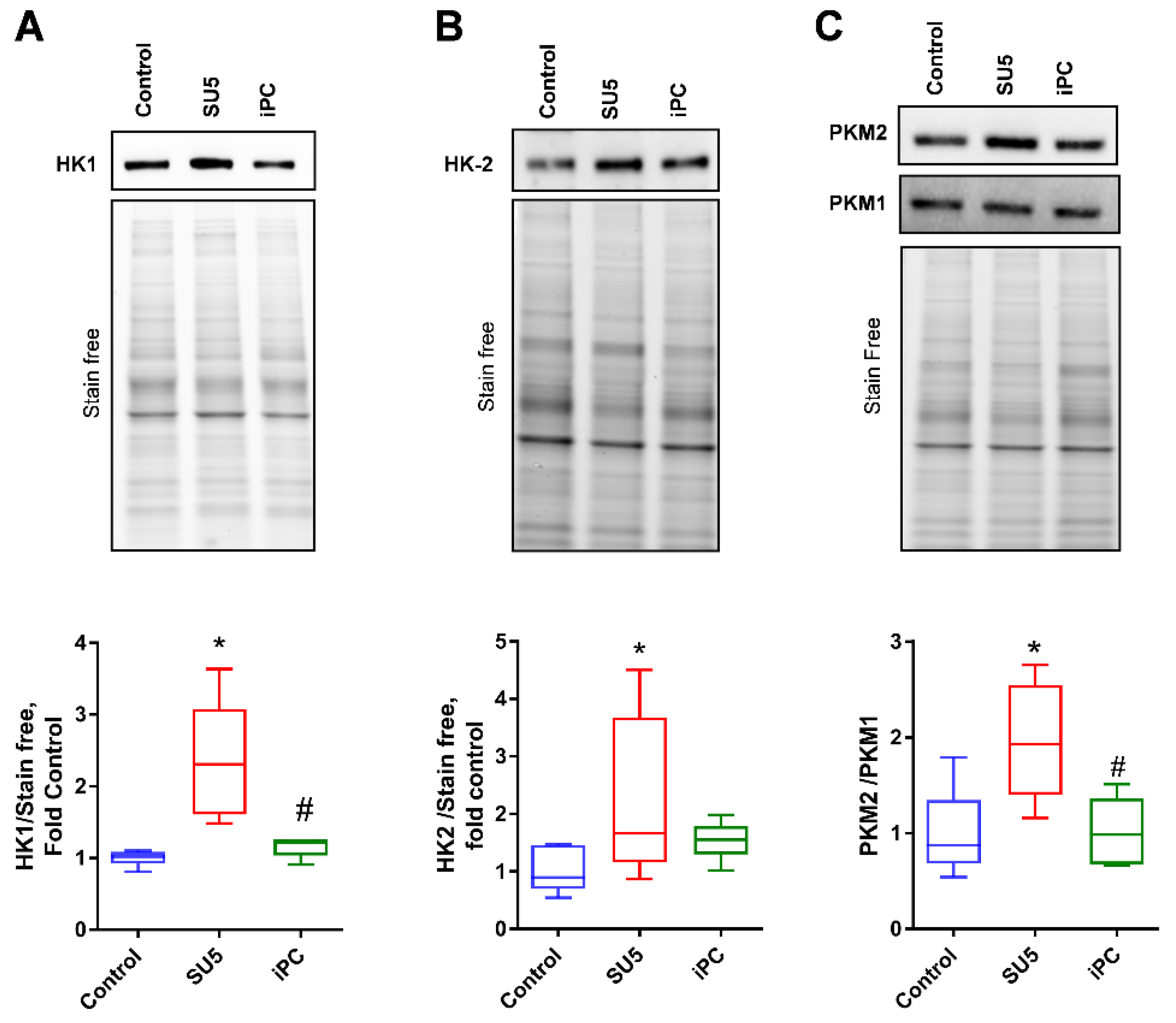
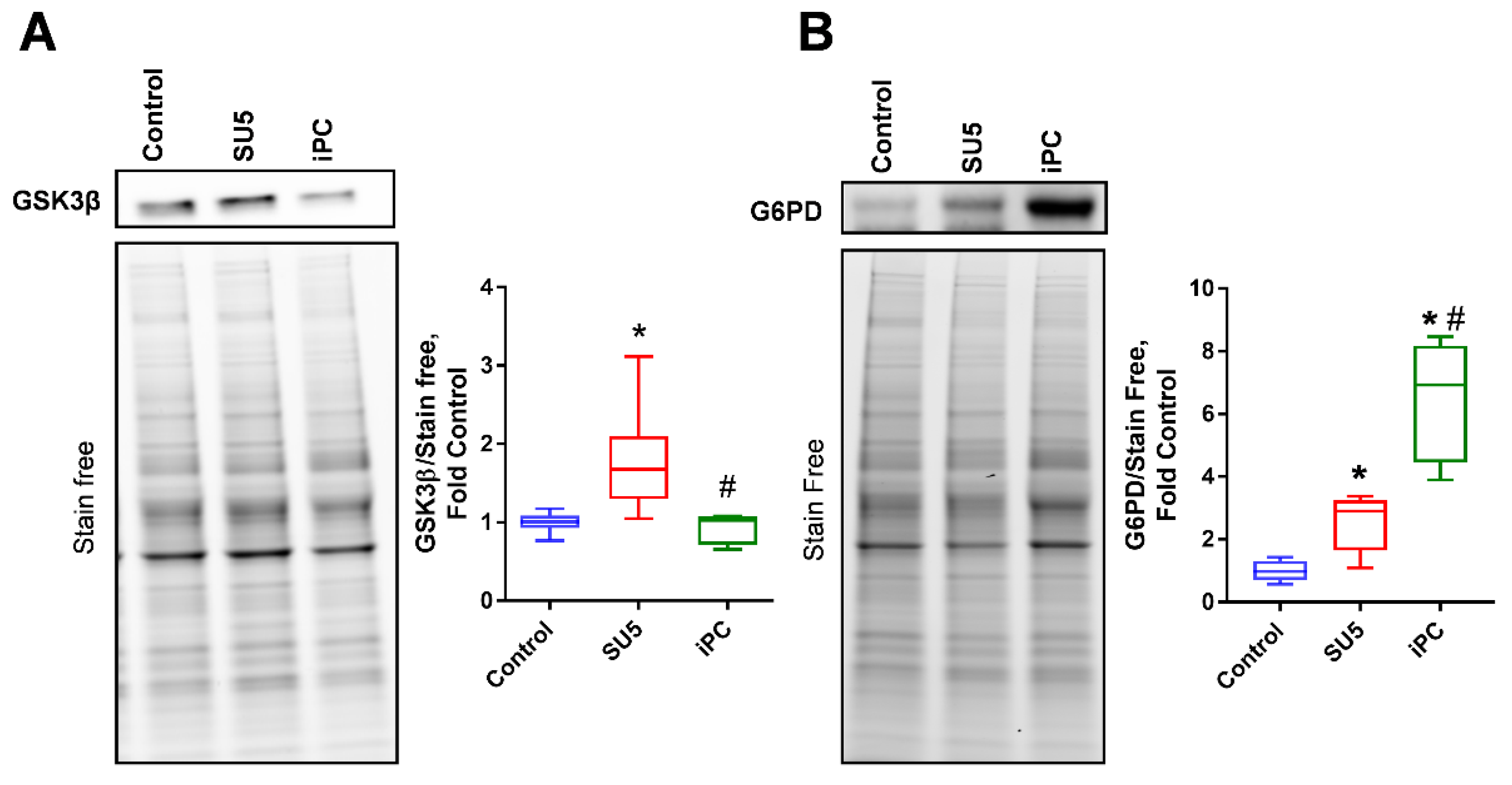
3.2. Inhibition of Akt Phosphorylation and Glucose Regulation by PC Inhibitor
3.3. PC Inhibition on Glycogen Synthesis and Pentose Phosphate Pathway
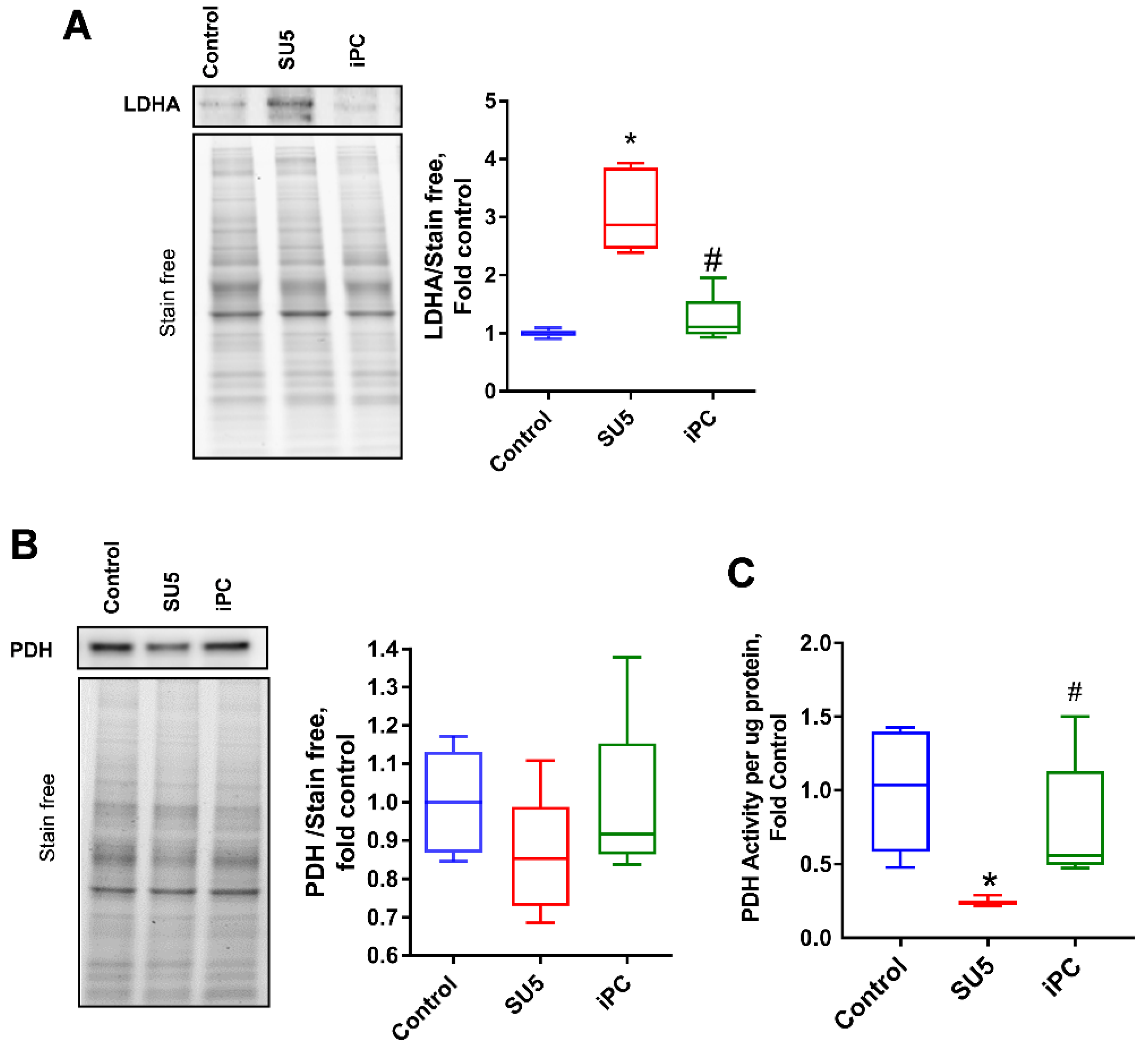
3.4. Inhibition of PC Inverted the Glycolytic Shift to Glucose Oxidation
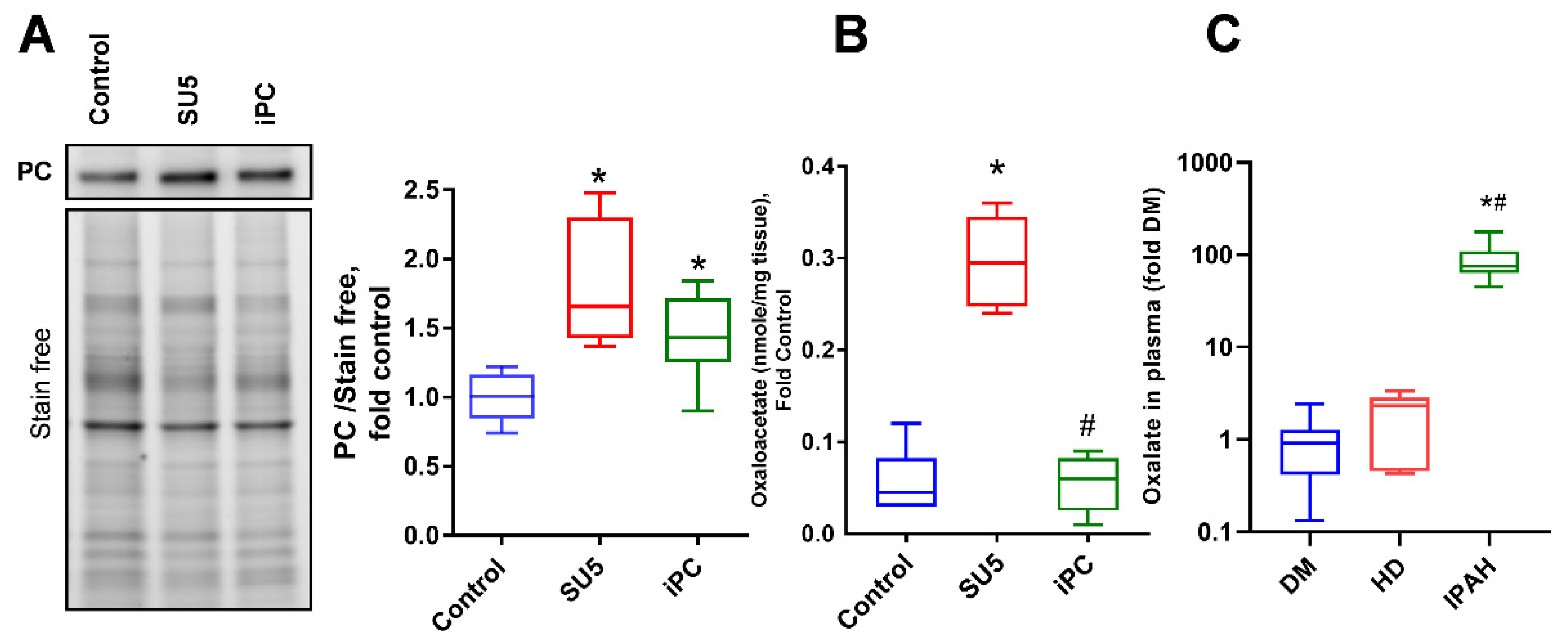
3.5. Anaplerotic Reprogramming was Controlled with PC Inhibition
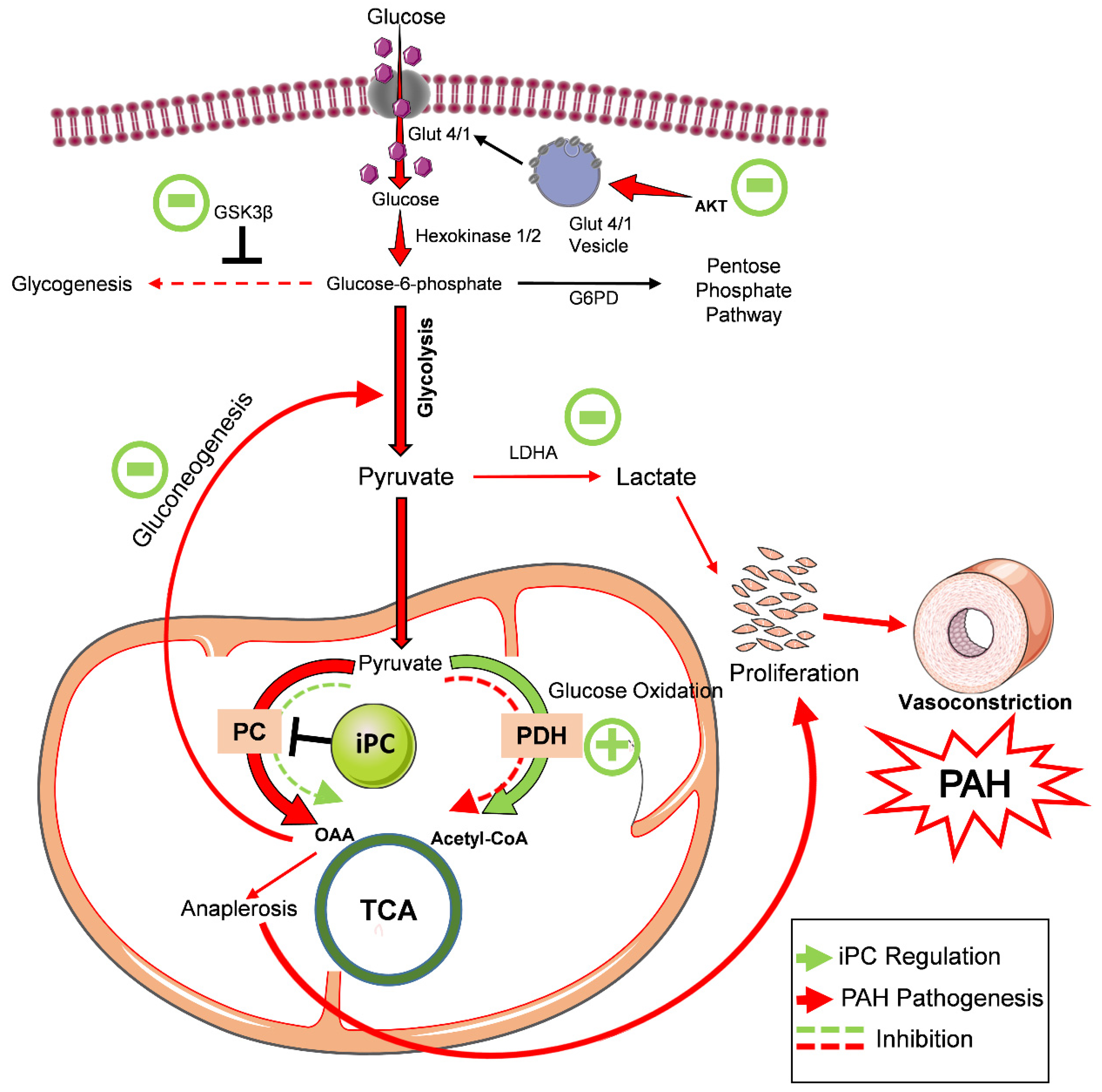
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sutendra, G.; Michelakis, E.D. Pulmonary Arterial Hypertension: Challenges in Translational Research and a Vision for Change. Sci. Transl. Med. 2013, 5, 208sr5. [Google Scholar] [CrossRef] [PubMed]
- Michelakis, E.D.; Gurtu, V.; Webster, L.; Barnes, G.; Watson, G.; Howard, L.; Cupitt, J.; Paterson, I.; Thompson, R.B.; Chow, K.; et al. Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients. Sci. Transl. Med. 2017, 9, eaao4583. [Google Scholar] [CrossRef] [PubMed]
- Rafikov, R.; Sun, X.; Rafikova, O.; Louise Meadows, M.; Desai, A.A.; Khalpey, Z.; Yuan, J.X.J.; Fineman, J.R.; Black, S.M. Complex I dysfunction underlies the glycolytic switch in pulmonary hypertensive smooth muscle cells. Redox Biol. 2015, 6, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Sellers, K.; Fox, M.P.; Ii, M.B.; Slone, S.P.; Higashi, R.M.; Miller, D.M.; Wang, Y.; Yan, J.; Yuneva, M.O.; Deshpande, R.; et al. Pyruvate carboxylase is critical for non–small-cell lung cancer proliferation. J. Clin. Investig. 2015, 125, 687–698. [Google Scholar] [CrossRef]
- Le, A.; Lane, A.N.; Hamaker, M.; Bose, S.; Gouw, A.; Barbi, J.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Zhang, H.; et al. Glucose-Independent Glutamine Metabolism via TCA Cycling for Proliferation and Survival in B Cells. Cell Metab. 2012, 15, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Rafikova, O.; Al Ghouleh, I.; Rafikov, R. Focus on Early Events: Pathogenesis of Pulmonary Arterial Hypertension Development. Antioxid. Redox Signal. 2019, 31, 933–953. [Google Scholar] [CrossRef] [PubMed]
- Gray, L.R.; Tompkins, S.C.; Taylor, E.B. Regulation of pyruvate metabolism and human disease. Cell. Mol. Life Sci. 2014, 71, 2577–2604. [Google Scholar] [CrossRef] [PubMed]
- Zeczycki, T.N.; St.Maurice, M.; Attwood, P.V. Inhibitors of Pyruvate Carboxylase. Open Enzym. Inhib. J. 2010, 3, 8–26. [Google Scholar] [CrossRef]
- Rafikova, O.; Srivastava, A.; Desai, A.A.; Rafikov, R.; Tofovic, S.P. Recurrent inhibition of mitochondrial complex III induces chronic pulmonary vasoconstriction and glycolytic switch in the rat lung. Respir. Res. 2018, 19, 69. [Google Scholar] [CrossRef]
- Rafikova, O.; Rafikov, R.; Meadows, M.L.; Kangath, A.; Jonigk, D.; Black, S.M. The Sexual Dimorphism Associated with Pulmonary Hypertension Corresponds to a Fibrotic Phenotype. Pulm. Circ. 2015, 5, 184–197. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Rafikova, O.; Rafikov, R.; Kangath, A.; Qu, N.; Aggarwal, S.; Sharma, S.; Desai, J.; Fields, T.; Ludewig, B.; Yuan, J.X.-Y.; et al. Redox regulation of epidermal growth factor receptor signaling during the development of pulmonary hypertension. Free Radic. Biol. Med. 2016, 95, 96–111. [Google Scholar] [CrossRef] [PubMed]
- Rivero-Gutiérrez, B.; Anzola, A.; Martínez-Augustin, O.; De Medina, F.S. Stain-free detection as loading control alternative to Ponceau and housekeeping protein immunodetection in Western blotting. Anal. Biochem. 2014, 467, 1–3. [Google Scholar]
- Ryan, J.J.; Archer, S.L. The right ventricle in pulmonary arterial hypertension: Disorders of metabolism, angiogenesis and adrenergic signaling in right ventricular failure. Circ. Res. 2014, 115, 176–188. [Google Scholar] [CrossRef]
- Cohen, G.; Raupachova, J.; Hörl, W.H. The uraemic toxin phenylacetic acid contributes to inflammation by priming polymorphonuclear leucocytes. Nephrol. Dial. Transplant. 2013. [Google Scholar] [CrossRef]
- Tang, H.; Chen, J.; Fraidenburg, D.R.; Song, S.; Sysol, J.R.; Drennan, A.R.; Offermanns, S.; Ye, R.D.; Bonini, M.G.; Minshall, R.D.; et al. Deficiency of Akt1, but not Akt2, attenuates the development of pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L208–L220. [Google Scholar] [CrossRef]
- Dong, G.; Mao, Q.; Xia, W.; Xu, Y.; Wang, J.; Xu, L.; Jiang, F. PKM2 and cancer: The function of PKM2 beyond glycolysis. Oncol. Lett. 2016, 11, 1980–1986. [Google Scholar] [CrossRef]
- Gharia, B.; Seegobin, K.; Mahida, H.; Shaikh, M.; Matthews Hew, T.; Pham, D. Fatal Type B Lactic Acidosis Associated With Metastatic Colorectal Cancer: A Case Report With Review of Literature, Pathogenesis, and Treatment. J. Investig. Med. High Impact Case Rep. 2018, 6. [Google Scholar] [CrossRef]
- Yamasaki, H.; Tada, H.; Kawano, S.; Aonuma, K. Reversible Pulmonary Hypertension, Lactic Acidosis, and Rapidly Evolving Multiple Organ Failure as Manifestations of Shoshin Beriberi. Circ. J. 2010, 74, 1983–1985. [Google Scholar] [CrossRef]
- Paulin, R.; Michelakis, E.D. The Metabolic Theory of Pulmonary Arterial Hypertension. Circ. Res. 2014, 115, 148–164. [Google Scholar] [CrossRef]
- Chan, S.Y.; Rubin, L.J. Metabolic dysfunction in pulmonary hypertension: From basic science to clinical practice. Eur. Respir. Rev. 2017, 26, 170094. [Google Scholar] [CrossRef] [PubMed]
- Bertero, T.; Oldham, W.M.; Cottrill, K.A.; Pisano, S.; Vanderpool, R.R.; Yu, Q.; Zhao, J.; Tai, Y.; Tang, Y.; Zhang, Y.-Y.; et al. Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J. Clin. Investig. 2016, 126, 3313–3335. [Google Scholar] [CrossRef] [PubMed]
- Sales Gil, R.; Vagnarelli, P. Ki-67: More Hidden behind a “Classic Proliferation Marker”. Trends Biochem. Sci. 2018, 43, 747–748. [Google Scholar] [CrossRef]
- Liu, P.; Begley, M.; Michowski, W.; Inuzuka, H.; Ginzberg, M.; Gao, D.; Tsou, P.; Gan, W.; Papa, A.; Kim, B.M.; et al. Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature 2014, 508, 541–545. [Google Scholar] [CrossRef] [PubMed]
- Hung, Y.P.; Teragawa, C.; Kosaisawe, N.; Gillies, T.E.; Pargett, M.; Minguet, M.; Distor, K.; Rocha-Gregg, B.L.; Coloff, J.L.; Keibler, M.A.; et al. Akt regulation of glycolysis mediates bioenergetic stability in epithelial cells. eLife 2017, 6, e27293. [Google Scholar] [CrossRef]
- Wieman, H.L.; Wofford, J.A.; Rathmell, J.C. Cytokine Stimulation Promotes Glucose Uptake via Phosphatidylinositol-3 Kinase/Akt Regulation of Glut1 Activity and Trafficking. Mol. Biol. Cell 2007, 18, 1437–1446. [Google Scholar] [CrossRef]
- Cottrill, K.A.; Chan, S.Y. Metabolic dysfunction in pulmonary hypertension: The expanding relevance of the Warburg effect. Eur. J. Clin. Investig. 2013, 43, 855–865. [Google Scholar] [CrossRef]
- Beg, M.; Abdullah, N.; Thowfeik, F.S.; Altorki, N.K.; McGraw, T.E. Distinct Akt phosphorylation states are required for insulin regulated Glut4 and Glut1-mediated glucose uptake. eLife 2017, 6, e26896. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Deberardinis, R.J. Leading Edge Review Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef]
- Kumashiro, N.; Beddow, S.A.; Vatner, D.F.; Majumdar, S.K.; Cantley, J.L.; Guebre-Egziabher, F.; Fat, I.; Guigni, B.; Jurczak, M.J.; Birkenfeld, A.L.; et al. Targeting Pyruvate Carboxylase Reduces Gluconeogenesis and Adiposity and Improves Insulin Resistance. Diabetes 2013, 62, 2183–2194. [Google Scholar] [CrossRef]
- Zhang, W.-H.; Qiu, M.-H.; Wang, X.-J.; Sun, K.; Zheng, Y.; Jing, Z.-C. Up-regulation of hexokinase1 in the right ventricle of monocrotaline induced pulmonary hypertension. Respir. Res. 2014, 15, 119. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wolf, A.; Agnihotri, S.; Micallef, J.; Mukherjee, J.; Sabha, N.; Cairns, R.; Hawkins, C.; Guha, A. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J. Exp. Med. 2011, 208, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Loberg, R.D.; Vesely, E.; Brosius, F.C. Enhanced glycogen synthase kinase-3beta activity mediates hypoxia-induced apoptosis of vascular smooth muscle cells and is prevented by glucose transport and metabolism. J. Biol. Chem. 2002, 277, 41667–41673. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-S.; Piao, S.; Lee, E.; Yoon, B.-Y.; Moon, H.R.; Lee, J.; Jung, Y.; Ha, N.-C. Development of Akt-activated GSK3β inhibitory peptide. Biochem. Biophys. Res. Commun. 2013, 434, 735–739. [Google Scholar] [CrossRef]
- Leopold, J.A.; Walker, J.; Scribner, A.W.; Voetsch, B.; Zhang, Y.-Y.; Loscalzo, A.J.; Stanton, R.C.; Loscalzo, J. Glucose-6-phosphate dehydrogenase modulates vascular endothelial growth factor-mediated angiogenesis. J. Biol. Chem. 2003, 278, 32100–32106. [Google Scholar] [CrossRef]
- Chettimada, S.; Gupte, R.; Rawat, D.; Gebb, S.A.; McMurtry, I.F.; Gupte, S.A. Hypoxia-induced glucose-6-phosphate dehydrogenase overexpression and -activation in pulmonary artery smooth muscle cells: Implication in pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014. [Google Scholar] [CrossRef]
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006, 9, 425–434. [Google Scholar] [CrossRef]
- Kim, J.; Dang, C.V. Multifaceted roles of glycolytic enzymes. Trends Biochem. Sci. 2005, 30, 142–150. [Google Scholar] [CrossRef]
- Kim, J.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valuparampil Varghese, M.; James, J.; Eccles, C.A.; Niihori, M.; Rafikova, O.; Rafikov, R. Inhibition of Anaplerosis Attenuated Vascular Proliferation in Pulmonary Arterial Hypertension. J. Clin. Med. 2020, 9, 443. https://doi.org/10.3390/jcm9020443
Valuparampil Varghese M, James J, Eccles CA, Niihori M, Rafikova O, Rafikov R. Inhibition of Anaplerosis Attenuated Vascular Proliferation in Pulmonary Arterial Hypertension. Journal of Clinical Medicine. 2020; 9(2):443. https://doi.org/10.3390/jcm9020443
Chicago/Turabian StyleValuparampil Varghese, Mathews, Joel James, Cody A Eccles, Maki Niihori, Olga Rafikova, and Ruslan Rafikov. 2020. "Inhibition of Anaplerosis Attenuated Vascular Proliferation in Pulmonary Arterial Hypertension" Journal of Clinical Medicine 9, no. 2: 443. https://doi.org/10.3390/jcm9020443
APA StyleValuparampil Varghese, M., James, J., Eccles, C. A., Niihori, M., Rafikova, O., & Rafikov, R. (2020). Inhibition of Anaplerosis Attenuated Vascular Proliferation in Pulmonary Arterial Hypertension. Journal of Clinical Medicine, 9(2), 443. https://doi.org/10.3390/jcm9020443