Is the Arginase Pathway a Novel Therapeutic Avenue for Diabetic Retinopathy?



Abstract
1. Introduction
2. Arginase
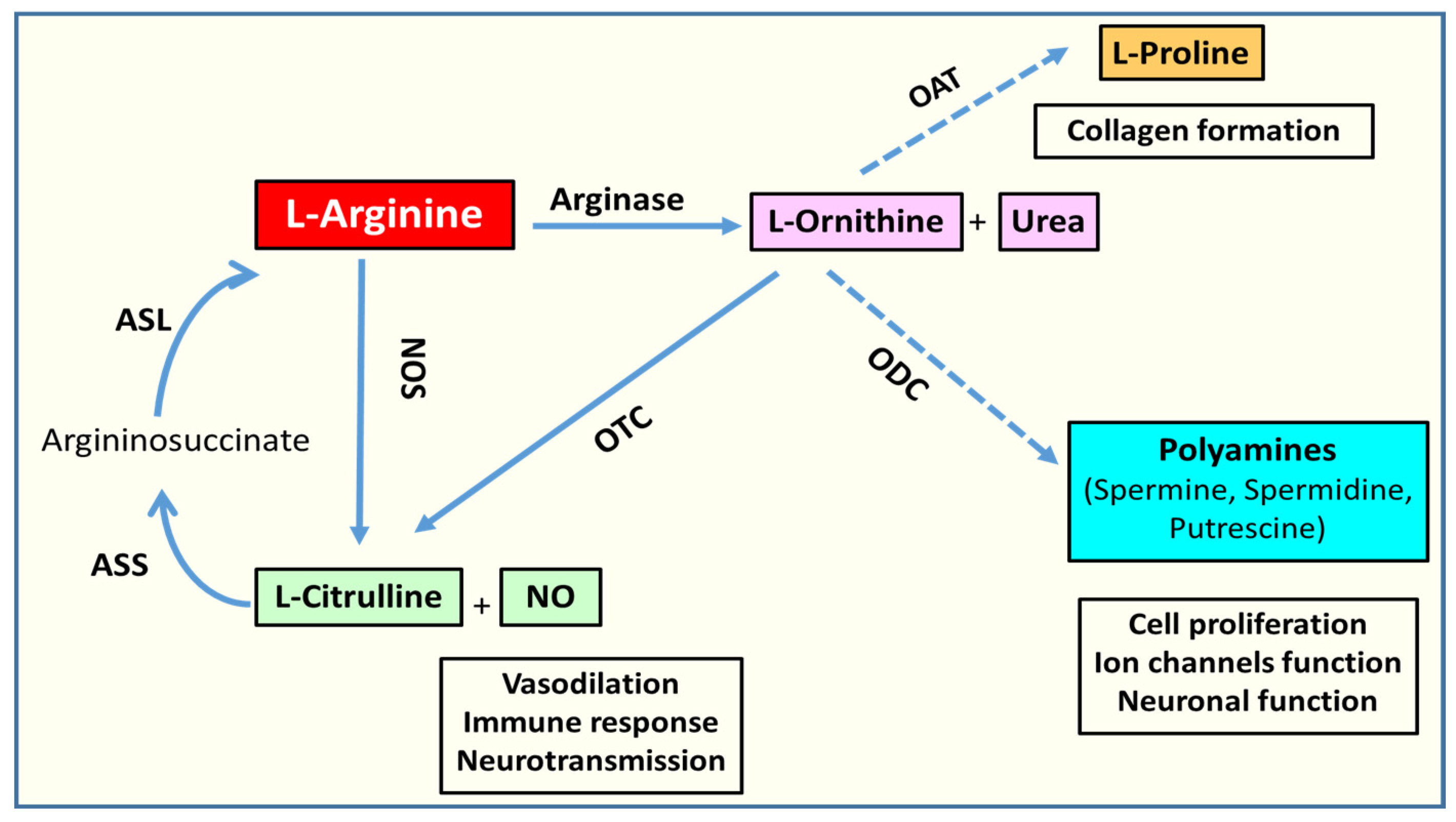
2.1. Arginase Isoforms and L-Arginine Metabolism
2.2. L-Arginine Paradox

2.3. Arginase, NOS Uncoupling, Oxidative/Nitrative Stress, and Inflammation
2.4. Arginase and Polyamine Metabolism
3. Arginase and Diabetic Retinopathy
3.1. Arginase in Diabetic Complications
3.2. Arginase in Other Ischemic Retinopathies
3.3. Arginase in Premature Cellular Senescence
3.4. Arginase and Inflammation
3.5. Polyamine Metabolism in Retinopathy
4. Conclusions and Future Directions
- 1-
- How is arginase regulated in DR?Further work is needed to address this question. Studies to date suggest that both isoforms are increased in retinal cells during DR but it is likely that these alterations occur in different cell types and that they are regulated by different mechanisms.
- 2-
- What are the underlying mechanisms of arginase-induced retinal injury?Studies in different models suggest that multiple mechanisms contribute to the pathology, ranging from eNOS uncoupling in EC due to upregulation of A1 expression to suppression of microglial/macrophage-mediated reparative functions due to upregulation of A2 and suppression of A1 function. Alterations in polyamine metabolism could also be involved as a downstream mechanism of arginase-induced retinal injury. Studies to address these issues are in progress.
- 3-
- What are the contradictory or complimentary roles of the two arginase isoforms?As has been outlined above, genetic studies have shown very different phenotypes with A1 vs. A2 deletion in that the former is lethal soon after birth whereas the latter produces only mild hypertension. Furthermore, studies in a variety of disease models have shown very different effects with A1 vs. A2 deletion in that A1 is prominently involved in promoting EC dysfunction whereas A2 is involved in microglia/macrophage-mediated inflammatory injury. Studies comparing the cell-specific effects of the two isoforms are needed to fully address this issue.
- 4-
- Is arginase deleterious or beneficial in DR? Does arginase administration offer a therapeutic benefit for DR?
Author Contributions
Funding
Patents
Conflicts of Interest
References
- Centers for Disease Control and Prevention. National Diabetes Statistics Report; Centers for Disease Control and Prevention: Atlanta, GA, USA; US Department of Health and Human Services: Washington, DC, USA, 2017.
- Economic costs of diabetes in the U.S. in 2012. Diabetes Care 2013, 36, 1033–1046. [CrossRef]
- Cheung, N.; Mitchell, P.; Wong, T.Y. Diabetic retinopathy. Lancet 2010, 376, 124–136. [Google Scholar] [CrossRef]
- Leasher, J.L.; Bourne, R.R.; Flaxman, S.R.; Jonas, J.B.; Keeffe, J.; Naidoo, K.; Pesudovs, K.; Price, H.; White, R.A.; Wong, T.Y.; et al. Global Estimates on the number of people blind or visually impaired by diabetic retinopathy: A meta-analysis from 1990 to 2010. Diabetes Care 2016, 39, 1643–1649. [Google Scholar] [CrossRef] [PubMed]
- Flaxman, S.R.; Bourne, R.R.A.; Resnikoff, S.; Ackland, P.; Braithwaite, T.; Cicinelli, M.V.; Das, A.; Jonas, J.B.; Keeffe, J.; Kempen, J.H.; et al. Global causes of blindness and distance vision impairment 1990–2020: A systematic review and meta-analysis. Lancet Glob. Health 2017, 5, e1221–e1234. [Google Scholar] [CrossRef]
- Heng, L.Z.; Comyn, O.; Peto, T.; Tadros, C.; Ng, E.; Sivaprasad, S.; Hykin, P.G. Diabetic retinopathy: Pathogenesis, clinical grading, management and future developments. Diabetes Med. 2013, 30, 640–650. [Google Scholar] [CrossRef]
- Di Leo, M.A.; Caputo, S.; Falsini, B.; Porciatti, V.; Greco, A.V.; Ghirlanda, G. Presence and further development of retinal dysfunction after 3-year follow up in IDDM patients without angiographically documented vasculopathy. Diabetologia 1994, 37, 911–916. [Google Scholar] [CrossRef] [PubMed]
- Ewing, F.M.; Deary, I.J.; Strachan, M.W.; Frier, B.M. Seeing beyond retinopathy in diabetes: Electrophysiological and psychophysical abnormalities and alterations in vision. Endocr. Rev. 1998, 19, 462–476. [Google Scholar] [CrossRef] [PubMed]
- Simo, R.; Stitt, A.W.; Gardner, T.W. Neurodegeneration in diabetic retinopathy: Does it really matter? Diabetologia 2018, 61, 1902–1912. [Google Scholar] [CrossRef]
- Mayo Clinic Patient Care and Health Information. Diabetic Retinopathy. Available online: https://www.mayoclinic.org/diseases-conditions/diabetic-retinopathy/symptoms-causes/syc-20371611 (accessed on 25 November 2019).
- Zhao, Y.; Singh, R.P. The role of anti-vascular endothelial growth factor (anti-VEGF) in the management of proliferative diabetic retinopathy. Drugs Context 2018, 7, 212532. [Google Scholar] [CrossRef]
- Touzani, F.; Geers, C.; Pozdzik, A. Intravitreal injection of anti-vegf antibody induces glomerular endothelial cells injury. Case Rep. Nephrol. 2019, 2019, 2919080. [Google Scholar] [CrossRef][Green Version]
- Porta, M.; Striglia, E. Intravitreal anti-VEGF agents and cardiovascular risk. Intern. Emerg. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Duh, E.J.; Sun, J.K.; Stitt, A.W. Diabetic retinopathy: Current understanding, mechanisms, and treatment strategies. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, P.E.; Freeman, S.C.; Janot, A.C. Novel stem cell and gene therapy in diabetic retinopathy, age related macular degeneration, and retinitis pigmentosa. Int. J. Retina Vitr. 2019, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Calderon, G.D.; Juarez, O.H.; Hernandez, G.E.; Punzo, S.M.; De la Cruz, Z.D. Oxidative stress and diabetic retinopathy: Development and treatment. Eye Lond 2017, 31, 1122–1130. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Kern, T.S. Inflammation in diabetic retinopathy. Prog. Retin. Eye Res. 2011, 30, 343–358. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Kern, T.S.; Song, B.; Stuebe, C. Mechanistic insights into pathological changes in the diabetic Retina: Implications for targeting diabetic retinopathy. Am. J. Pathol. 2017, 187, 9–19. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Mishra, M. Oxidative stress, mitochondrial damage and diabetic retinopathy. Biochim. Biophys. Acta 2015, 1852, 2474–2483. [Google Scholar] [CrossRef]
- Madsen-Bouterse, S.A.; Kowluru, R.A. Oxidative stress and diabetic retinopathy: Pathophysiological mechanisms and treatment perspectives. Rev. Endocr. Metab. Disord. 2008, 9, 315–327. [Google Scholar] [CrossRef]
- Semeraro, F.; Morescalchi, F.; Cancarini, A.; Russo, A.; Rezzola, S.; Costagliola, C. Diabetic retinopathy, a vascular and inflammatory disease: Therapeutic implications. Diabetes Metab. 2019, 45, 517–527. [Google Scholar] [CrossRef]
- Guerrero-Romero, F.; Rodríguez-Morán, M. Complementary therapies for diabetes: The case for chromium, magnesium, and antioxidants. Arch. Med. Res. 2005, 36, 250–257. [Google Scholar] [CrossRef]
- Vincent, A.M.; Russell, J.W.; Low, P.; Feldman, E.L. Oxidative stress in the pathogenesis of diabetic neuropathy. Endocr. Rev. 2004, 25, 612–628. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.M.; Mohammad, G.; Zhong, Q.; Kowluru, R.A. Diabetic retinopathy, superoxide damage and antioxidants. Curr. Pharm. Biotechnol. 2011, 12, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Kowluru, A.; Mishra, M.; Kumar, B. Oxidative stress and epigenetic modifications in the pathogenesis of diabetic retinopathy. Prog. Retin. Eye Res. 2015, 48, 40–61. [Google Scholar] [CrossRef] [PubMed]
- Durante, W.; Johnson, F.K.; Johnson, R.A. Arginase: A critical regulator of nitric oxide synthesis and vascular function. Clin. Exp. Pharm. Physiol. 2007, 34, 906–911. [Google Scholar] [CrossRef] [PubMed]
- Pernow, J.; Jung, C. Arginase as a potential target in the treatment of cardiovascular disease: Reversal of arginine steal? Cardiovasc. Res. 2013, 98, 334–343. [Google Scholar] [CrossRef]
- Kovamees, O.; Shemyakin, A.; Checa, A.; Wheelock, C.E.; Lundberg, J.O.; Ostenson, C.G.; Pernow, J. Arginase inhibition improves microvascular endothelial function in patients with type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2016, 101, 3952–3958. [Google Scholar] [CrossRef]
- Pernow, J.; Jung, C. The emerging role of arginase in endothelial dysfunction in diabetes. Curr. Vasc. Pharmacol. 2016, 14, 155–162. [Google Scholar] [CrossRef]
- Caldwell, R.W.; Rodriguez, P.C.; Toque, H.A.; Narayanan, S.P.; Caldwell, R.B. Arginase: A multifaceted enzyme important in health and disease. Physiol. Rev. 2018, 98, 641–665. [Google Scholar] [CrossRef]
- Paris, L.P.; Johnson, C.H.; Aguilar, E.; Usui, Y.; Cho, K.; Hoang, L.T.; Feitelberg, D.; Benton, H.P.; Westenskow, P.D.; Kurihara, T.; et al. Global metabolomics reveals metabolic dysregulation in ischemic retinopathy. Metabolomics 2016, 12, 15. [Google Scholar] [CrossRef]
- Sumarriva, K.; Uppal, K.; Ma, C.; Herren, D.J.; Wang, Y.; Chocron, I.M.; Warden, C.; Mitchell, S.L.; Burgess, L.G.; Goodale, M.P.; et al. Arginine and carnitine metabolites are altered in diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2019, 60, 3119–3126. [Google Scholar] [CrossRef]
- Zhu, X.R.; Yang, F.Y.; Lu, J.; Zhang, H.R.; Sun, R.; Zhou, J.B.; Yang, J.K. Plasma metabolomic profiling of proliferative diabetic retinopathy. Nutr. Metab. 2019, 16, 37. [Google Scholar] [CrossRef] [PubMed]
- Ash, D.E.; Cox, J.D.; Christianson, D.W. Arginase: A binuclear manganese metalloenzyme. Met. Ions Biol. Syst. 2000, 37, 407–428. [Google Scholar] [PubMed]
- Morris, S.M., Jr. Regulation of enzymes of the urea cycle and arginine metabolism. Annu. Rev. Nutr. 2002, 22, 87–105. [Google Scholar] [CrossRef] [PubMed]
- Miyanaka, K.; Gotoh, T.; Nagasaki, A.; Takeya, M.; Ozaki, M.; Iwase, K.; Takiguchi, M.; Iyama, K.I.; Tomita, K.; Mori, M. Immunohistochemical localization of arginase II and other enzymes of arginine metabolism in rat kidney and liver. Histochem. J. 1998, 30, 741–751. [Google Scholar] [CrossRef]
- Patel, C.; Rojas, M.; Narayanan, S.P.; Zhang, W.; Xu, Z.; Lemtalsi, T.; Jittiporn, K.; Caldwell, R.W.; Caldwell, R.B. Arginase as a mediator of diabetic retinopathy. Front. Immunol. 2013, 4, 173. [Google Scholar] [CrossRef]
- Narayanan, S.P.; Rojas, M.; Suwanpradid, J.; Toque, H.A.; Caldwell, R.W.; Caldwell, R.B. Arginase in retinopathy. Prog. Retin. Eye Res. 2013, 36, 260–280. [Google Scholar] [CrossRef]
- Crombez, E.A.; Cederbaum, S.D. Hyperargininemia due to liver arginase deficiency. Mol. Genet. Metab. 2005, 84, 243–251. [Google Scholar] [CrossRef]
- Iyer, R.K.; Yoo, P.K.; Kern, R.M.; Rozengurt, N.; Tsoa, R.; O’Brien, W.E.; Yu, H.; Grody, W.W.; Cederbaum, S.D. Mouse model for human arginase deficiency. Mol. Cell Biol. 2002, 22, 4491–4498. [Google Scholar] [CrossRef]
- Shi, O.; Morris, S.M., Jr.; Zoghbi, H.; Porter, C.W.; O’Brien, W.E. Generation of a mouse model for arginase II deficiency by targeted disruption of the arginase II gene. Mol. Cell Biol. 2001, 21, 811–813. [Google Scholar] [CrossRef]
- Huynh, N.N.; Andrews, K.L.; Head, G.A.; Khong, S.M.; Mayorov, D.N.; Murphy, A.J.; Lambert, G.; Kiriazis, H.; Xu, Q.; Du, X.J.; et al. Arginase II knockout mouse displays a hypertensive phenotype despite a decreased vasoconstrictory profile. Hypertension 2009, 54, 294–301. [Google Scholar] [CrossRef]
- Morris, S.M., Jr. Arginine metabolism revisited. J. Nutr. 2016, 146, 2579s–2586s. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, R.B.; Toque, H.A.; Narayanan, S.P.; Caldwell, R.W. Arginase: An old enzyme with new tricks. Trends Pharmacol. Sci. 2015, 36, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Morris, S.M., Jr. Arginine metabolism: Nitric oxide and beyond. Biochem. J. 1998, 336, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Griffith, O.W.; Stuehr, D.J. Nitric oxide synthases: Properties and catalytic mechanism. Annu. Rev. Physiol. 1995, 57, 707–736. [Google Scholar] [CrossRef] [PubMed]
- Bode-Boger, S.M.; Boger, R.H.; Creutzig, A.; Tsikas, D.; Gutzki, F.M.; Alexander, K.; Frolich, J.C. L-arginine infusion decreases peripheral arterial resistance and inhibits platelet aggregation in healthy subjects. Clin. Sci. 1994, 87, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Gold, M.E.; Bush, P.A.; Ignarro, L.J. Depletion of arterial L-arginine causes reversible tolerance to endothelium-dependent relaxation. Biochem. Biophys. Res. Commun. 1989, 164, 714–721. [Google Scholar] [CrossRef]
- Elms, S.; Chen, F.; Wang, Y.; Qian, J.; Askari, B.; Yu, Y.; Pandey, D.; Iddings, J.; Caldwell, R.B.; Fulton, D.J. Insights into the arginine paradox: Evidence against the importance of subcellular location of arginase and eNOS. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H651–H666. [Google Scholar] [CrossRef]
- Boger, R.H.; Bode-Boger, S.M. The clinical pharmacology of L-arginine. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 79–99. [Google Scholar] [CrossRef]
- Dioguardi, F.S. To give or not to give? Lessons from the arginine paradox. Lifestyle Genom. 2011, 4, 90–98. [Google Scholar] [CrossRef]
- Martens, C.R.; Kuczmarski, J.M.; Lennon-Edwards, S.; Edwards, D.G. Impaired L-arginine uptake but not arginase contributes to endothelial dysfunction in rats with chronic kidney disease. J. Cardiovasc. Pharmacol. 2014, 63, 40–48. [Google Scholar] [CrossRef]
- Hatzoglou, M.; Fernandez, J.; Yaman, I.; Closs, E. Regulation of cationic amino acid transport: The story of the CAT-1 transporter. Annu. Rev. Nutr. 2004, 24, 377–399. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.M.; Harada, R.; Nair, N.; Balasubramanian, N.; Cooke, J.P. L-arginine supplementation in peripheral arterial disease: No benefit and possible harm. Circulation 2007, 116, 188–195. [Google Scholar] [CrossRef]
- Kuiper, M.A.; Teerlink, T.; Visser, J.J.; Bergmans, P.L.; Scheltens, P.; Wolters, E.C. L-glutamate, L-arginine and L-citrulline levels in cerebrospinal fluid of Parkinson’s disease, multiple system atrophy, and Alzheimer’s disease patients. J. Neural Transm. 2000, 107, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Wiesinger, H. Arginine metabolism and the synthesis of nitric oxide in the nervous system. Prog. Neurobiol. 2001, 64, 365–391. [Google Scholar] [CrossRef]
- Lee, J.; Ryu, H.; Ferrante, R.J.; Morris, S.M., Jr.; Ratan, R.R. Translational control of inducible nitric oxide synthase expression by arginine can explain the arginine paradox. Proc. Natl. Acad. Sci. USA 2003, 100, 4843–4848. [Google Scholar] [CrossRef]
- SMART SERVIER MEDICAL ART. Available online: https://smart.servier.com/ (accessed on 3 February 2020).
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef]
- Calabrese, V.; Mancuso, C.; Calvani, M.; Rizzarelli, E.; Butterfield, D.A.; Stella, A.M.G. Nitric oxide in the central nervous system: Neuroprotection versus neurotoxicity. Nat. Rev. Neurosci. 2007, 8, 766–775. [Google Scholar] [CrossRef]
- Romero, M.J.; Platt, D.H.; Tawfik, H.E.; Labazi, M.; El-Remessy, A.B.; Bartoli, M.; Caldwell, R.B.; Caldwell, R.W. Diabetes-induced coronary vascular dysfunction involves increased arginase activity. Circ. Res. 2008, 102, 95–102. [Google Scholar] [CrossRef]
- White, A.R.; Ryoo, S.; Li, D.; Champion, H.C.; Steppan, J.; Wang, D.; Nyhan, D.; Shoukas, A.A.; Hare, J.M.; Berkowitz, D.E. Knockdown of arginase I restores NO signaling in the vasculature of old rats. Hypertension 2006, 47, 245–251. [Google Scholar] [CrossRef]
- Amendola, R.; Bellini, A.; Cervelli, M.; Degan, P.; Marcocci, L.; Martini, F.; Mariottini, P. Direct oxidative DNA damage, apoptosis and radio sensitivity by spermine oxidase activities in mouse neuroblastoma cells. Biochim. Biophys. Acta 2005, 1755, 15–24. [Google Scholar] [CrossRef]
- Moghe, A.; Ghare, S.; Lamoreau, B.; Mohammad, M.; Barve, S.; McClain, C.; Joshi-Barve, S. Molecular mechanisms of acrolein toxicity: Relevance to human disease. Toxicol. Sci. 2015, 143, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Furuhata, A.; Nakamura, M.; Osawa, T.; Uchida, K. Thiolation of protein-bound carcinogenic aldehyde. An electrophilic acrolein-lysine adduct that covalently binds to thiols. J. Biol. Chem. 2002, 277, 27919–27926. [Google Scholar] [CrossRef] [PubMed]
- Shemyakin, A.; Kovamees, O.; Rafnsson, A.; Bohm, F.; Svenarud, P.; Settergren, M.; Jung, C.; Pernow, J. Arginase inhibition improves endothelial function in patients with coronary artery disease and type 2 diabetes mellitus. Circulation 2012, 126, 2943–2950. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Sun, C.B.; Liu, C.; Fan, Y.; Zhu, H.Y.; Wu, X.W.; Hu, L.; Li, Q.P. Upregulation of arginase activity contributes to intracellular ROS production induced by high glucose in H9c2 cells. Int. J. Clin. Exp. Pathol. 2015, 8, 2728–2736. [Google Scholar]
- Romero, M.J.; Iddings, J.A.; Platt, D.H.; Ali, M.I.; Cederbaum, S.D.; Stepp, D.W.; Caldwell, R.B.; Caldwell, R.W. Diabetes-induced vascular dysfunction involves arginase I. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H159–H166. [Google Scholar] [CrossRef]
- You, H.; Gao, T.; Cooper, T.K.; Morris, S.M., Jr.; Awad, A.S. Arginase inhibition mediates renal tissue protection in diabetic nephropathy by a nitric oxide synthase 3-dependent mechanism. Kidney Int. 2013, 84, 1189–1197. [Google Scholar] [CrossRef]
- Toque, H.A.; Tostes, R.C.; Yao, L.; Xu, Z.; Webb, R.C.; Caldwell, R.B.; Caldwell, R.W. Arginase II deletion increases corpora cavernosa relaxation in diabetic mice. J. Sex. Med. 2011, 8, 722–733. [Google Scholar] [CrossRef]
- Morris, S.M., Jr.; Gao, T.; Cooper, T.K.; Kepka-Lenhart, D.; Awad, A.S. Arginase-2 mediates diabetic renal injury. Diabetes 2011, 60, 3015–3022. [Google Scholar] [CrossRef]
- Bhatta, A.; Yao, L.; Xu, Z.; Toque, H.A.; Chen, J.; Atawia, R.T.; Fouda, A.Y.; Bagi, Z.; Lucas, R.; Caldwell, R.B.; et al. Obesity-induced vascular dysfunction and arterial stiffening requires endothelial cell arginase 1. Cardiovasc. Res. 2017, 113, 1664–1676. [Google Scholar] [CrossRef]
- Atawia, R.T.; Toque, H.A.; Meghil, M.M.; Benson, T.W.; Yiew, N.K.H.; Cutler, C.W.; Weintraub, N.L.; Caldwell, R.B.; Caldwell, R.W. Role of Arginase 2 in Systemic Metabolic Activity and Adipose Tissue Fatty Acid Metabolism in Diet-Induced Obese Mice. Int. J. Mol. Sci. 2019, 20, 1462. [Google Scholar] [CrossRef]
- Beleznai, T.; Feher, A.; Spielvogel, D.; Lansman, S.L.; Bagi, Z. Arginase 1 contributes to diminished coronary arteriolar dilation in patients with diabetes. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H777–H783. [Google Scholar] [CrossRef] [PubMed]
- Elms, S.C.; Toque, H.A.; Rojas, M.; Xu, Z.; Caldwell, R.W.; Caldwell, R.B. The role of arginase I in diabetes-induced retinal vascular dysfunction in mouse and rat models of diabetes. Diabetologia 2013, 56, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Atawia, R.T.; Bunch, K.L.; Fouda, A.Y.; Lemtalsi, T.; Eldahshan, W.S.; Xu, Z.; Saul, A.B.; Elmasry, K.; Al-Shabrawey, M.; Caldwell, R.B.; et al. Role of arginase 2 in retinopathy associated with western diet-induced obesity. J. Clin. Med. 2020, 92, 317. [Google Scholar] [CrossRef] [PubMed]
- Kern, T.S.; Antonetti, D.A.; Smith, L.E.H. Pathophysiology of diabetic retinopathy: Contribution and limitations of laboratory research. Ophthalmic Res. 2019, 62, 191–197. [Google Scholar] [CrossRef]
- Smith, L.E.; Wesolowski, E.; McLellan, A.; Kostyk, S.K.; D’Amato, R.; Sullivan, R.; D’Amore, P.A. Oxygen-induced retinopathy in the mouse. Investig. Ophthalmol. Vis. Sci. 1994, 35, 101–111. [Google Scholar]
- Narayanan, S.P.; Suwanpradid, J.; Saul, A.; Xu, Z.; Still, A.; Caldwell, R.W.; Caldwell, R.B. Arginase 2 deletion reduces neuro-glial injury and improves retinal function in a model of retinopathy of prematurity. PLoS ONE 2011, 6, e22460. [Google Scholar] [CrossRef]
- Suwanpradid, J.; Rojas, M.; Behzadian, M.A.; Caldwell, R.W.; Caldwell, R.B. Arginase 2 deficiency prevents oxidative stress and limits hyperoxia-induced retinal vascular degeneration. PLoS ONE 2014, 9, e110604. [Google Scholar] [CrossRef]
- Wang, L.; Bhatta, A.; Toque, H.A.; Rojas, M.; Yao, L.; Xu, Z.; Patel, C.; Caldwell, R.B.; Caldwell, R.W. Arginase inhibition enhances angiogenesis in endothelial cells exposed to hypoxia. Microvasc. Res. 2015, 98, 1–8. [Google Scholar] [CrossRef]
- Morbidelli, L.; Chang, C.H.; Douglas, J.G.; Granger, H.J.; Ledda, F.; Ziche, M. Nitric oxide mediates mitogenic effect of VEGF on coronary venular endothelium. Am. J. Physiol. 1996, 270, H411–H415. [Google Scholar] [CrossRef]
- Ziche, M.; Morbidelli, L.; Choudhuri, R.; Zhang, H.T.; Donnini, S.; Granger, H.J.; Bicknell, R. Nitric oxide synthase lies downstream from vascular endothelial growth factor-induced but not basic fibroblast growth factor-induced angiogenesis. J. Clin. Investig. 1997, 99, 2625–2634. [Google Scholar] [CrossRef]
- Murohara, T.; Horowitz, J.R.; Silver, M.; Tsurumi, Y.; Chen, D.; Sullivan, A.; Isner, J.M. Vascular endothelial growth factor/vascular permeability factor enhances vascular permeability via nitric oxide and prostacyclin. Circulation 1998, 97, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.M.; Huang, Q.; Yuan, Y.; Granger, H.J. VEGF induces NO-dependent hyperpermeability in coronary venules. Am. J. Physiol. 1996, 271, H2735–H2739. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Chen, Q.; Ye, Z.; Li, X.; Ju, R. VEGFR1 Signaling Regulates IL-4-Mediated Arginase 1 Expression in Macrophages. Curr. Mol. Med. 2017, 17, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Gong, B.; Hatala, D.A.; Kern, T.S. Retinal ischemia and reperfusion causes capillary degeneration: Similarities to diabetes. Investig. Ophthalmol. Vis. Sci. 2007, 48, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Shosha, E.; Xu, Z.; Yokota, H.; Saul, A.; Rojas, M.; Caldwell, R.W.; Caldwell, R.B.; Narayanan, S.P. Arginase 2 promotes neurovascular degeneration during ischemia/reperfusion injury. Cell Death Dis. 2016, 7, e2483. [Google Scholar] [CrossRef]
- Fouda, A.Y.; Xu, Z.; Shosha, E.; Lemtalsi, T.; Chen, J.; Toque, H.A.; Tritz, R.; Cui, X.; Stansfield, B.K.; Huo, Y.; et al. Arginase 1 promotes retinal neurovascular protection from ischemia through suppression of macrophage inflammatory responses. Cell Death Dis. 2018, 9, 1001. [Google Scholar] [CrossRef]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Perez-Mancera, P.A.; Young, A.R.; Narita, M. Inside and out: The activities of senescence in cancer. Nat. Rev. Cancer 2014, 14, 547–558. [Google Scholar] [CrossRef]
- Oubaha, M.; Miloudi, K.; Dejda, A.; Guber, V.; Mawambo, G.; Germain, M.A.; Bourdel, G.; Popovic, N.; Rezende, F.A.; Kaufman, R.J.; et al. Senescence-associated secretory phenotype contributes to pathological angiogenesis in retinopathy. Sci. Transl. Med. 2016, 8, 362ra144. [Google Scholar] [CrossRef]
- Lamoke, F.; Shaw, S.; Yuan, J.; Ananth, S.; Duncan, M.; Martin, P.; Bartoli, M. Increased Oxidative and Nitrative Stress Accelerates Aging of the Retinal Vasculature in the Diabetic Retina. PLoS ONE 2015, 10, e0139664. [Google Scholar] [CrossRef]
- Thounaojam, M.C.; Jadeja, R.N.; Warren, M.; Powell, F.L.; Raju, R.; Gutsaeva, D.; Khurana, S.; Martin, P.M.; Bartoli, M. MicroRNA-34a (miR-34a) mediates retinal endothelial cell premature senescence through mitochondrial dysfunction and loss of antioxidant activities. Antioxidants 2019, 8, 328. [Google Scholar] [CrossRef] [PubMed]
- Shosha, E.; Xu, Z.; Narayanan, S.P.; Lemtalsi, T.; Fouda, A.Y.; Rojas, M.; Xing, J.; Fulton, D.; Caldwell, R.W.; Caldwell, R.B. Mechanisms of diabetes-induced endothelial cell senescence: Role of Arginase 1. Int. J. Mol. Sci. 2018, 19, 1215. [Google Scholar] [CrossRef] [PubMed]
- Rojas, M.; Lemtalsi, T.; Toque, H.A.; Xu, Z.; Fulton, D.; Caldwell, R.W.; Caldwell, R.B. NOX2-induced activation of arginase and diabetes-induced retinal endothelial cell senescence. Antioxidants 2017, 6, 43. [Google Scholar] [CrossRef]
- Koga, T.; Koshiyama, Y.; Gotoh, T.; Yonemura, N.; Hirata, A.; Tanihara, H.; Negi, A.; Mori, M. Coinduction of nitric oxide synthase and arginine metabolic enzymes in endotoxin-induced uveitis rats. Exp. Eye Res. 2002, 75, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Baban, B.; Rojas, M.; Tofigh, S.; Virmani, S.K.; Patel, C.; Behzadian, M.A.; Romero, M.J.; Caldwell, R.W.; Caldwell, R.B. Arginase activity mediates retinal inflammation in endotoxin-induced uveitis. Am. J. Pathol. 2009, 175, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Jablonski, K.A.; Amici, S.A.; Webb, L.M.; Ruiz-Rosado Jde, D.; Popovich, P.G.; Partida-Sanchez, S.; Guerau-de-Arellano, M. Novel markers to delineate murine M1 and M2 macrophages. PLoS ONE 2015, 10, e0145342. [Google Scholar] [CrossRef]
- Yang, Z.; Ming, X.F. Functions of arginase isoforms in macrophage inflammatory responses: Impact on cardiovascular diseases and metabolic disorders. Front. Immunol. 2014, 5, 533. [Google Scholar] [CrossRef]
- Ming, X.F.; Rajapakse, A.G.; Yepuri, G.; Xiong, Y.; Carvas, J.M.; Ruffieux, J.; Scerri, I.; Wu, Z.; Popp, K.; Li, J.; et al. Arginase II promotes macrophage inflammatory responses through mitochondrial reactive oxygen species, contributing to insulin resistance and atherogenesis. J. Am. Heart Assoc. 2012, 1, e000992. [Google Scholar] [CrossRef]
- Xu, H.; Chen, M. Diabetic retinopathy and dysregulated innate immunity. Vis. Res. 2017, 139, 39–46. [Google Scholar] [CrossRef]
- Canataroglu, H.; Varinli, I.; Ozcan, A.A.; Canataroglu, A.; Doran, F.; Varinli, S. Interleukin (IL)-6, interleukin (IL)-8 levels and cellular composition of the vitreous humor in proliferative diabetic retinopathy, proliferative vitreoretinopathy, and traumatic proliferative vitreoretinopathy. Ocul. Immunol. Inflamm. 2005, 13, 375–381. [Google Scholar] [CrossRef]
- Urbancic, M.; Stunf, S.; Milutinovic Zivin, A.; Petrovic, D.; GlobocnikPetrovic, M. Epiretinal membrane inflammatory cell density might reflect the activity of proliferative diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2014, 55, 8576–8582. [Google Scholar] [CrossRef]
- Sanchez-Jimenez, F.; Medina, M.A.; Villalobos-Rueda, L.; Urdiales, J.L. Polyamines in mammalian pathophysiology. Cell Mol. Life Sci. 2019, 76, 3987–4008. [Google Scholar] [CrossRef]
- Takano, K.; Ogura, M.; Nakamura, Y.; Yoneda, Y. Neuronal and glial responses to polyamines in the ischemic brain. Curr. Neurovasc. Res. 2005, 2, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Paik, M.J.; Ahn, Y.H.; Lee, P.H.; Kang, H.; Park, C.B.; Choi, S.; Lee, G. Polyamine patterns in the cerebrospinal fluid of patients with Parkinson’s disease and multiple system atrophy. Clin. Chim. Acta 2010, 411, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, S.; Batliwalla, F.; Mocco, J.; Kiss, S.; Huang, J.; Mack, W.; Coon, A.; Eaton, J.W.; Al-Abed, Y.; Gregersen, P.K.; et al. Neuroprotection in cerebral ischemia by neutralization of 3-aminopropanal. Proc. Natl. Acad. Sci. USA 2002, 99, 5579–5584. [Google Scholar] [CrossRef]
- Nicoletti, R.; Venza, I.; Ceci, G.; Visalli, M.; Teti, D.; Reibaldi, A. Vitreous polyamines spermidine, putrescine, and spermine in human proliferative disorders of the retina. Br. J. Ophthalmol. 2003, 87, 1038–1042. [Google Scholar] [CrossRef][Green Version]
- Seiler, N. Oxidation of polyamines and brain injury. Neurochem. Res. 2000, 25, 471–490. [Google Scholar] [CrossRef]
- Pichavaram, P.; Palani, C.D.; Patel, C.; Xu, Z.; Shosha, E.; Fouda, A.Y.; Caldwell, R.B.; Narayanan, S.P. Targeting Polyamine Oxidase to Prevent Excitotoxicity-Induced Retinal Neurodegeneration. Front. Neurosci. 2018, 12, 956. [Google Scholar] [CrossRef]
- Narayanan, S.P.; Shosha, E.; Palani, C.D. Spermine oxidase: A promising therapeutic target for neurodegeneration in diabetic retinopathy. Pharmacol. Res. 2019, 147, 104299. [Google Scholar] [CrossRef]
- Liu, F.; Saul, A.B.; Pichavaram, P.; Xu, Z.; Rudraraju, M.; Somanath, P.R.; Smith, S.B.; Caldwell, R.B.; Narayanan, S.P. Pharmacological inhibition of spermine oxidase reduces neurodegeneration and improves retinal function in diabetic mice. J. Clin. Med. 2020, 9, 340. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, S.P.; Xu, Z.; Putluri, N.; Sreekumar, A.; Lemtalsi, T.; Caldwell, R.W.; Caldwell, R.B. Arginase 2 deficiency reduces hyperoxia-mediated retinal neurodegeneration through the regulation of polyamine metabolism. Cell Death Dis. 2014, 5, e1075. [Google Scholar] [CrossRef]
- Patel, C.; Xu, Z.; Shosha, E.; Xing, J.; Lucas, R.; Caldwell, R.W.; Caldwell, R.B.; Narayanan, S.P. Treatment with polyamine oxidase inhibitor reduces microglial activation and limits vascular injury in ischemic retinopathy. Biochim. Biophys. Acta 2016, 1862, 1628–1639. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Noda, K.; Murata, M.; Yoshida, S.; Saito, W.; Kanda, A.; Ishida, S. Localization of acrolein-lysine adduct in fibrovascular tissues of proliferative diabetic retinopathy. Curr. Eye Res. 2017, 42, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Yong, P.H.; Zong, H.; Medina, R.J.; Limb, G.A.; Uchida, K.; Stitt, A.W.; Curtis, T.M. Evidence supporting a role for N-(3-formyl-3,4-dehydropiperidino) lysine accumulation in Muller glia dysfunction and death in diabetic retinopathy. Mol. Vis. 2010, 16, 2524–2538. [Google Scholar] [PubMed]
- Roy, S.; Kim, D.; Sankaramoorthy, A. Mitochondrial Structural Changes in the Pathogenesis of Diabetic Retinopathy. J. Clin. Med. 2019, 8, 1363. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A. Mitochondrial stability in diabetic retinopathy: Lessons learned from epigenetics. Diabetes 2019, 68, 241–247. [Google Scholar] [CrossRef]
- Xiong, Y.; Yu, Y.; Montani, J.P.; Yang, Z.; Ming, X.F. Arginase-II induces vascular smooth muscle cell senescence and apoptosis through p66Shc and p53 independently of its l-arginine ureahydrolase activity: Implications for atherosclerotic plaque vulnerability. J. Am. Heart Assoc. 2013, 2, e000096. [Google Scholar] [CrossRef]
- Koo, B.-H.; Yi, B.-G.; Jeong, M.-S.; Kwon, S.-H.; Hoe, K.-L.; Kwon, Y.-G.; Won, M.-H.; Kim, Y.-M.; Ryoo, S. Arginase II inhibition prevents interleukin-8 production through regulation of p38 MAPK phosphorylation activated by loss of mitochondrial membrane potential in nLDL-stimulated hAoSMCs. Exp. Mol. Med. 2018, 50, e438. [Google Scholar] [CrossRef]
- Liang, X.; Arullampalam, P.; Yang, Z.; Ming, X.F. Hypoxia enhances endothelial intercellular adhesion molecule 1 protein level through upregulation of arginase type II and mitochondrial oxidative stress. Front. Physiol. 2019, 10, 1003. [Google Scholar] [CrossRef]
- Xu, W.; Ghosh, S.; Comhair, S.A.A.; Asosingh, K.; Janocha, A.J.; Mavrakis, D.A.; Bennett, C.D.; Gruca, L.L.; Graham, B.B.; Queisser, K.A.; et al. Increased mitochondrial arginine metabolism supports bioenergetics in asthma. J. Clin. Investig. 2016, 126, 2465–2481. [Google Scholar] [CrossRef]
- Han, W.H.; Gotzmann, J.; Kuny, S.; Huang, H.; Chan, C.B.; Lemieux, H.; Sauvé, Y. Modifications in retinal mitochondrial respiration precede type 2 diabetes and protracted microvascular retinopathy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3826–3839. [Google Scholar] [CrossRef] [PubMed]
- Joyal, J.S.; Sun, Y.; Gantner, M.L.; Shao, Z.; Evans, L.P.; Saba, N.; Fredrick, T.; Burnim, S.; Kim, J.S.; Patel, G.; et al. Retinal lipid and glucose metabolism dictates angiogenesis through the lipid sensor ffar1. Nat. Med. 2016, 22, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Wang, Z.; Liu, C.H.; Gong, Y.; Cakir, B.; Liegl, R.; Sun, Y.; Meng, S.S.; Burnim, S.B.; Arellano, I.; et al. Fibroblast growth factor 21 protects photoreceptor function in type 1 diabetic mice. Diabetes 2018, 67, 974–985. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Animal Model | Main Findings | References |
|---|---|---|
| STZ Mice and Rats | Heterozygous deletion of A1 or arginase inhibition protected against the diabetes-induced retinal vascular dysfunction. | [75] |
| STZ Mice | A1 expression and activity are increased in 2-month diabetic retinas. The double knockout of A1 (one copy) and A2 (both copies) reduced superoxide production. Arginase inhibition enhanced NO formation and decreased leukocyte adhesion. | [37] |
| STZ Mice | Diabetes-induced activation of NOX2 and NOX2-derived ROS is linked to EC senescence through arginase activation. A1 gene deletion protected against the increased activity of senescence-associated β-galactosidase (SA β-gal) in isolated retinal vessels. The pharmacological inhibition of arginase activity decreased the expression of senescence-associated mediators. | [95,96] |
| STZ Mice | The expression of SMOX is increased in retinas of diabetic mice. The inhibition of SMOX activity improved retinal function and protected against the loss of inner retinal neurons. | [113] |
| HFHS Mice | A2 levels in the retina are increased after 16 weeks of Western diet. A2 deletion protected against the obesity-induced abnormalities in the ERG responses. | [76] |
| OIR Mice | A2 retinal levels are increased during the ischemic phase of OIR. A2 deletion ameliorated OIR-induced neurovascular alterations. | [79] |
| OIR Mice | The SMOX expression is increased during the hyperoxia phase of OIR, which was associated with neurovascular degeneration. A2 deletion prevented increases in SMOX expression/activity. The inhibition of SMOX activity protected against the hyperoxia-induced vascular injury through the inhibition of microglia-mediated endothelial cell injury. | [114,115] |
| I/R Mice | A2 expression was increased within 3 h after I/R. A1 expression and activity were significantly decreased after I/R. A2 deletion protected against I/R-induced neurovascular dysfunction. Conversely, the heterozygous deletion of A1 worsened the neurovascular injury. | [88,89] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shosha, E.; Fouda, A.Y.; Narayanan, S.P.; Caldwell, R.W.; Caldwell, R.B. Is the Arginase Pathway a Novel Therapeutic Avenue for Diabetic Retinopathy? J. Clin. Med. 2020, 9, 425. https://doi.org/10.3390/jcm9020425
Shosha E, Fouda AY, Narayanan SP, Caldwell RW, Caldwell RB. Is the Arginase Pathway a Novel Therapeutic Avenue for Diabetic Retinopathy? Journal of Clinical Medicine. 2020; 9(2):425. https://doi.org/10.3390/jcm9020425
Chicago/Turabian StyleShosha, Esraa, Abdelrahman Y. Fouda, S. Priya Narayanan, R. William Caldwell, and Ruth B. Caldwell. 2020. "Is the Arginase Pathway a Novel Therapeutic Avenue for Diabetic Retinopathy?" Journal of Clinical Medicine 9, no. 2: 425. https://doi.org/10.3390/jcm9020425
APA StyleShosha, E., Fouda, A. Y., Narayanan, S. P., Caldwell, R. W., & Caldwell, R. B. (2020). Is the Arginase Pathway a Novel Therapeutic Avenue for Diabetic Retinopathy? Journal of Clinical Medicine, 9(2), 425. https://doi.org/10.3390/jcm9020425