Peripheral Blood Mononuclear Cells and Platelets Mitochondrial Dysfunction, Oxidative Stress, and Circulating mtDNA in Cardiovascular Diseases

 , ,
, ,  , and
, and
Abstract
1. Introduction
2. Is Mitochondrial Function Accessible in all Circulating Cells in the Blood?
2.1. Classification of Circulating Cells
2.2. Isolation and Mitochondrial Respiratory Chain Activities’ Determination in PBMCs and Platelets
3. Mitochondrial Respiratory Chain Complex Activities of PBMCs and Platelets in Patients with Cardiovascular Diseases
3.1. PBMCs Mitochondrial Respiratory Chain Activity in Cardiovascular Diseases
3.2. Platelets’ Mitochondrial Respiratory Chain Activity in Cardiovascular Diseases
4. Mitochondrial ROS Production and Antioxidant Defense of PBMCs and Platelets in Patients with Cardiovascular Diseases
4.1. Measurements of ROS in Circulating Cells
4.2. Mitochondrial ROS in PBMCs in CVDs
4.2.1. Mitochondrial ROS in PBMCs in Heart Failure
4.2.2. Mitochondrial ROS in Arterial Hypertension, Coronary Artery Disease, and Stroke
4.3. Mitochondrial ROS in Circulating Platelets in CVDs
5. Circulating Mitochondrial DNA (mtDNA) Originating from PBMCs and Platelets in Patients with Cardiovascular Diseases
5.1. Circulating Mitochondrial DNA (mtDNA) Originating from PBMCs in Patients with Cardiovascular Diseases
5.2. Circulating Mitochondrial DNA (mtDNA) Originating from Platelets in Patients with Cardiovascular Diseases
6. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Santulli, G. Epidemiology of Cardiovascular Disease in the 21st Century: Updated Numbers and Updated Facts. J. Cardiovasc. Dis. 2013, 2, 1–2. [Google Scholar]
- Heil, B.; Tang, W.H.W. Biomarkers: Their potential in the diagnosis and treatment of heart failure. Clevel. Clin. J. Med. 2015, 82, S28–S35. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef] [PubMed]
- Townsend, N.; Wilson, L.; Bhatnagar, P.; Wickramasinghe, K.; Rayner, M.; Nichols, M. Cardiovascular disease in Europe: Epidemiological update 2016. Eur. Heart. J. 2016, 37, 3232–3245. [Google Scholar] [CrossRef]
- World Health Organization. World health statistics overview 2019: Monitoring health for the SDGs, Sustainable Development Goals; World Health Organization: Geneva, Switzerland, 2019; Available online: https://apps.who.int/iris/handle/10665/311696 (accessed on 22 January 2020).
- Yin, W.H.; Chen, J.W.; Lin, S.J. Prognostic value of combining echocardiography and natriuretic peptide levels in patients with heart failure. Curr. Heart. Fail. Rep. 2012, 9, 148–153. [Google Scholar] [CrossRef]
- Taylor, C.; Hobbs, R. Diagnosing Heart Failure—Experience and ‘Best Pathways’. Available online: https://www.ecrjournal.com/articles/diagnosing-hf-experience (accessed on 16 August 2019).
- Bárány, T.; Simon, A.; Szabó, G.; Benkő, R.; Mezei, Z.; Molnár, L.; Becker, D.; Merkely, B.; Zima, E.; Horváth, E.M. Oxidative Stress-Related Parthanatos of Circulating Mononuclear Leukocytes in Heart Failure. Oxid. Med. Cell. Longev. 2017, 2017, 1249614. [Google Scholar] [CrossRef]
- Lugnier, C.; Meyer, A.; Charloux, A.; Andrès, E.; Gény, B.; Talha, S. The Endocrine Function of the Heart: Physiology and Involvements of Natriuretic Peptides and Cyclic Nucleotide Phosphodiesterases in Heart Failure. J. Clin. Med. 2019, 8, 1746. [Google Scholar] [CrossRef]
- Rose, S.; Carvalho, E.; Diaz, E.C.; Cotter, M.; Bennuri, S.C.; Azhar, G.; Frye, R.E.; Adams, S.H.; Børsheim, E. A comparative study of mitochondrial respiration in circulating blood cells and skeletal muscle fibers in women. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E503–E512. [Google Scholar] [CrossRef]
- Petrus, A.T.; Lighezan, D.L.; Danila, M.D.; Duicu, O.M.; Sturza, A.; Muntean, D.M.; Ionita, I. Assessment of platelet respiration as emerging biomarker of disease. Physiol. Res. 2019, 68, 347–363. [Google Scholar] [CrossRef]
- Ost, M.; Doerrier, C.; Gama-Perez, P.; Moreno-Gomez, S. Analysis of mitochondrial respiratory function in tissue biopsies and blood cells. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 336–342. [Google Scholar] [CrossRef]
- Kramer, P.A.; Ravi, S.; Chacko, B.; Johnson, M.S.; Darley-Usmar, V.M. A review of the mitochondrial and glycolytic metabolism in human platelets and leukocytes: Implications for their use as bioenergetic biomarkers. Redox Biol. 2014, 2, 206–210. [Google Scholar] [CrossRef]
- Maestraggi, Q.; Lebas, B.; Clere-Jehl, R.; Ludes, P.O.; Chamaraux-Tran, T.N.; Schneider, F.; Diemunsch, P.; Geny, B.; Pottecher, J. Skeletal Muscle and Lymphocyte Mitochondrial Dysfunctions in Septic Shock Trigger ICU-Acquired Weakness and Sepsis-Induced Immunoparalysis. BioMed. Res. Int. 2017, 2017, 7897325. [Google Scholar] [CrossRef]
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Invest. 2018, 128, 3716–3726. [Google Scholar] [CrossRef]
- Pizzimenti, M.; Riou, M.; Charles, A.L.; Talha, S.; Meyer, A.; Andres, E.; Chakfé, N.; Lejay, A.; Geny, B. The Rise of Mitochondria in Peripheral Arterial Disease Physiopathology: Experimental and Clinical Data. J. Clin. Med. 2019, 8, 2125. [Google Scholar] [CrossRef]
- Rosca, M.G.; Hoppel, C.L. Mitochondria in heart failure. Cardiovasc. Res. 2010, 88, 40–50. [Google Scholar] [CrossRef]
- Brown, D.A.; Perry, J.B.; Allen, M.E.; Sabbah, H.N.; Stauffer, B.L.; Shaikh, S.R.; Cleland, J.G.; Colucci, W.S.; Butler, J.; Voors, A.A.; et al. Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017, 14, 238–250. [Google Scholar] [CrossRef]
- Weiss, S.L.; Selak, M.A.; Tuluc, F.; Perales Villarroel, J.; Nadkarni, V.M.; Deutschman, C.S.; Becker, L.B. Mitochondrial Dysfunction in Peripheral Blood Mononuclear Cells in Pediatric Septic Shock. Pediatr. Crit. Care Med. 2015, 16, e4–e12. [Google Scholar] [CrossRef]
- Muntean, D.M.; Sturza, A.; Dănilă, M.D.; Borza, C.; Duicu, O.M.; Mornoș, C. The Role of Mitochondrial Reactive Oxygen Species in Cardiovascular Injury and Protective Strategies. Oxid. Med. Cell. Longev. 2016, 2016, 8254942. [Google Scholar] [CrossRef]
- Martin-Ventura, J.L.; Rodrigues-Diez, R.; Martinez-Lopez, D.; Salaices, M.; Blanco-Colio, L.M.; Briones, A.M. Oxidative Stress in Human Atherothrombosis: Sources, Markers and Therapeutic Targets. Int. J. Mol. Sci. 2017, 18, 2315. [Google Scholar] [CrossRef]
- Shirakawa, R.; Yokota, T.; Nakajima, T.; Takada, S.; Yamane, M.; Furihata, T.; Maekawa, S.; Nambu, H.; Katayama, T.; Fukushima, A.; et al. Mitochondrial reactive oxygen species generation in blood cells is associated with disease severity and exercise intolerance in heart failure patients. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef]
- Maynard, S.; Keijzers, G.; Gram, M.; Desler, C.; Bendix, L.; Budtz-Jørgensen, E.; Molbo, D.; Croteau, D.L.; Osler, M.; Stevnsner, T.; et al. Relationships between human vitality and mitochondrial respiratory parameters, reactive oxygen species production and dNTP levels in peripheral blood mononuclear cells. Aging 2013, 5, 850–864. [Google Scholar] [CrossRef] [PubMed]
- Ederlé, C.; Charles, A.L.; Khayath, N.; Poirot, A.; Meyer, A.; Clere-Jehl, R.; Andres, E.; De Blay, F.; Geny, B. Mitochondrial Function in Peripheral Blood Mononuclear Cells (PBMC) is Enhanced, Together with Increased Reactive Oxygen Species, in Severe Asthmatic Patients in Exacerbation. J. Clin. Med. 2019, 8, 1613. [Google Scholar] [CrossRef] [PubMed]
- Stier, A.; Bize, P.; Schull, Q.; Zoll, J.; Singh, F.; Geny, B.; Gros, F.; Royer, C.; Massemin, S.; Criscuolo, F. Avian erythrocytes have functional mitochondria, opening novel perspectives for birds as animal models in the study of ageing. Front. Zool. 2013, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Melchinger, H.; Jain, K.; Tyagi, T.; Hwa, J. Role of Platelet Mitochondria: Life in a Nucleus-Free Zone. Front. Cardiovasc. Med. 2019, 6, 153. [Google Scholar] [CrossRef] [PubMed]
- Gregg, D.; Goldschmidt-Clermont, P.J. Cardiology patient page. Platelets and cardiovascular disease. Circulation 2003, 108, e88–e90. [Google Scholar] [CrossRef] [PubMed]
- Braganza, A.; Annarapu, G.K.; Shiva, S. Blood-based bioenergetics: An emerging translational and clinical tool. Mol. Aspects Med. 2019, 100835. [Google Scholar] [CrossRef]
- Li, P.; Wang, B.; Sun, F.; Li, Y.; Li, Q.; Lang, H.; Zhao, Z.; Gao, P.; Zhao, Y.; Shang, Q.; et al. Mitochondrial respiratory dysfunctions of blood mononuclear cells link with cardiac disturbance in patients with early-stage heart failure. Sci. Rep. 2015, 5, 10229. [Google Scholar] [CrossRef]
- Spinazzi, M.; Casarin, A.; Pertegato, V.; Salviati, L.; Angelini, C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat. Protoc. 2012, 7, 1235–1246. [Google Scholar] [CrossRef]
- Hsiao, C.P.; Hoppel, C. Analyzing mitochondrial function in human peripheral blood mononuclear cells. Anal. Biochem. 2018, 549, 12–20. [Google Scholar] [CrossRef]
- Horan, M.P.; Pichaud, N.; Ballard, J.W.O. Review: Quantifying mitochondrial dysfunction in complex diseases of aging. J. Gerontol. A Biol. Sci. Med. Sci. 2012, 67, 1022–1035. [Google Scholar] [CrossRef]
- Salabei, J.K.; Gibb, A.A.; Hill, B.G. Comprehensive measurement of respiratory activity in permeabilized cells using extracellular flux analysis. Nat. Protoc. 2014, 9, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Zharikov, S.; Shiva, S. Platelet mitochondrial function: From regulation of thrombosis to biomarker of disease. Biochem. Soc. Trans. 2013, 41, 118–123. [Google Scholar] [CrossRef]
- Raffa, S.; Chin, X.L.D.; Stanzione, R.; Forte, M.; Bianchi, F.; Cotugno, M.; Marchitti, S.; Micaloni, A.; Gallo, G.; Schirone, L.; et al. The reduction of NDUFC2 expression is associated with mitochondrial impairment in circulating mononuclear cells of patients with acute coronary syndrome. Int. J. Cardiol. 2019, 286, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Li, T.; Chen, S.; Yang, D.; Luo, L.; Wang, T.; Han, X.; Bai, L.; Ma, A. Correlations between MTP and ROS Levels of Peripheral Blood Lymphocytes and Readmission in Patients with Chronic Heart Failure. Heart Lung Circ. 2016, 25, 296–302. [Google Scholar] [CrossRef]
- Kong, C.W.; Hsu, T.G.; Lu, F.J.; Chan, W.L.; Tsai, K. Leukocyte mitochondria depolarization and apoptosis in advanced heart failure: Clinical correlations and effect of therapy. J. Am. Coll. Cardiol. 2001, 38, 1693–1700. [Google Scholar] [CrossRef]
- Coluccia, R.; Raffa, S.; Ranieri, D.; Micaloni, A.; Valente, S.; Salerno, G.; Scrofani, C.; Testa, M.; Gallo, G.; Pagannone, E.; et al. Chronic heart failure is characterized by altered mitochondrial function and structure in circulating leucocytes. Oncotarget 2018, 9, 35028–35040. [Google Scholar] [CrossRef][Green Version]
- Akkerman, J.W. Regulation of carbohydrate metabolism in platelets. A review. Thromb. Haemost. 1978, 39, 712–724. [Google Scholar]
- Guppy, M.; Abas, L.; Neylon, C.; Whisson, M.E.; Whitham, S.; Pethick, D.W.; Niu, X. Fuel choices by human platelets in human plasma. Eur. J. Biochem. 1997, 244, 161–167. [Google Scholar] [CrossRef]
- Daniel, J.L.; Molish, I.R.; Holmsen, H. Radioactive labeling of the adenine nucleotide pool of cells as a method to distinguish among intracellular compartments. Studies on human platelets. Biochim. Biophys. Acta 1980, 632, 444–453. [Google Scholar] [CrossRef]
- Verhoeven, A.J.; Mommersteeg, M.E.; Akkerman, J.W. Quantification of energy consumption in platelets during thrombin-induced aggregation and secretion. Tight coupling between platelet responses and the increment in energy consumption. Biochem. J. 1984, 221, 777–787. [Google Scholar] [CrossRef]
- Protti, A.; Fortunato, F.; Artoni, A.; Lecchi, A.; Motta, G.; Mistraletti, G.; Novembrino, C.; Comi, G.P.; Gattinoni, L. Platelet mitochondrial dysfunction in critically ill patients: Comparison between sepsis and cardiogenic shock. Crit. Care 2015, 19, 39. [Google Scholar] [CrossRef] [PubMed]
- Penniall, R. The effects of salicylic acid on the respiratory activity of mitochondria. Biochim. Biophys. Acta 1958, 30, 247–251. [Google Scholar] [CrossRef]
- Nguyen, Q.L.; Wang, Y.; Helbling, N.; Simon, M.A.; Shiva, S. Alterations in platelet bioenergetics in Group 2 PH-HFpEF patients. PLoS ONE 2019, 14, e0220490. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.M.; Sparagna, G.C.; Phillips, E.K.; Miyano, C.A.; Nunley, K.; Chatfield, K.C.; Stauffer, B.L.; Sucharov, C.; Miyamoto, S.D. Reactive Oxygen Species Accumulation and Mitochondrial Dysfunction in Peripheral Blood Mononuclear Cells Are Associated With Heart Failure in Patients With Single Ventricle Congenital Heart Disease. Circulation 2019, 140, 15615. [Google Scholar]
- Mondal, N.K.; Sorensen, E.; Hiivala, N.; Feller, E.; Griffith, B.; Wu, Z.J. Oxidative Stress, DNA Damage and Repair in Heart Failure Patients after Implantation of Continuous Flow Left Ventricular Assist Devices. Int. J. Med. Sci. 2013, 10, 883–893. [Google Scholar] [CrossRef]
- Ijsselmuiden, A.J.; Musters, R.J.; de Ruiter, G.; van Heerebeek, L.; Alderse-Baas, F.; van Schilfgaarde, M.; Leyte, A.; Tangelder, G.J.; Laarman, G.J.; Paulus, W.J.l. Circulating white blood cells and platelets amplify oxidative stress in heart failure. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, 811–820. [Google Scholar] [CrossRef]
- Wenzel, P.; Kossmann, S.; Münzel, T.; Daiber, A. Redox regulation of cardiovascular inflammation—Immunomodulatory function of mitochondrial and Nox-derived reactive oxygen and nitrogen species. Free Radic. Biol. Med. 2017, 109, 48–60. [Google Scholar] [CrossRef]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Rubattu, S.; Forte, M.; Raffa, S. Circulating Leukocytes and Oxidative Stress in Cardiovascular Diseases: A State of the Art. Oxid. Med. Cell. Longev. 2019, 2019, 2650429. [Google Scholar] [CrossRef]
- Forte, M.; Palmerio, S.; Yee, D.; Frati, G.; Sciarretta, S. Functional Role of Nox4 in Autophagy. Adv. Exp. Med. Biol. 2017, 982, 307–326. [Google Scholar]
- Forte, M.; Nocella, C.; De Falco, E.; Palmerio, S.; Schirone, L.; Valenti, V.; Frati, G.; Carnevale, R.; Sciarretta, S. The Pathophysiological Role of NOX2 in Hypertension and Organ Damage. High Blood Press. Cardiovasc. Prev. 2016, 23, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Senoner, T.; Dichtl, W. Oxidative Stress in Cardiovascular Diseases: Still a Therapeutic Target? Nutrients 2019, 11, 2090. [Google Scholar] [CrossRef] [PubMed]
- Marrocco, I.; Altieri, F.; Peluso, I. Measurement and Clinical Significance of Biomarkers of Oxidative Stress in Humans. Oxid. Med. Cell. Longev. 2017, 2017, 32. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zou, M.H. Measurement of Reactive Oxygen Species (ROS) and Mitochondrial ROS in AMPK Knockout Mice Blood Vessels. Methods Mol. Biol. Clifton. N.J. 2018, 1732, 507–517. [Google Scholar]
- Ito, F.; Sono, Y.; Ito, T. Measurement and Clinical Significance of Lipid Peroxidation as a Biomarker of Oxidative Stress: Oxidative Stress in Diabetes, Atherosclerosis, and Chronic Inflammation. Antioxidants 2019, 8, 72. [Google Scholar] [CrossRef]
- Griendling, K.K.; Touyz, R.M.; Zweier, J.L.; Dikalov, S.; Chilian, W.; Chen, Y.R.; Harrison, D.G.; Bhatnagar, A. American Heart Association Council on Basic Cardiovascular Sciences. Measurement of Reactive Oxygen Species, Reactive Nitrogen Species, and Redox-Dependent Signaling in the Cardiovascular System: A Scientific Statement from the American Heart Association. Circ. Res. 2016, 119, e39–e75. [Google Scholar] [CrossRef]
- Kalyanaraman, B.; Darley-Usmar, V.; Davies, K.J.; Dennery, P.A.; Forman, H.J.; Grisham, M.B.; Mann, G.E.; Moore, K.; Roberts, L.J.; Ischiropoulos, H. Measuring reactive oxygen and nitrogen species with fluorescent probes: Challenges and limitations. Free Radic. Biol. Med. 2012, 52, 1–6. [Google Scholar] [CrossRef]
- Dikalov, S.I.; Harrison, D.G. Methods for detection of mitochondrial and cellular reactive oxygen species. Antioxid. Redox. Signal. 2014, 20, 372–382. [Google Scholar] [CrossRef]
- Grieve, D.J.; Shah, A.M. Oxidative stress in heart failure. More than just damage. Eur. Heart J. 2003, 24, 2161–2163. [Google Scholar] [CrossRef]
- Tang, W.H.; Tong, W.; Troughton, R.W.; Martin, M.G.; Shrestha, K.; Borowski, A.; Jasper, S.; Hazen, S.L.; Klein, A.L. Prognostic value and echocardiographic determinants of plasma myeloperoxidase levels in chronic heart failure. J. Am. Coll. Cardiol. 2007, 49, 2364–2370. [Google Scholar] [CrossRef]
- Van der Pol, A.; van Gilst, W.H.; Voors, A.A.; van der Meer, P. Treating oxidative stress in heart failure: Past, present and future. Eur. J. Heart Fail. 2019, 21, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Rosca, M.G.; Hoppel, C.L. Mitochondrial dysfunction in heart failure. Heart Fail. Rev. 2013, 18, 607–622. [Google Scholar] [CrossRef] [PubMed]
- White, M.; Ducharme, A.; Ibrahim, R.; Whittom, L.; Lavoie, J.; Guertin, M.C.; Racine, N.; He, Y.; Yao, G.; Rouleau, J.L.; et al. Increased systemic inflammation and oxidative stress in patients with worsening congestive heart failure: Improvement after short-term inotropic support. Clin. Sci. 2006, 110, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Tousoulis, D.; Oikonomou, E.; Siasos, G.; Chrysohoou, C.; Charakida, M.; Trikas, A.; Siasou, Z.; Limperi, M.; Papadimitriou, E.D.; Papavassiliou, A.G.; et al. Predictive value of biomarkers in patients with heart failure. Curr. Med. Chem. 2012, 19, 2534–2547. [Google Scholar] [CrossRef]
- Ribeiro-Samora, G.A.; Rabelo, L.A.; Ferreira, A.C.C.; Favero, M.; Guedes, G.S.; Pereira, L.S.M.; Parreira, V.F.; Britto, R.R. Inflammation and oxidative stress in heart failure: Effects of exercise intensity and duration. Braz. J. Med. Biol. Res. 2017, 50, e6393. [Google Scholar] [CrossRef]
- Dhiman, M.; Thakur, S.; Upadhyay, S.; Kaur, A.; Mantha Anil, K. Oxidative Stress and Inflammation in Cardiovascular Diseases: Two Sides of the Same Coin. In Free Radicals in Human Health and Disease; Rani, V., Yadav, U.C.S., Eds.; Springer: New Delhi, India, 2015; pp. 259–278. [Google Scholar]
- Yasunari, K.; Maeda, K.; Nakamura, M.; Yoshikawa, J. Oxidative Stress in Leukocytes Is a Possible Link between Blood Pressure, Blood Glucose, and C-Reacting Protein. Hypertension 2002, 39, 777–780. [Google Scholar] [CrossRef]
- Aizawa, H.; Makita, Y.; Sumitomo, K.; Aburakawa, Y.; Katayama, T.; Nakatani-Enomoto, S.; Suzuki, Y.; Fujiwara, K.; Enomoto, H.; Kuroda, K.; et al. Edaravone diminishes free radicals from circulating neutrophils in patients with ischemic brain attack. Intern. Med. 2006, 45, 1–4. [Google Scholar] [CrossRef]
- Boudreau, L.H.; Duchez, A.C.; Cloutier, N.; Soulet, D.; Martin, N.; Bollinger, J.; Paré, A.; Rousseau, M.; Naika, G.S.; Lévesque, T.; et al. Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood 2014, 124, 2173–2183. [Google Scholar] [CrossRef]
- Jobe, S.M.; Wilson, K.M.; Leo, L.; Raimondi, A.; Molkentin, J.D.; Lentz, S.R.; Di Paola, J. Critical role for the mitochondrial permeability transition pore and cyclophilin D in platelet activation and thrombosis. Blood 2008, 111, 1257–1265. [Google Scholar] [CrossRef]
- Liu, F.; Gamez, G.; Myers, D.R.; Clemmons, W.; Lam, W.A.; Jobe, S.M. Mitochondrially Mediated Integrin αIIbβ3 Protein Inactivation Limits Thrombus Growth. J. Biol. Chem. 2013, 288, 30672–30681. [Google Scholar] [CrossRef]
- Yamagishi, S.I.; Edelstein, D.; Du, X.L.; Brownlee, M. Hyperglycemia potentiates collagen-induced platelet activation through mitochondrial superoxide overproduction. Diabetes 2001, 50, 1491–1494. [Google Scholar] [CrossRef] [PubMed]
- Avila, C.; Huang, R.J.; Stevens, M.V.; Aponte, A.M.; Tripodi, D.; Kim, K.Y.; Sack, M.N. Platelet mitochondrial dysfunction is evident in type 2 diabetes in association with modifications of mitochondrial anti-oxidant stress proteins. Exp. Clin. Endocrinol. Diabetes 2012, 120, 248–251. [Google Scholar] [CrossRef]
- Xin, G.; Wei, Z.; Ji, C.; Zheng, H.; Gu, J.; Ma, L.; Huang, W.; Morris-Natschke, S.L.; Yeh, J.L.; Zhang, R.; et al. Metformin Uniquely Prevents Thrombosis by Inhibiting Platelet Activation and mtDNA Release. Sci. Rep. 2016, 6, 36222. [Google Scholar] [CrossRef] [PubMed]
- Knez, J.; Cauwenberghs, N.; Thijs, L.; Winckelmans, E.; Brguljan-Hitij, J.; Yang, W.Y.; Staessen, J.A.; Nawrot, T.S.; Kuznetsova, T. Association of left ventricular structure and function with peripheral blood mitochondrial DNA content in a general population. Int. J. Cardiol. 2016, 214, 180–188. [Google Scholar] [CrossRef]
- Bayeva, M.; Gheorghiade, M.; Ardehali, H. Mitochondria as a therapeutic target in heart failure. J. Am. Coll. Cardiol. 2013, 61, 599–610. [Google Scholar] [CrossRef]
- Ashar, F.N.; Zhang, Y.; Longchamps, R.J.; Lane, J.; Moes, A.; Grove, M.L.; Mychaleckyj, J.C.; Taylor, K.D.; Coresh, J.; Rotter, J.I.; et al. Association of Mitochondrial DNA Copy Number With Cardiovascular Disease. JAMA Cardiol. 2017, 2, 1247–1255. [Google Scholar] [CrossRef]
- Gahan, M.E.; Miller, F.; Lewin, S.R.; Cherry, C.L.; Hoy, J.F.; Mijch, A.; Rosenfeldt, F.; Wesselingh, S.L. Quantification of mitochondrial DNA in peripheral blood mononuclear cells and subcutaneous fat using real-time polymerase chain reaction. J. Clin. Virol. 2001, 22, 241–247. [Google Scholar] [CrossRef]
- Yue, P.; Jing, S.; Liu, L.; Ma, F.; Zhang, Y.; Wang, C.; Duan, H.; Zhou, K.; Hua, Y.; Wu, G.; et al. Association between mitochondrial DNA copy number and cardiovascular disease: Current evidence based on a systematic review and meta-analysis. PLoS ONE 2018, 13, e0206003. [Google Scholar] [CrossRef]
- Liu, L.P.; Cheng, K.; Ning, M.A.; Li, H.H.; Wang, H.C.; Li, F.; Chen, S.Y.; Qu, F.L.; Guo, W.Y. Association between peripheral blood cells mitochondrial DNA content and severity of coronary heart disease. Atherosclerosis 2017, 261, 105–110. [Google Scholar] [CrossRef]
- Bliksøen, M.; Mariero, L.H.; Ohm, I.K.; Haugen, F.; Yndestad, A.; Solheim, S.; Seljeflot, I.; Ranheim, T.; Andersen, G.Ø.; Aukrust, P.; et al. Increased circulating mitochondrial DNA after myocardial infarction. Int. J. Cardiol. 2012, 158, 132–134. [Google Scholar] [CrossRef]
- Berezin, A.E. The Cell-Free Mitochondrial DNA: A Novel Biomarker of Cardiovascular Risk? Transl. Biomed. 2016, 7, 68–71. [Google Scholar] [CrossRef]
- Yu, E.P.K.; Bennett, M.R. The role of mitochondrial DNA damage in the development of atherosclerosis. Free Radic. Biol. Med. 2016, 100, 223–230. [Google Scholar] [CrossRef]
- Fetterman, J.L.; Holbrook, M.; Westbrook, D.G.; Brown, J.A.; Feeley, K.P.; Bretón-Romero, R.; Linder, E.A.; Berk, B.D.; Weisbrod, R.M.; Widlansky, M.E.; et al. Mitochondrial DNA damage and vascular function in patients with diabetes mellitus and atherosclerotic cardiovascular disease. Cardiovasc. Diabetol. 2016, 15, 53. [Google Scholar] [CrossRef] [PubMed]
- Sudakov, N.; Apartsin, K.A.; Lepekhova, S.A.; Nikiforov, S.B.; Katyshev, A.I.; Lifshits, G.I.; Vybivantseva, A.V.; Konstantinov, Y.M. The level of free circulating mitochondrial DNA in blood as predictor of death in case of acute coronary syndrome. Eur. J. Med. Res. 2017, 22, 1. [Google Scholar] [CrossRef]
- Chen, S.; Xie, X.; Wang, Y.; Gao, Y.; Xie, X.; Yang, J.; Ye, J. Association between leukocyte mitochondrial DNA content and risk of coronary heart disease: A case-control study. Atherosclerosis 2014, 237, 220–226. [Google Scholar] [CrossRef]
- Huang, J.; Tan, L.; Shen, R.; Zhang, L.; Zuo, H.; Wang, D.W. Decreased Peripheral Mitochondrial DNA Copy Number is Associated with the Risk of Heart Failure and Long-term Outcomes. Medicine (Baltimore) 2016, 95, e3323. [Google Scholar] [CrossRef]
- Huang, C.H.; Kuo, C.L.; Huang, C.S.; Liu, C.S.; Chang, C.C. Depleted Leukocyte Mitochondrial DNA Copy Number Correlates With Unfavorable Left Ventricular Volumetric and Spherical Shape Remodeling in Acute Myocardial Infarction After Primary Angioplasty. Circ. J. 2017, 81, 1901–1910. [Google Scholar] [CrossRef]
- Lien, L.M.; Chiou, H.Y.; Yeh, H.L.; Chiu, S.Y.; Jeng, J.S.; Lin, H.J.; Hu, C.J.; Hsieh, F.I.; Wei, Y.H. Significant Association Between Low Mitochondrial DNA Content in Peripheral Blood Leukocytes and Ischemic Stroke. J. Am. Heart Assoc. 2017, 6, e006157. [Google Scholar] [CrossRef]
- Zhang, Y.; Guallar, E.; Ashar, F.N.; Longchamps, R.J.; Castellani, C.A.; Lane, J.; Grove, M.L.; Coresh, J.; Sotoodehnia, N.; Ilkhanoff, L.; et al. Association between mitochondrial DNA copy number and sudden cardiac death: Findings from the Atherosclerosis Risk in Communities study (ARIC). Eur. Heart J. 2017, 38, 3443–3448. [Google Scholar] [CrossRef]
- Cohen, Z.; Gonzales, R.F.; Davis-Gorman, G.F.; Copeland, J.G.; McDonagh, P.F. Thrombin activity and platelet microparticle formation are increased in type 2 diabetic platelets: A potential correlation with caspase activation. Thromb. Res. 2002, 107, 217–221. [Google Scholar] [CrossRef]
- Sjövall, F.; Morota, S.; Hansson, M.J.; Friberg, H.; Gnaiger, E.; Elmér, E. Temporal increase of platelet mitochondrial respiration is negatively associated with clinical outcome in patients with sepsis. Crit. Care 2010, 14, R214. [Google Scholar] [CrossRef] [PubMed]
- Baccarelli, A.A.; Byun, H.M. Platelet mitochondrial DNA methylation: A potential new marker of cardiovascular disease. Clin. Epigenetics 2015, 7, 44. [Google Scholar] [CrossRef] [PubMed]
- Yu, E.; Calvert, P.A.; Mercer, J.R.; Harrison, J.; Baker, L.; Figg, N.L.; Kumar, S.; Wang, J.C.; Hurst, L.A.; Obaid, D.R.; et al. Mitochondrial DNA damage can promote atherosclerosis independently of reactive oxygen species through effects on smooth muscle cells and monocytes and correlates with higher-risk plaques in humans. Circulation 2013, 128, 702–712. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
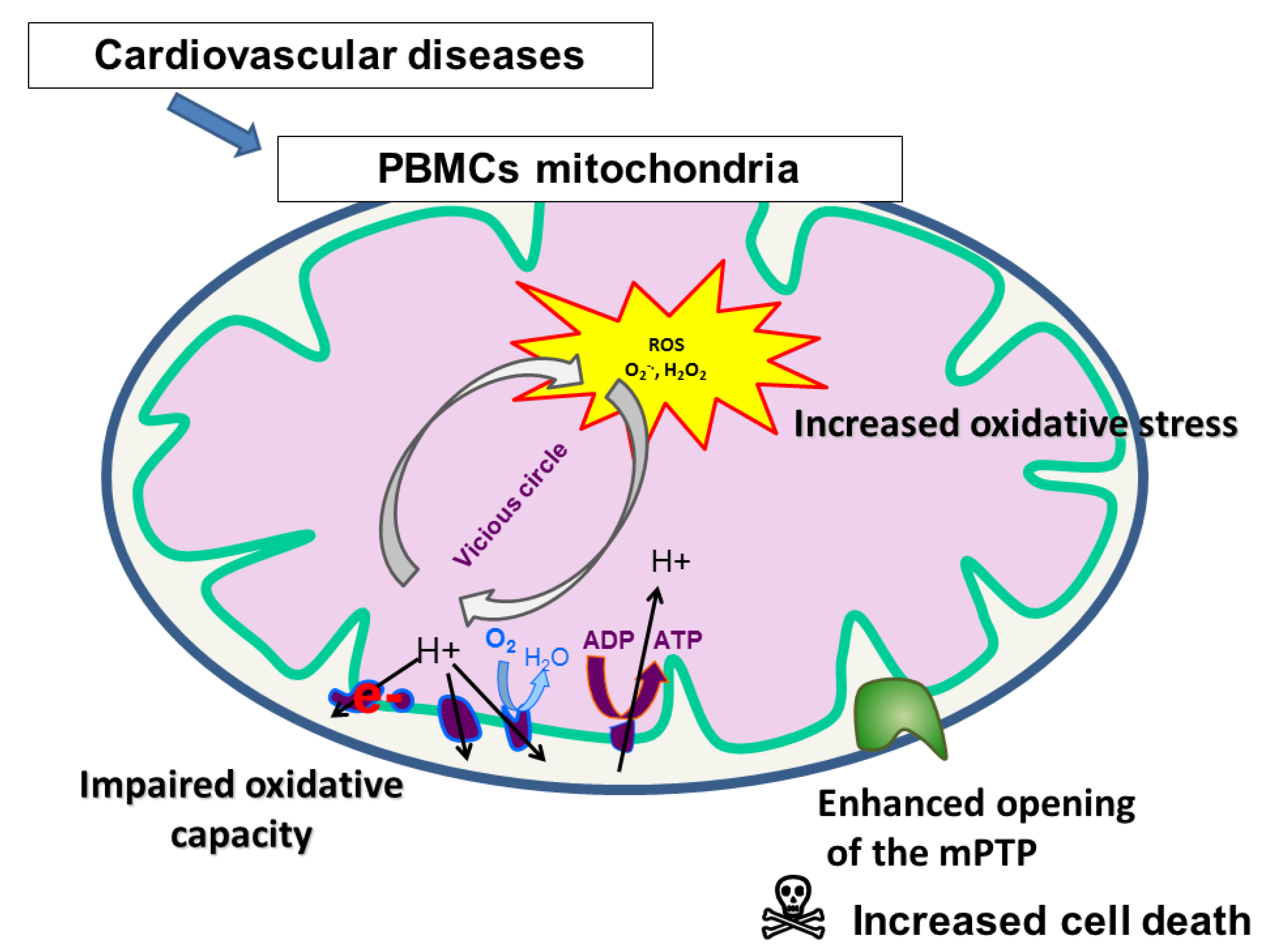{kind=link}
| Population Characteristics | Study Design/ Cells Analyzed | Mitochondrial Function | Oxidative Stress ROS Production/ Antioxidant Level | Cell Viability/Apoptosis | Results | References |
|---|---|---|---|---|---|---|
| HF pediatric patients with single ventricle (SV) congenital heart disease | PBMCs | -Oxygen consumption rate (Seahorse) -Mitochondrial respiration (oroboros) | ROS (Amplex red dye) | NA | -Respiratory capacity, coupling efficiency and mitochondrial oxygen flux were reduced in SV patients. -ROS was higher in SV patients | Garcia Anastacia et al., 2019, Circulation (Abstract) [46]. |
| -Mild Congestive Heart Failure patient (CHF) (Class I-II) n = 15, 14 male, 1 female Age: 63 ± 13 yo EF: 44.3 ± 14.5 % -Moderate-to-severe CHF (Class III) n = 16, 15 male, 1 female Age: 61 ± 14 yo EF: 26.9 ± 6% | PBMCs | -Mitochondrial respiration (oroboros) -Maximal electron transfer system capacity (ETS) | Assessment of ROS generation in permeabilized PBMCs before and after addition of mitochondrial oxidative phosphorylation uncoupler (FCCP) urinary 8-OHdG, a biomarker of oxidative DNA damage | N/A | Mitochondrial respiratory capacity of class III HF was lower than class II patients. -ETS capacity was significantly reduced in class III compared to class I–II -Mitochondrial ROS level was higher in class III CHF compared to class I–II patients, before and after FCCP. | Shirakawa et al., 2019, Scientific Report. [22] |
| Chronic HF patients n = 15, 12 male, 3 female Age: 56.6 ± 10.8 yo EF: 28 ± 8% Control group n = 10, 8 male, 2 female Age: 49.3 ± 8 yo EF: 65 ± 2% | PBMCs Basal and modulation by LPS | Mitochondrial membrane potential (TMRM and JC-1 staining). | -For cytoplasmic oxidative stress evaluation: PBMCs were incubate with 5 µM 2′,7′-dichlorofluorescein diacetate at 37 °C for 10 min. -For mitochondrial oxidative stress evaluation: (MitoSOX™ Red mitochondrial superoxide) -For antioxidant system (SOD GPx levels) | Assessment of overall cell damage Mitochondrial area percentage of intact cristae, and loss of inner mitochondrial membrane (IMM) -Cell damage (Annexin-v and P1 staining by flow cytometric analysis) -Assessment of mitophagy flux (gene expression by RT-PCR quantitation). | Baseline -Cytoplasmic ROS: no difference between HF-PBMCs and healthy subject. -Mitochondrial ROS: increased in HF-PBMCs as compared to controls -Index associated with the loss of inner mitochondrial membrane was lower in HF patients -mitophagy flux: increased autophagy genes in HF-PBMCs After LPS -Mitochondrial membrane potential: depolarization in PBMCs of HF patients (p < 0.05). -Antioxidant system: reduced SOD (P < 0.05 and <0.01) and GPx (p < 0.05) activity in HF-PBMCs -Cytoplasmic ROS: HF-PBMCs shows marked increase cytoplasmic ROS than control group. (p < 0.05) -Mitochondrial ROS: increased in HF patients (p < 0.05). - Index associated with the loss of inner mitochondrial membrane was more deteriorated after stimulation, and reduction of mitochondrial area with intact cristae in HF-PBMCs than in healthy group (p < 0.01) -Cell damage: apoptotic cell percentage was increased in HF patients. (p < 0.05) -Mitophagy flux: the response in HF-PBMCs was increased much more after stimulation. | Coluccia et al., 2018, Oncotarget. [38] |
| Congestive heart patients (CHF) n = 20, 16 male, 4 female Age: 68.9 ± 8 yo EF: 24.9 ± 5.9% -Control group n = 15, 13 male, 2 female Age:63.3 ± 9.4 yo EF: 60.0 ± 5.3% | Leukocyte were isolated by gradient centrifugation to measure cellular lipid, protein, PARP & AIF Modulation: Activation of PARP | N/A | C-reactive protein, N-terminal probrain-type natriuretic peptide, oxidative nitrative stress, plasma total peroxide level (PRX), total plasma antioxidant capacity (TAC)and oxidative stress index (OSI), Leukocyte lipid peroxidation, and protein tyrosine nitration (NT)were evaluated. PRX was determined by Oxystat and TAC was detected by OxiSelect™ TAC Assay kit | poly (ADP-ribose) polymerase (PARP), and apoptosis inducing factor (AIF) was measured | In CHF patients, plasma PRX level was markedly increased suggesting the increase of oxidative stress in this group. Oxidative stress of leucocytes increased in CHF group. PARP activity and AIF in circulating mononuclear cells of CHF group was higher than in the control group. A positive correlation was demonstrated between oxidative stress (Plasma PRX level, OSI) and PARP activation in circulating leukocytes with pro-BNP levels of CHF. | Bárány et al., 2017 Oxidative Medicine and Cellular Longevity. [8] |
| Pulmonary hypertension patients (PH group classified as WHO Group 2) n = 20, 10 male, 10 female Age: 69 ± 7.4 Control group n = 20, 10 male, 10 female Age: 69.4 ± 17.6 | Platelets | Oxygen consumption (Seahorse) Extracellular acidification rate (Seahorse) | ROS level analyzed using MitoSOX | N/A | Maximal oxygen consumption rate was significantly increased compared to controls Activity of complex II tended to increase in Group 2 PH platelets compared to controls (p = 0.09). Enhanced maximal capacity correlates negatively with right ventricular stroke work index No change with administration of inhaled nitrite, a modulator of pulmonary hemodynamics. | Nguyen et al., 2019, Plos one. [45] |
| CHFn = 54, male Age: 60 ± 10 EF% 33.3 ± 7.7 Control group n = 30, male Age: 61 ± 10 EF% 65.1 ± 7.3 | PMBCs (peripheral blood Lymphocyte Serum NT-ProBNP level were assessed | Mitochondrial transmembrane potential (MTP) Analyzed by flow cytometry described as JC-1 fluorescence ratio | ROS level of PBMCs were investigated. Described as DCF fluorescence intensity. | CHF patients experienced decreased MTP, (and increase level of ROS of lymphocytes (intensity 11.12) than the control group. -CHF patients had higher Serum NT-ProBNP level -Study conclude that patients with CHF, the MTP and ROS level of PBMCs are correlated with the changes in serum NT-ProBNP level | Song et al., 2016, Heart, Lung and circulation. [36] | |
| Early stage HF patients n = 25, 12 male, 13 female Age: 49 ± 3 years EF: 67.40 ± 0.83 Control group n = 24, 11 male, 13 female Age: 47 ± 3 yearsEF: 69.63 ± 0.99 | PBMCs sample | Mitochondrial respiration (Oroboros) | Measurement of inflammatory factors: High sensitivity C-reactive protein (hs-CRP), IL6, and TNF-⍺ -Oxidative stress biomarker: MDA Antioxidant system: SOD By using ELISA | Decreased mitochondrial oxygen consumption in HF compared to control group. -Inflammatory factors were significantly higher in patients with early stage HF. -SOD reduced, but MDA stayed unchanged in diseased patients. | Li et al., 2015 Scientific Report. [29] | |
| HF patients with left ventricular assist device n = 10, 8 male, 2 female Age, median (range): 65 (57–69) EF% (median (range): 15 (10–20) Control group n = 10, 8 male, 2 female Age, median (range): 63 (26–74) EF %: NA | PBMCs (Circulating blood leukocyte) | N/A | -Detection of ROS in leukocyte by flow cytometry, and immunofluorescence microscopy -Antioxidant defense system; SOD in erythrocyte was measured by spectrophotometry. -oxidized low density (oxLDL) lipoproteins were analyzed in plasma, by ELISA. -DNA damage markers were assessed in blood lymphocyte, and measured by immunofluorescence microscopy | N/A | -In HF patients, the mean fluorescence intensity (MFI) of DCF-DA exhibited increased level of ROS in peripheral blood leukocyte than in control group. Post-operative value (1 week): Neutrophils ROS (+51%) Lymphocytes ROS (+37%) Monocytes ROS (+54%) -Quantity of ROS reach the highest 3 months later (value not specified) -SOD level decreased in HF patient than in control. And continue to decrease to reach the minimum at 3 months post-operative. -oxLDL were markedly higher in HF than in control group. These results suggested increased oxidative stress among HF patients which leads to mitochondria dysfunction. -Markers used to express DNA damage, reveals abnormal DNA repair. | Mondal et al., 2013, International Journal of Medical Sciences. [47] |
| Congestive heart patients (CHF) n = 15 9 Male, 6 female Age: 79 ± 9 EF% =37 ± 17 Control group n = 9 6 male, 3 female Age: 49 ± 22EF% =63 ± 5 | WBC and Platelets blood sampling from radial artery, brachial vein and coronary sinus | N/A | Oxidative stress (immunofluorescence microscopy analysis of nitrotyrosine) -cytoplasmic oxidative stress (incubation of resuspended buffy coat with 5-6 CM-DCF). -7 CHF and 6 health individuals were evaluated for Mitochondrial oxidative stress, (Mitotracker red CM-H2 XRos M7513 Probe). -Both cytoplasmic and mitochondrial oxidative stress (live- cells fluorescence microscopy and FACS) | N/A | CHF exhibited increased protein nitosylation in arterial and venous WBC than control. -Cytoplasmic oxidative stress in CHF was increased in venous and arterially localized WBC and platelets. -For coronary sinus sampling, the number of ROS was higher than in venous (946 ± 475 vs. 659 ± 428 per 10,000 cells). -CHF patients had elevated mitochondrial ROS in WBC and platelets than healthy group. The number of ROS-positive venous WBC and platelets is (478 ± 261 per 10,000 cells vs. 162 ± 81 per 10,000 cells for control group). While, ROS-positive arterial WBC and platelets is 471 ± 211 per 10,000 cells vs. 85 ± 42 per 10,000 cells for healthy group. This increased number of circulating ROS suggesting increase oxidative stress in HF patients. | IJsselmuiden et al., 2008, (CardiovascularMedicine. [48] |
| Acute CHF Edematous n = 15 male 9 female 7 Age: 72.6 ± 3.7 EF% 36.2 ± 5.1 Non-edematous n = 15 male 10 female 5 Age: 78.5 ± 2.8 EF% 35.3 ± 2.7 Control group n = 20 male 18 female 2 Age: 68.5 ± 1.6 | PBMCs (Peripheral blood leukocyte) 10 mL venous blood sample was collected, 5 mL was anticoagulated and assayed for fluorescence staining | Mitochondrial transmembrane potential (MTP) in leukocyte was analyzed by flow cytometry | Intracellular oxidants formation was examined by DCF for 20 min at 37°. -Fluorescence was Detected by flow cytometry -Analyzing plasma factors nitrogen metabolites. -Lipid peroxides including (MDA, HNE) -inflammatory factors: IL6, and TNF-⍺ using ELISA. | -Cell apoptosis was measured by tunnel assay | In CHF, MTP of PBMCs was markedly decreased, with the weakening in edematous HF patients more than in non-edematous specifically in lymphocyte. -Intracellular oxidants of PBMCs were increased, with the highest was in monocytes. -Edematous CHF had higher DCF fluorescence level than the other CHF group. -Apoptotic cells percentage was higher in polymorphonuclear leukocyte (PMN) than PBMCs. -edematous leukocyte presented with higher percentage of apoptosis than another CHF group. -plasma nitrogen level, lipid peroxide, and inflammatory factors was higher in CHF than control. | Kong et al., 2001, Journal of the American College of Cardiology. [37] |
| Population Characteristics | Study Design | Mitochondrial Function/mtDNA Copy Number | Oxidative Stress | Cell Viability/Apoptosis | Results | Reference |
|---|---|---|---|---|---|---|
| Ischemic stroke patients Total n = 350 Age: 60.9 ± 9.1 Male n = 246 Female n = 104 Control group N = 350 Age:60.4 ± 9.1 Male n = 246 Female n = 104 | mtDNA in Peripheral Blood Leukocyte | -mtDNA content (rt PCR) -The ratio of mtDNA to NuclearDNA is used to estimate the number of mtDNA per cell | -oxidized glutathione (GSSG), and reduced glutathione (GSH), (enzymatic (method) -8-hydroxy-2’-deoxyguanosine (biomarker of oxidative DNA damage, ELISA) | NA | mtDNA content in peripheral leukocyte for ischemic stroke patients was significantly lower than the control group. P < 0.0001 mtDNA content evaluated for 150 ischemic stroke patients = 0.90, while in 50 control individuals = 1.20 -The level of GSSG and 8-hydroxy-2’-deoxyguanosine were higher in patients with ischemic stroke than on the control group. GSSG Ischemic stroke = 1.83 Control = 0.79 8-hydroxy-2´-deoxyguanosine ischemic stroke = 6.33 Control = 4.87 These results exhibited that oxidative stress was higher in patients with ischemic stroke than in control group | Lien et al., 2017, Journal of American Heart Association [91] |
| 3 cohort study with a risk factor of CVD 1st: Cardiovascular Health Study (CHS) n = 4830 Age: >65 years 2nd: Atherosclerosis Risk in Communities (ARIC) n = 11153 Age: Between 45 to 65 years 3rd: Multiethnic Study of Atherosclerosis (MESA) n = 5887 Age: 45 to 85 years Control: NA | In CHS: DNA was extracted from the buffy coat using salt precipitation following proteinase K digestion In ARIC: DNA was extracted from the buffy coat of whole blood using (Qiagen) In MESA: DNA was extracted from leukocyte using (Qiagen) | In ARIC and MESA, mtDNA copy number was measured by using prob intensities of mitochondrial single nucleotide polymorphisms (SNP) on the Affymetrix Genome-Wide Human SNP Array 6.0 IN CHS: mtDNA was calculated using multiplexed TaqMan-based PCR | NA | NA | -The effect of mtDNA copy number on the incidence of coronary heart disease was higher than in stroke and in other CVDs In all 3 cohort groups, the mtDNA copy number was inversely associated with CVD events | Ashar et al., 2017, JAMA Cardiology [79] |
| Coronary Heart Disease (CHD) classified in 4 groups according to Gensini score 1-Gensini score: 0-–22 n = 99, Male 72, Age: 57.3 2-Gensini score: 22–55 n = 98, Male 73, Age:57.9 3-Gensini score: 55–96 n = 102, Male 79, Age: 58.3 4-Gensini score:96–254 n = 101, Male 86, Age: 58.8 -Control groupn = 110Age: 58.1 | mtDNA of Leukocytes for CHF categorized by Gensini score | -genomic DNA was isolated from peripheral blood cells by E.Z.N.A blood DNA Midi Kit. -mtDNA quantification (Quantitative real time PCR). | NA | NA | mtDNA content of PBMCs was lower in CHD patients than in the control group. -mtDNA was reduced significantly, while Gensini score was increased suggesting the level of circulating mtDNA correlates with presence and severity of CHD. | Liu et al. 2017, Atherosclerosis [82] |
| Acute coronary syndrome (ACS) Total n = 14 Divided into 2 groups 1st group: (Survivor) who survive during 30 day of hospitalization n = 11, male 9, female 2 Age: 53 2nd group: (deceased) who died due to ACS during time of analysis n = 3 female Age: 87 | Blood samples were collected from platelet poor plasma | -To evaluate mtDNA. Isolation performed with PROBA-NK reagent kit. -quantitation of mtDNA was performed by PCR | NA | NA | -Deceased group: the level of mtDNA level was higher (5900 copies/mL) than the survived group (36 copies/mL) p = 0.049 -increased level of mtDNA in plasma suggest a probability of death of 50% for ACS patients | Sudakov et al., 2017, European Journal of Medical Research [87] |
| Patients from the Atherosclerosis Risk in Communities (ARIC) n = 11093 male n = 4971 female n = 6122 Age: 57.9 ± 6.0 | mtDNA in peripheral blood buffy coat | mtDNA copy number was measured by using prob intensities of mitochondrial single nucleotide polymorphisms (SNP) on the Affymetrix Genome-Wide Human SNP Array 6.0 | NA | -Inverse association between mtDNA copy number and sudden cardiac death | Zhang et al., 2017, Eur Heart Journal [92] | |
| Acute myocardial infarction patient undergoing primary angioplasty n = 55 male n = 47 female n = 8 Age: 57.4 ± 11.4 years Control group: n = 54 male n = 44 female n = 10 age: 55.3 ± 7.4 | Peripheral blood leukocyte | Leukocyte mitochondrial DNA copy number (MCN) was measured from venous blood using PCR -AMI patients were divided into two groups according to median baseline leukocyte mtDNA copy number = 82/cell 1st group MCN ≥ 82 2nd group MCN < 82 | NA | NA | -Baseline characteristics: In AMI patients the plasma leukocyte mtDNA copy number was significantly lower than in the control group. 122.7 ± 109.3 vs. 194.9 ± 119.5/cell p = 0.003 -AMI patients with lower MCN, had higher left ventricle shape sphericity index (SI), at 1,3,6 months after angioplasty and higher left ventricle diastolic and systolic volume at 6 months after angioplasty. | Huang et al., 2017, Circulatiog Journal, [90] |
| Patients with diabetes mellitus and atherosclerosis cardiovascular disease Total n = 275 -only Atherosclerosis: N = 55 Female 18 Age:60 ± 10 -only DM: N = 74 Female 47 Age: 55 ± 10 -Atherosclerosis and DM N = 48 Female 31 Age: 62 ± 8 Control group n = 98 Female 49 Age: 55 ± 7 | PBMCs | Measuring mitochondrial DNA damage in PBMCs by PCR. -Total DNA was separated using QIAmp DNA mini kit and quantification determined by using Pico-green assay kit | Oxidative stress of arterial pulsatility (increased baseline pulse amplitude p = 0.009) | NA | Mitochondrial DNA damage was higher in all 3 diseased group, as compared with controls, with the highest in the group combining atherosclerosis and diabetes. -mtDNA measured in DM alone (0.65 ± 1.0) -mtDNA measured in atherosclerosis alone (0.55 ± 0.65) -mtDNA measured in both atherosclerosis and DM (0.89 ± 1.32) p < 0.05 mtDNA damage correlated with baseline pulse amplitude | Fetterman et al., 2016, (Cardiovascular Diabetology) [86] |
| General population Total n = 701 Divided by 3 tertiles of mtDNA content -Tertile 1 mtDNA content 0.39–0.86 N = 233 Female 103 Age: 51.6 ± 16.8 EF% 61.3 ± 7.0 -Tertile 2 mtDNA content 0.86–1.10 N = 234 Female126 Age:53.5 ± 14.7 EF%: 63.3 ± 6.56 -Tertile 3 mtDNA content 1.11–3.06 N = 234 Female 128 Age:54.3 ± 14.2 EF%: 62.9 ± 6.65 | Peripheral blood cells | To assess the circulating mtDNA content, PCR was used. Total DNA was extracted from peripheral blood sample using QIAmp DNA Mini Kit. | NA | NA | There is a relation between peripheral blood mtDNA copy number and left ventricular function. Higher mtDNA content was associated with better systolic and diastolic left ventricular function | Knez et al. 2016, International Journal of Cardiology [77] |
| Chronic Heart Failure Total N = 1700 -Ischemic HF N = 790 Male 543 Age: 62.6 ± 10.4 EF% 57 -Nonischemic HF N = 910 Male 572 Age: 53.8 ± 14.3 EF% 40 Control group n = 1700 male 1115 Age: 57.7 ± 11.0 EF%: NA | Circulating Leukocyte -Blood sample were drawn, and leukocyte were isolated in K2-EDTA tubes. | Total DNA was extracted by using QG-Mini80 workflow with a DB-S kit. And DNAs of cardiac tissues were isolated by using QIAmp DNA Mini Kit. And copy number ratio was evaluated. | ROS were quantified in heart tissues using Dihydroethidium (DHE) staining. -In lymphocyte intracellular ROS was analyzed by flow cytometry using DCFH-DA -LDL was detected | HF patients presented a low mtDNA content compared to control group. Median 0.83, IQR: 0.60–1.16 vs. median 1.00, IQR: 0.47–2.20)P < 0.001. Ischemic HF patients are more susceptible to lower mt DNA copy number(Median 0.77, IQR: 0.56–1.08) than non-ischemic HF median 0.91, IQR 0.63–1.22 -mtDNA content of leukocyte was not correlated with LV diameter p = 0.988 -in HF group, LDL was associated with the mtDNA copy number p = 0.007 -Lower circulating mtDNA was correlated with increased risk of HF, p < 0.001 -In HF patients, the level of ROS was higher than in control group in heart tissues and in lymphocytes. | Huang et al., 2016, Medicine [89] | |
| Coronary heart Disease Patients N = 378 Male 279 Female 99 Age: 57.9 -Control group n = 378 male 279 female 99 Age: 58.9 | Peripheral Blood Leukocytes -5 mL of venous blood was drawn from each individual and anticoagulated into sodium citrate tube. | -DNA was separated from peripheral blood leukocyte using E.Z.N.A blood DNA Midi Kit. -DNA content was measured using PCR | NA | NA | -mtDNA content was inversely related to increased risk of CHD -CHD group shows marked lower mtDNA content, compared to controls, p < 0.001, -CHF had higher neutrophils counts compared to controls (5.10 ± 1.66 vs. 4.50 ± 1.51) but no difference in WBC count p = 0.154 | Chen et al. 2014 (Atherosclerosis) [88] |
| Myocardial infarction ST segment elevation MI (STEMI) n = 20, 5 femaleStable angina pectoris (SAP) n = 10, 1 female Both undergoing percutaneous coronary intervention (PCI) and categorized as transmural or non-transmural Age: between 30 and 75 years | Platelet poor plasma | Venous blood sample were gathered, and DNA was extracted from platelet poor plasma using QIAmp DNA blood Mini Kit -Quantification of mtDNA using real time PCR | NA | NA | -Baseline characteristics: Both groups were similar except SAP group which received more PCI treatment than the other group. -After PCI: 3 h later, mtDNA plasma level of NADH dehydrogenase subunit 1 (ND1) were increased in STEMI compared to SAP. p = 0.01 -patients with transmural: ND1 levels were greater in STEMI patients n = 10, than STEMI patients with non-transmural n = 6 -positive correlation between the severity of myocardial damage and the level of mtDNA, mtDNA being increased in myocardial infarction. | Bliksøen et al., 2012, [83] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alfatni, A.; Riou, M.; Charles, A.-L.; Meyer, A.; Barnig, C.; Andres, E.; Lejay, A.; Talha, S.; Geny, B. Peripheral Blood Mononuclear Cells and Platelets Mitochondrial Dysfunction, Oxidative Stress, and Circulating mtDNA in Cardiovascular Diseases. J. Clin. Med. 2020, 9, 311. https://doi.org/10.3390/jcm9020311
Alfatni A, Riou M, Charles A-L, Meyer A, Barnig C, Andres E, Lejay A, Talha S, Geny B. Peripheral Blood Mononuclear Cells and Platelets Mitochondrial Dysfunction, Oxidative Stress, and Circulating mtDNA in Cardiovascular Diseases. Journal of Clinical Medicine. 2020; 9(2):311. https://doi.org/10.3390/jcm9020311
Chicago/Turabian StyleAlfatni, Abrar, Marianne Riou, Anne-Laure Charles, Alain Meyer, Cindy Barnig, Emmanuel Andres, Anne Lejay, Samy Talha, and Bernard Geny. 2020. "Peripheral Blood Mononuclear Cells and Platelets Mitochondrial Dysfunction, Oxidative Stress, and Circulating mtDNA in Cardiovascular Diseases" Journal of Clinical Medicine 9, no. 2: 311. https://doi.org/10.3390/jcm9020311
APA StyleAlfatni, A., Riou, M., Charles, A.-L., Meyer, A., Barnig, C., Andres, E., Lejay, A., Talha, S., & Geny, B. (2020). Peripheral Blood Mononuclear Cells and Platelets Mitochondrial Dysfunction, Oxidative Stress, and Circulating mtDNA in Cardiovascular Diseases. Journal of Clinical Medicine, 9(2), 311. https://doi.org/10.3390/jcm9020311