The Biomarker Potential of miRNAs in Myotonic Dystrophy Type I


 ,
,
Abstract
1. Introduction
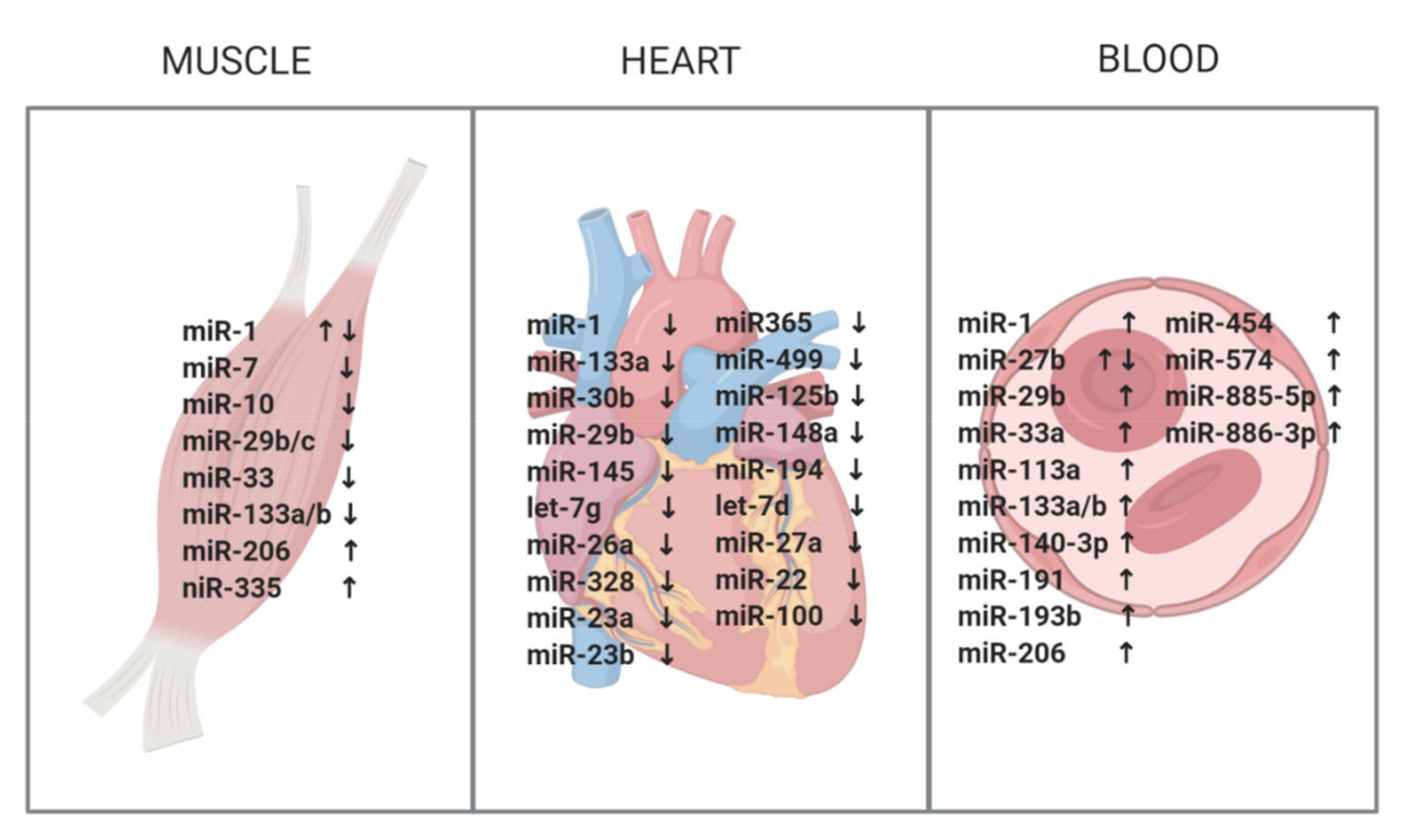
2. miRNAs in Skeletal Muscle
3. miRNAs in the Heart
4. miRNAs in Blood and Serum
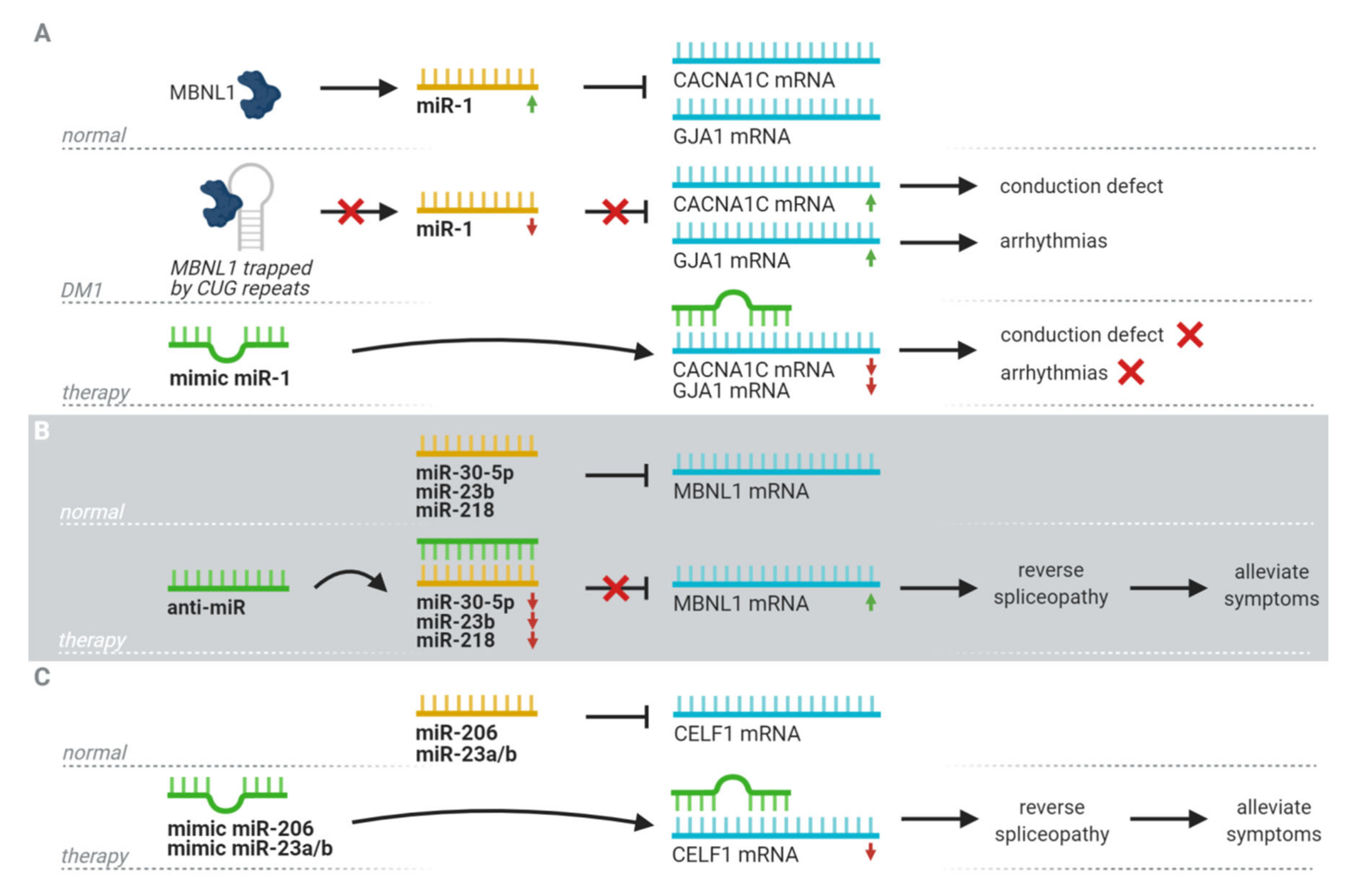
5. Therapeutic Potential
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Guo, H.; Ingolia, N.T.; Weissman, J.S.; Bartel, D.P. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 2010, 466, 835–840. [Google Scholar] [CrossRef]
- Berezikov, E.; Guryev, V.; van de Belt, J.; Wienholds, E.; Plasterk, R.H.A.; Cuppen, E. Phylogenetic Shadowing and Computational Identification of Human microRNA Genes. Cell 2005, 120, 21–24. [Google Scholar] [CrossRef]
- Shivdasani, R.A. MicroRNAs: Regulators of gene expression and cell differentiation. Blood 2006, 108, 3646–3653. [Google Scholar] [CrossRef] [PubMed]
- Condrat, C.E.; Thompson, D.C.; Barbu, M.G.; Bugnar, O.L.; Boboc, A.; Cretoiu, D.; Suciu, N.; Cretoiu, S.M.; Voinea, S.C. miRNAs as Biomarkers in Disease: Latest Findings Regarding Their Role in Diagnosis and Prognosis. Cells 2020, 9, 276. [Google Scholar] [CrossRef] [PubMed]
- Makarova, J.A.; Shkurnikov, M.U.; Wicklein, D.; Lange, T.; Samatov, T.R.; Turchinovich, A.A.; Tonevitsky, A.G. Intracellular and extracellular microRNA: An update on localization and biological role. Prog. Histochem. Cytochem. 2016, 51, 33–49. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Shi, K.; Yang, S.; Liu, J.; Zhou, Q.; Wang, G.; Song, J.; Li, Z.; Zhang, Z.; Yuan, W. Effect of exosomal miRNA on cancer biology and clinical applications. Mol. Cancer 2018, 17. [Google Scholar] [CrossRef]
- Wiedrick, J.T.; Phillips, J.I.; Lusardi, T.A.; McFarland, T.J.; Lind, B.; Sandau, U.S.; Harrington, C.A.; Lapidus, J.A.; Galasko, D.R.; Quinn, J.F.; et al. Validation of MicroRNA Biomarkers for Alzheimer’s Disease in Human Cerebrospinal Fluid. J. Alzheim. Dis. 2019, 67, 875–891. [Google Scholar] [CrossRef] [PubMed]
- Pitkänen, A.; Ekolle Ndode-Ekane, X.; Lapinlampi, N.; Puhakka, N. Epilepsy biomarkers—Toward etiology and pathology specificity. Neurobiol. Dis. 2019, 123, 42–58. [Google Scholar] [CrossRef]
- Kingsley, S.M.K.; Bhat, B.V. Role of microRNAs in sepsis. Inflamm. Res. 2017, 66, 553–569. [Google Scholar] [CrossRef]
- Viswambharan, V.; Thanseem, I.; Vasu, M.M.; Poovathinal, S.A.; Anitha, A. miRNAs as biomarkers of neurodegenerative disorders. Biomark. Med. 2017, 11, 151–167. [Google Scholar] [CrossRef]
- Coenen-Stass, A.M.L.; Wood, M.J.A.; Roberts, T.C. Biomarker Potential of Extracellular miRNAs in Duchenne Muscular Dystrophy. Trends Mol. Med. 2017, 23, 989–1001. [Google Scholar] [CrossRef] [PubMed]
- Harper, P.S. Major Problems in Neurology: Myotonic Dystrophy, 3rd ed.; WB Saunders: London, UK, 2001. [Google Scholar]
- Campbell, C.; Sherlock, R.; Jacob, P.; Blayney, M. Congenital myotonic dystrophy: Assisted ventilation duration and outcome. Pediatrics 2004, 113, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Douniol, M.; Jacquette, A.; Cohen, D.; Bodeau, N.; Rachidi, L.; Angeard, N.; Cuisset, J.-M.; Vallée, L.; Eymard, B.; Plaza, M.; et al. Psychiatric and cognitive phenotype of childhood myotonic dystrophy type 1. Dev. Med. Child Neurol. 2012, 54, 905–911. [Google Scholar] [CrossRef] [PubMed]
- De Antonio, M.; Dogan, C.; Hamroun, D.; Mati, M.; Zerrouki, S.; Eymard, B.; Katsahian, S.; Bassez, G. French Myotonic Dystrophy Clinical Network Unravelling the myotonic dystrophy type 1 clinical spectrum: A systematic registry-based study with implications for disease classification. Rev. Neurol. (Paris) 2016, 172, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, J.; Allard, P.; Potvin, L.; Prévost, C.; Bégin, P. A 10-year study of mortality in a cohort of patients with myotonic dystrophy. Neurology 1999, 52, 1658–1662. [Google Scholar] [CrossRef]
- De Die-Smulders, C.E.; Höweler, C.J.; Thijs, C.; Mirandolle, J.F.; Anten, H.B.; Smeets, H.J.; Chandler, K.E.; Geraedts, J.P. Age and causes of death in adult-onset myotonic dystrophy. Brain 1998, 121, 1557–1563. [Google Scholar] [CrossRef]
- Groh, W.J.; Groh, M.R.; Saha, C.; Kincaid, J.C.; Simmons, Z.; Ciafaloni, E.; Pourmand, R.; Otten, R.F.; Bhakta, D.; Nair, G.V.; et al. Electrocardiographic abnormalities and sudden death in myotonic dystrophy type 1. N. Engl. J. Med. 2008, 358, 2688–2697. [Google Scholar] [CrossRef]
- Groh, W.J.; Groh, M.R.; Shen, C.; Monckton, D.G.; Bodkin, C.L.; Pascuzzi, R.M. Survival and CTG repeat expansion in adults with myotonic dystrophy type 1. Muscle Nerv. 2011, 43, 648–651. [Google Scholar] [CrossRef]
- Logigian, E.L.; Moxley, R.T.; Blood, C.L.; Barbieri, C.A.; Martens, W.B.; Wiegner, A.W.; Thornton, C.A.; Moxley, R.T. Leukocyte CTG repeat length correlates with severity of myotonia in myotonic dystrophy type 1. Neurology 2004, 62, 1081–1089. [Google Scholar] [CrossRef]
- Meola, G.; Cardani, R. Myotonic dystrophies: An update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 594–606. [Google Scholar] [CrossRef]
- López-Martínez, A.; Soblechero-Martín, P.; De-La-puente-ovejero, L.; Nogales-Gadea, G.; Arechavala-Gomeza, V. An overview of alternative splicing defects implicated in myotonic dystrophy type i. Genes 2020, 11, 1109. [Google Scholar] [CrossRef]
- Kuyumcu-Martinez, N.M.; Wang, G.S.; Cooper, T.A. Increased Steady-State Levels of CUGBP1 in Myotonic Dystrophy 1 Are Due to PKC-Mediated Hyperphosphorylation. Mol. Cell 2007, 28, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Mankodi, A.; Takahashi, M.P.; Jiang, H.; Beck, C.L.; Bowers, W.J.; Moxley, R.T.; Cannon, S.C.; Thornton, C.A. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol. Cell 2002, 10, 35–44. [Google Scholar] [CrossRef]
- Sicot, G.; Gourdon, G.; Gomes-Pereira, M. Myotonic dystrophy, when simple repeats reveal complex pathogenic entities: New findings and future challenges. Hum. Mol. Genet. 2011, 20, R116–R123. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, M.S. Myotonic dystrophy: Is a narrow focus obscuring the rest of the field? Curr. Opin. Neurol. 2012, 25, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Gudde, A.E.E.G.; van Heeringen, S.J.; de Oude, A.I.; van Kessel, I.D.G.; Estabrook, J.; Wang, E.T.; Wieringa, B.; Wansink, D.G. Antisense transcription of the myotonic dystrophy locus yields low-abundant RNAs with and without (CAG)n repeat. RNA Biol. 2017, 14, 1374–1388. [Google Scholar] [CrossRef] [PubMed]
- Zu, T.; Gibbens, B.; Doty, N.S.; Gomes-Pereira, M.; Huguet, A.; Stone, M.D.; Margolis, J.; Peterson, M.; Markowski, T.W.; Ingram, M.A.C.; et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. USA 2011, 108, 260–265. [Google Scholar] [CrossRef]
- Wahbi, K.; Babuty, D.; Probst, V.; Wissocque, L.; Labombarda, F.; Porcher, R.; Bécane, H.M.; Lazarus, A.; Béhin, A.; Laforêt, P.; et al. Incidence and predictors of sudden death, major conduction defects and sustained ventricular tachyarrhythmias in 1388 patients withmyotonic dystrophy type 1. Eur. Heart J. 2017, 38, 751–758. [Google Scholar] [CrossRef]
- Falsaperla, R.; Parano, E.; Romano, C.; Praticò, A.D.; Pavone, P. HyperCKemia as a biomarker for muscular diseases. Clin. Ter. 2010, 161, 185–187. [Google Scholar]
- Koch, A.J.; Pereira, R.; Machado, M. The creatine kinase response to resistance exercise. J. Musculoskelet. Neu. Interact. 2014, 14, 68–77. [Google Scholar]
- Nakamori, M.; Sobczak, K.; Puwanant, A.; Welle, S.; Eichinger, K.; Pandya, S.; Dekdebrun, J.; Heatwole, C.R.; McDermott, M.P.; Chen, T.; et al. Splicing biomarkers of disease severity in myotonic dystrophy. Ann. Neurol. 2013, 74, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Perfetti, A.; Greco, S.; Bugiardini, E.; Cardani, R.; Gaia, P.; Gaetano, C.; Meola, G.; Martelli, F. Plasma microRNAs as biomarkers for myotonic dystrophy type 1. Neuromuscul. Disord. 2014, 24, 509–515. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Koutsoulidou, A.; Kyriakides, T.C.; Papadimas, G.K.; Christou, Y.; Kararizou, E.; Papanicolaou, E.Z.; Phylactou, L.A. Elevated muscle-specific miRNAs in serum of myotonic dystrophy patients relate to muscle disease progress. PLoS ONE 2015, 10, e0125341. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, C.; Heatwole, C.; Hébert, L.J.; Hogrel, J.-Y.; Laberge, L.; Leone, M.; Meola, G.; Richer, L.; Sansone, V.; Kierkegaard, M. Report of the third outcome measures in myotonic dystrophy type 1 (OMMYD-3) international workshop. J. Neuromuscul. Dis. 2018, 5, 523–537. [Google Scholar] [CrossRef] [PubMed]
- Alfano, L.; Lowes, L.; Berry, K.; Flanigan, K.; Cripe, L.; Mendell, J. Role of motivation on performance of the 6-minute walk test in boys with Duchenne muscular dystrophy. Dev. Med. Child Neurol. 2015, 57, 57–58. [Google Scholar] [CrossRef]
- Wu, N.; Gu, T.; Lu, L.; Cao, Z.; Song, Q.; Wang, Z.; Zhang, Y.; Chang, G.; Xu, Q.; Chen, G. Roles of miRNA-1 and miRNA-133 in the proliferation and differentiation of myoblasts in duck skeletal muscle. J. Cell. Physiol. 2019, 234, 3490–3499. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Mandel, E.M.; Thomson, J.M.; Wu, Q.; Callis, T.E.; Hammond, S.M.; Conlon, F.L.; Wang, D.Z. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 2006. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Niu, L.L.; Wei, W.; Zhang, W.Y.; Li, X.Y.; Cao, J.H.; Zhao, S.H. A feedback circuit between miR-133 and the ERK1/2 pathway involving an exquisite mechanism for regulating myoblast proliferation and differentiation. Cell Death Dis. 2013, 4. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Cardiac-specific miRNA in cardiogenesis, heart function, and cardiac pathology (with focus on myocardial infarction). J. Mol. Cell. Cardiol. 2016, 94, 107–121. [Google Scholar] [CrossRef]
- Hak, K.K.; Yong, S.L.; Sivaprasad, U.; Malhotra, A.; Dutta, A. Muscle-specific microRNA miR-206 promotes muscle differentiation. J. Cell Biol. 2006, 174, 677–687. [Google Scholar] [CrossRef]
- Van Rooij, E.; Quiat, D.; Johnson, B.A.; Sutherland, L.B.; Qi, X.; Richardson, J.A.; Kelm, R.J.; Olson, E.N. A Family of microRNAs Encoded by Myosin Genes Governs Myosin Expression and Muscle Performance. Dev. Cell 2009, 17, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Small, E.M.; O’Rourke, J.R.; Moresi, V.; Sutherland, L.B.; McAnally, J.; Gerard, R.D.; Richardson, J.A.; Olson, E.N. Regulation of PI3-kinase/Akt signaling by muscle-enriched microRNA-486. Proc. Natl. Acad. Sci. USA 2010, 107, 4218–4223. [Google Scholar] [CrossRef]
- Carè, A.; Catalucci, D.; Felicetti, F.; Bonci, D.; Addario, A.; Gallo, P.; Bang, M.L.; Segnalini, P.; Gu, Y.; Dalton, N.D.; et al. MicroRNA-133 controls cardiac hypertrophy. Nat. Med. 2007, 13, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Lin, H.; Xiao, J.; Lu, Y.; Luo, X.; Li, B.; Zhang, Y.; Xu, C.; Bai, Y.; Wang, H.; et al. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat. Med. 2007, 13, 486–491. [Google Scholar] [CrossRef]
- Greco, S.; De Simone, M.; Colussi, C.; Zaccagnini, G.; Fasanaro, P.; Pescatori, M.; Cardani, R.; Perbellini, R.; Isaia, E.; Sale, P.; et al. Common micro-RNA signature in skeletal muscle damage and regeneration induced by Duchenne muscular dystrophy and acute ischemia. FASEB J. 2009, 23, 3335–3346. [Google Scholar] [CrossRef]
- Eisenberg, I.; Eran, A.; Nishino, I.; Moggio, M.; Lamperti, C.; Amato, A.A.; Lidov, H.G.; Kang, P.B.; North, K.N.; Mitrani-Rosenbaum, S.; et al. Distinctive patterns of microRNA expression in primary muscular disorders. Proc. Natl. Acad. Sci. USA 2007, 104, 17016–17021. [Google Scholar] [CrossRef]
- Gambardella, S.; Rinaldi, F.; Lepore, S.M.; Viola, A.; Loro, E.; Angelini, C.; Vergani, L.; Novelli, G.; Botta, A. Overexpression of microRNA-206 in the skeletal muscle from myotonic dystrophy type 1 patients. J. Transl. Med. 2010, 8, 48. [Google Scholar] [CrossRef]
- Hon, L.S.; Zhang, Z. The roles of binding site arrangement and combinatorial targeting in microRNA repression of gene expression. Genome Biol. 2007, 8. [Google Scholar] [CrossRef]
- Yuasa, K.; Hagiwara, Y.; Ando, M.; Nakamura, A.; Takeda, S.; Hijikata, T. MicroRNA-206 is highly expressed in newly formed muscle fibers: Implications regarding potential for muscle regeneration and maturation in muscular dystrophy. Cell Struct. Funct. 2008, 33, 163–169. [Google Scholar] [CrossRef]
- Perbellini, R.; Greco, S.; Sarra-Ferraris, G.; Cardani, R.; Capogrossi, M.C.; Meola, G.; Martelli, F. Dysregulation and cellular mislocalization of specific miRNAs in myotonic dystrophy type 1. Neuromuscul. Disord. 2011, 21, 81–88. [Google Scholar] [CrossRef]
- Fernandez-Costa, J.M.; Garcia-Lopez, A.; Zuñiga, S.; Fernandez-Pedrosa, V.; Felipo-Benavent, A.; Mata, M.; Jaka, O.; Aiastui, A.; Hernandez-Torres, F.; Aguado, B.; et al. Expanded CTG repeats trigger miRNA alterations in Drosophila that are conserved in myotonic dystrophy type 1 patients. Hum. Mol. Genet. 2013, 22, 704–716. [Google Scholar] [CrossRef] [PubMed]
- Fritegotto, C.; Ferrati, C.; Pegoraro, V.; Angelini, C. Micro-RNA expression in muscle and fiber morphometry in myotonic dystrophy type 1. Neurol. Sci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.C.; Coenen-Stass, A.M.L.; Wood, M.J.A. Assessment of RT-qPCR normalization strategies for accurate quantification of extracellular microRNAs in murine Serum. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Rau, F.; Freyermuth, F.; Fugier, C.; Villemin, J.P.; Fischer, M.C.; Jost, B.; Dembele, D.; Gourdon, G.; Nicole, A.; Duboc, D.; et al. Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nat. Struct. Mol. Biol. 2011, 18, 840–845. [Google Scholar] [CrossRef] [PubMed]
- Kalsotra, A.; Singh, R.K.; Gurha, P.; Ward, A.J.; Creighton, C.J.; Cooper, T.A. The Mef2 transcription network is disrupted in myotonic dystrophy heart tissue, dramatically altering miRNA and mRNA expression. Cell Rep. 2014, 6, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Kalsotra, A.; Wang, K.; Li, P.F.; Cooper, T.A. MicroRNAs coordinate an alternative splicing network during mouse postnatal heart development. Genes Dev. 2010, 24, 653–658. [Google Scholar] [CrossRef]
- Perfetti, A.; Greco, S.; Cardani, R.; Fossati, B.; Cuomo, G.; Valaperta, R.; Ambrogi, F.; Cortese, A.; Botta, A.; Mignarri, A.; et al. Validation of plasma microRNAs as biomarkers for myotonic dystrophy type 1. Sci. Rep. 2016, 6, 38174. [Google Scholar] [CrossRef] [PubMed]
- Koutsoulidou, A.; Photiades, M.; Kyriakides, T.C.; Georgiou, K.; Prokopi, M.; Kapnisis, K.; Lusakowska, A.; Nearchou, M.; Christou, Y.; Papadimas, G.K.; et al. Identification of exosomal muscle-specific miRNAs in serum of myotonic dystrophy patients relating to muscle disease progress. Hum. Mol. Genet. 2017, 26, 3285–3302. [Google Scholar] [CrossRef]
- Lötvall, J.; Hill, A.F.; Hochberg, F.; Buzás, E.I.; Di Vizio, D.; Gardiner, C.; Gho, Y.S.; Kurochkin, I.V.; Mathivanan, S.; Quesenberry, P.; et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: A position statement from the International Society for Extracellular Vesicles. J. Extracell. Vesicles 2014, 1, 1–6. [Google Scholar] [CrossRef]
- Karttunen, J.; Heiskanen, M.; Navarro-Ferrandis, V.; Das Gupta, S.; Lipponen, A.; Puhakka, N.; Rilla, K.; Koistinen, A.; Pitkänen, A. Precipitation-based extracellular vesicle isolation from rat plasma co-precipitate vesicle-free microRNAs. J. Extracell. Vesicles 2019, 8. [Google Scholar] [CrossRef]
- Fernandez-Costa, J.M.; Llamusi, B.; Bargiela, A.; Zulaica, M.; Alvarez-Abril, M.C.; Perez-Alonso, M.; De Munain, A.L.; Lopez-Castel, A.; Artero, R. Six serum miRNAs fail to validate as myotonic dystrophy type 1 biomarkers. PLoS ONE 2016, 11, e0150501. [Google Scholar] [CrossRef] [PubMed]
- Pegoraro, V.; Cudia, P.; Baba, A.; Angelini, C. MyomiRNAs and myostatin as physical rehabilitation biomarkers for myotonic dystrophy. Neurol. Sci. 2020, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, K.K.; Ishak, T.; Lian, L.H.; Goh, K.J.; Wong, K.T.; Ahmad-Annuar, A.; Thong, M.K. Deregulation of microRNAs in blood and skeletal muscles of myotonic dystrophy type 1 patients. Neurol. India 2017, 65, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.; Meese, E. Can circulating miRNAs live up to the promise of being minimal invasive biomarkers in clinical settings? Wiley Interdiscip. Rev. RNA 2016, 7, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Vignier, N.; Amor, F.; Fogel, P.; Duvallet, A.; Poupiot, J.; Charrier, S.; Arock, M.; Montus, M.; Nelson, I.; Richard, I.; et al. Distinctive Serum miRNA Profile in Mouse Models of Striated Muscular Pathologies. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Coenen-Stass, A.M.L.; Betts, C.A.; Lee, Y.F.; Mäger, I.; Turunen, M.P.; El Andaloussi, S.; Morgan, J.E.; Wood, M.J.A.; Roberts, T.C. Selective release of muscle-specific, extracellular microRNAs during myogenic differentiation. Hum. Mol. Genet. 2016, 25, 3960–3974. [Google Scholar] [CrossRef]
- Coenen-Stass, A.M.L.; Sork, H.; Gatto, S.; Godfrey, C.; Bhomra, A.; Krjutškov, K.; Hart, J.R.; Westholm, J.O.; O’Donovan, L.; Roos, A.; et al. Comprehensive RNA-Sequencing Analysis in Serum and Muscle Reveals Novel Small RNA Signatures with Biomarker Potential for DMD. Mol. Ther. Nucl. Acids 2018, 13, 1–15. [Google Scholar] [CrossRef]
- Zhang, B.W.; Cai, H.F.; Wei, X.F.; Sun, J.J.; Lan, X.Y.; Lei, C.Z.; Lin, F.P.; Qi, X.L.; Plath, M.; Chen, H. mir-30-5p regulates muscle differentiation and alternative splicing of muscle-related genes by targeting MBNL. Int. J. Mol. Sci. 2016, 17, 182. [Google Scholar] [CrossRef]
- Cerro-Herreros, E.; Sabater-Arcis, M.; Fernandez-Costa, J.M.; Moreno, N.; Perez-Alonso, M.; Llamusi, B.; Artero, R. MiR-23b and miR-218 silencing increase Muscleblind-like expression and alleviate myotonic dystrophy phenotypes in mammalian models. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Cerro-Herreros, E.; González-Martínez, I.; Moreno-Cervera, N.; Overby, S.; Pérez-Alonso, M.; Llamusí, B.; Artero, R. Therapeutic Potential of AntagomiR-23b for Treating Myotonic Dystrophy. Mol. Ther. Nucleic Acids 2020, 21, 837–849. [Google Scholar] [CrossRef]
- Koutalianos, D.; Koutsoulidou, A.; Mastroyiannopoulos, N.P.; Furling, D.; Phylactou, L.A. MyoD transcription factor induces myogenesis by inhibiting Twist-1 through miR-206. J. Cell Sci. 2015, 128, 3631–3645. [Google Scholar] [CrossRef] [PubMed]
- Sabater-Arcis, M.; Bargiela, A.; Furling, D.; Artero, R. miR-7 Restores Phenotypes in Myotonic Dystrophy Muscle Cells by Repressing Hyperactivated Autophagy. Mol. Ther. Nucleic Acids 2020, 19, 278–292. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Dong, L.; Zhang, J.; Zhao, Y.; Li, Z. Recent advances in microRNA detection. Analyst 2018, 143, 1758–1774. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Study | miRNAs | Muscle Biopsy | Sample Size | Normalization | Targets | Associated Pathological Signs and Mechanisms | |
|---|---|---|---|---|---|---|---|
| Gambardella 2010 [48] | miR-206 | ↑ | Vastus lateralis | 7 DM1 4 ctrls | Hsa-let-7a | ||
| Perbellini 2011 [51] | miR-1 * miR-29b/c ** miR-335 miR-33 | ↑ ↓ ↑ ↑ | Biceps branchi | 15 DM1 14 ctrls | miR-16 | MEF2a *,1 MET *,1 GATA6 *,1 HAND2 *,1 | muscle development 1 |
| KCNE1 *,2 KCNJ2 *,2 CALM2 *,2 | Arrhythmia 2 | ||||||
| SFRS9 *,3 | Splicing 3 | ||||||
| DAG1 **,4 DIABLO **,4 RET **TRIM63 **,4 TGFB3 **,4 | Atrophy 4 | ||||||
| Fernandez-Costa 2012 [52] | miR-1 *** miR-7 **** miR-10 ***** | ↓ ↓ ↓ | Vastus lateralis Biceps branchi Deltoid | 5 DM1 3 ctrls | Sno-RNA RNU48 | SOD1 ***,5 SMARCA4 ***,6 NET 1 ***,7 CTSB ****,8 ATG4 ****,9 VCL ****,10 UBE11 *****,11 | Free radical removal 5 Transcription 6 Apoptosis and signal transduction 7 Proteolysis 8 Autophagy 9 Cytoskeleton 10 Protein localization/activity regulation 11 |
| Fritegotto 2017 [53] | miR-1 miR-133a/b miR206 | ↓ ↓ ↑ | Vastus lateralis | 12 DM1 6 ctrls | Hsa-let-7a | ||
| Ambrose 2017 [52] | miR-1 miR-133a miR-29c | ↓ ↓ ↓ | Biceps branchi Gastrocnemius Deltoid | 9 DM1 9 ctrls | Hsa-let-7a | ||
| Study | miRNAs | Heart | Sample Size | Normalization | Targets | Associated Pathological Signs | |
|---|---|---|---|---|---|---|---|
| Rau 2011 [55] | miR-1 | ↓ | Left ventricle | 5 DM1 8 ctrls | U6 snRNA | CACNA1C 1 GJA1 2 | Arrhythmias 1 Conduction 2 |
| Kalsotra 2014 [56] | miR-1 * miR-133 ** miR-30b ** miR-29b ** miR-145 let-7g miR-26a miR-328 miR-23a miR-23b miR365 miR-499 miR-125b miR-148a miR-194 let-7d miR-27a miR-22 miR-100 | ↓ ↓ ↓ ↓ ↓ ↓ ↓ ↓ ↓ ↓ ↓ ↓ ↓ ↓ ↓ ↓ ↓ ↓ ↓ | unknown | 8 DM1 4 ctrls | MammU6 | Conduction * Fibrosis ** | |
| Study | miRNAs | Blood | Sample Size | Normalization | Associated Clinical Characteristics | |
|---|---|---|---|---|---|---|
| Perfetti 2014 [33] | miR-113a miR-193b miR-191 miR-454 miR-574 miR-885-5p miR-886-3p miR-27b | ↑ ↑ ↑ ↑ ↑ ↑ ↑ ↓ | Plasma | 36 DM1 36 ctrls | Cel-miR-39 miR-17-5p miR-106a | Negative correlation to MRC score Positive correlation to MIRS scale |
| Koutsoulidou 2015 [34] | miR-1 miR-133a/b miR-206 | ↑ ↑ ↑ | Serum | 23 DM1 23 ctrls | miR-16 | Increased levels in progressive muscle wasting compared to stable muscle wasting |
| Perfetti 2016 [58] | miR-1 miR-133a/b miR-206 miR-140-3p miR-454 miR-574 miR-27b | ↑ ↑ ↑ ↑ ↑ ↑ ↓ | Plasma | 103 DM1 111 ctrls | Cel-miR-39 miR-17-5p miR-106a | Negative correlation to muscle strength Non-significant correlation to CK levels |
| Koutsoulidou 2017 [59] | miR-1 miR-133a/b miR-206 miR-113a miR-193b miR-191 miR-574 miR-885-5p miR-886-3p miR-27b | ↑ ↑ ↑ ↑ ↑ ↑ ↑ ↑ ↑ ↑ | Serum | 63 DM1 63 ctrls | miR-16 | Increased levels in progressive muscle wasting compared to stable muscle wasting |
| Pegoraro 2020 [51] | miR-1 miR-133a/b miR-206 | ↑ ↑* ↑ | Serum | 9 DM1 7 ctrls | miR-39-3p C. elegans | After a six-week exercise training the 4 miRNAs significantly decreased in parallel with improved muscle function |
| Ambrose 2017 [52] | miR-133a miR-29b miR-33a | ↑ ↑ ↑ | Whole blood | 10 DM1 10 ctrls | Hsa-miR-183 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koehorst, E.; Ballester-Lopez, A.; Arechavala-Gomeza, V.; Martínez-Piñeiro, A.; Nogales-Gadea, G. The Biomarker Potential of miRNAs in Myotonic Dystrophy Type I. J. Clin. Med. 2020, 9, 3939. https://doi.org/10.3390/jcm9123939
Koehorst E, Ballester-Lopez A, Arechavala-Gomeza V, Martínez-Piñeiro A, Nogales-Gadea G. The Biomarker Potential of miRNAs in Myotonic Dystrophy Type I. Journal of Clinical Medicine. 2020; 9(12):3939. https://doi.org/10.3390/jcm9123939
Chicago/Turabian StyleKoehorst, Emma, Alfonsina Ballester-Lopez, Virginia Arechavala-Gomeza, Alicia Martínez-Piñeiro, and Gisela Nogales-Gadea. 2020. "The Biomarker Potential of miRNAs in Myotonic Dystrophy Type I" Journal of Clinical Medicine 9, no. 12: 3939. https://doi.org/10.3390/jcm9123939
APA StyleKoehorst, E., Ballester-Lopez, A., Arechavala-Gomeza, V., Martínez-Piñeiro, A., & Nogales-Gadea, G. (2020). The Biomarker Potential of miRNAs in Myotonic Dystrophy Type I. Journal of Clinical Medicine, 9(12), 3939. https://doi.org/10.3390/jcm9123939