Difficult Cases of Autoimmune Hemolytic Anemia: A Challenge for the Internal Medicine Specialist



Abstract
1. Introduction
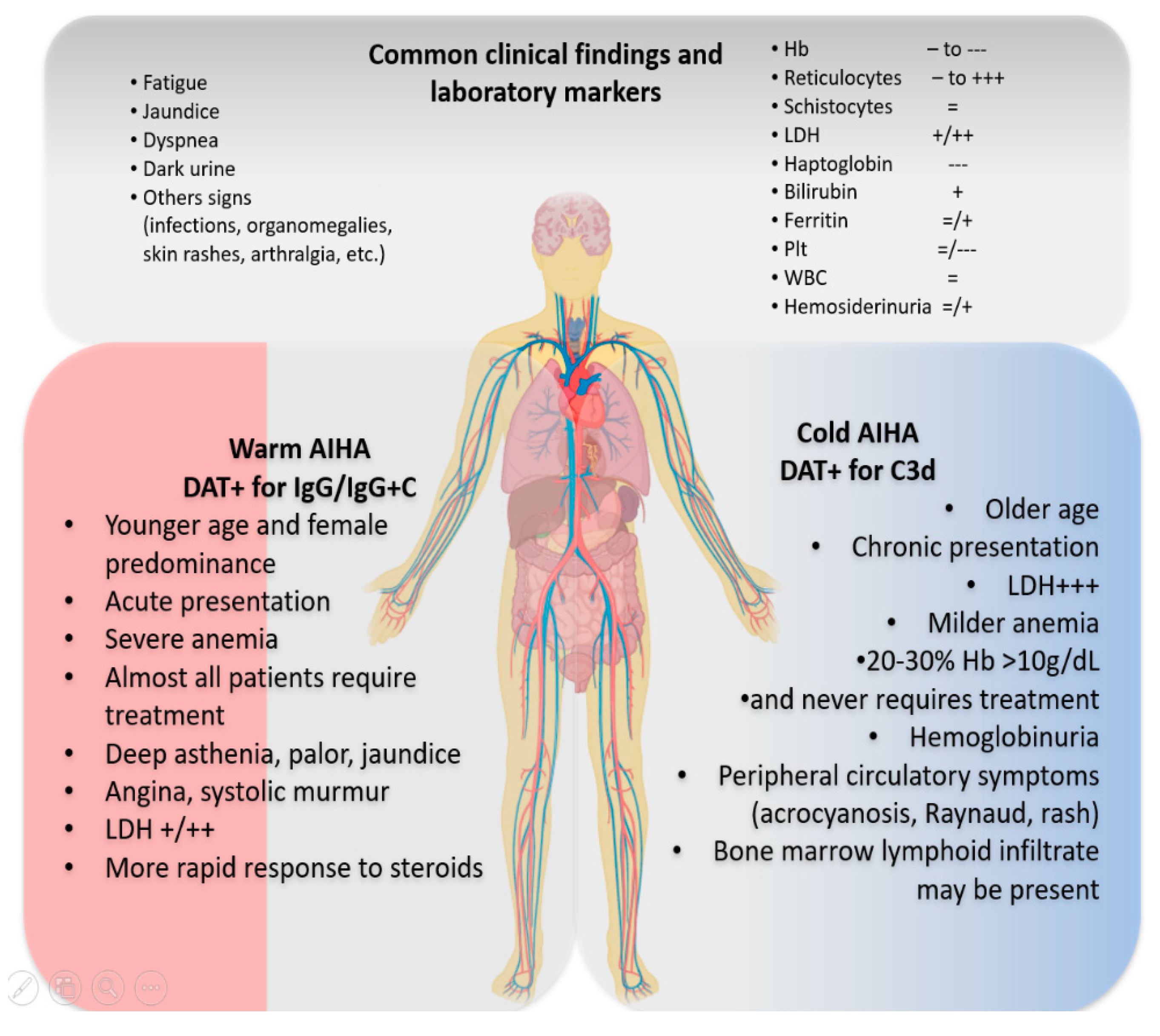
2. Typical AIHA Presentation and Differential Diagnosis
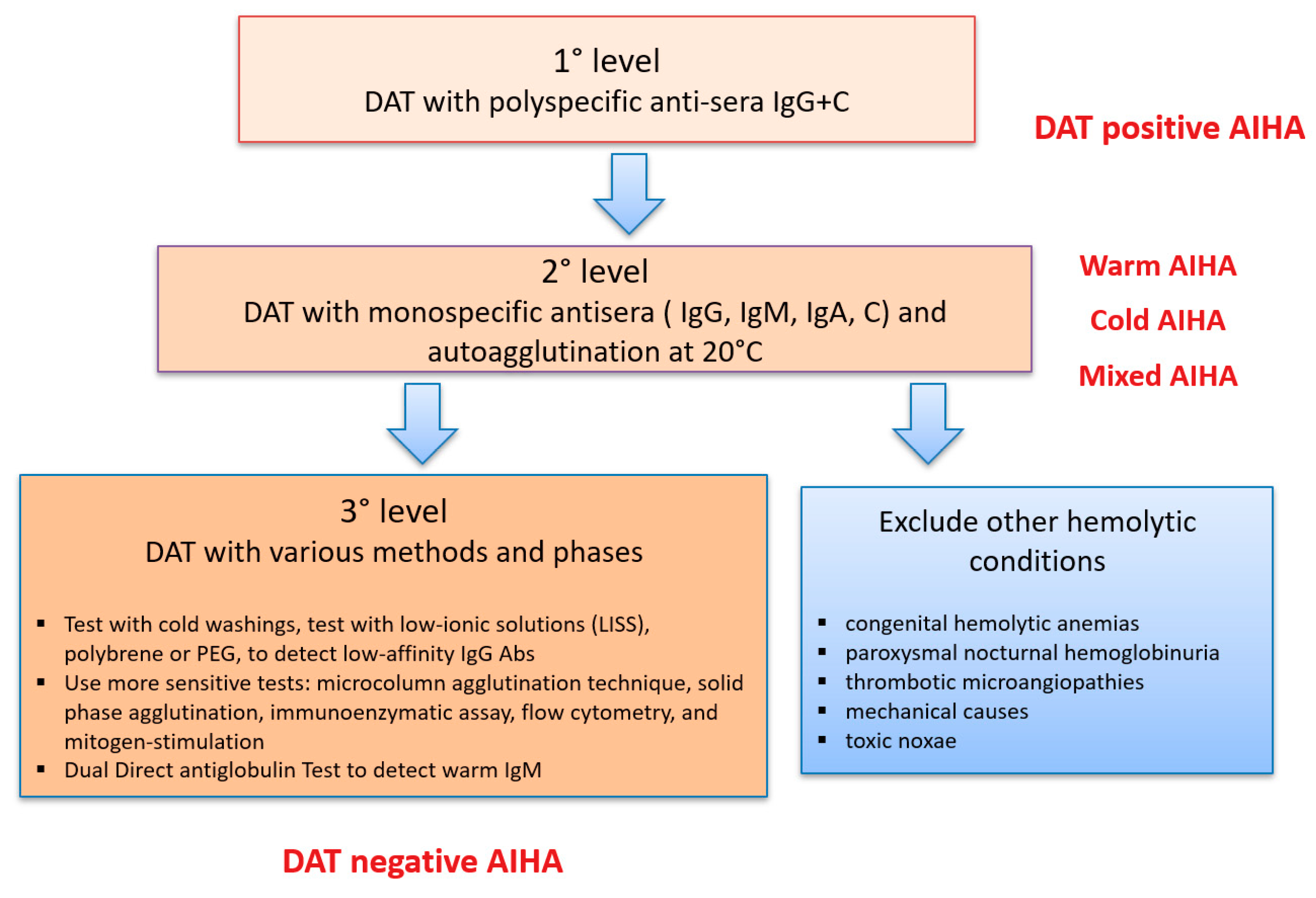
3. What If DAT is Negative?
4. What Secondary Settings Should Be Taken into Account?
5. Peculiar AIHA Presentations Deserving Specific Attention and Therapy
5.1. AIHA in the Intensive Care Unit (ICU) and Hyperacute Hemolysis
5.2. AIHA with Reticulocytopenia
5.3. AIHA in Pregnancy
5.4. AIHA after Novel Anti-Cancer Therapies
5.5. Post-Transplant AIHA
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gehrs, B.C.; Friedberg, R.C. Autoimmune hemolytic anemia. Am. J. Hematol. 2002, 69, 258–271. [Google Scholar] [CrossRef] [PubMed]
- Sokol, R.J.; Hewitt, S.; Stamps, B.K. Autoimmune haemolysis: An 18-year study of 865 cases referred to a regional transfusion centre. Br. Med. J. (Clin. Res. Ed.) 1981, 282, 2023–2027. [Google Scholar] [CrossRef] [PubMed]
- Berentsen, S. How I manage patients with cold agglutinin disease. Br. J. Haematol. 2018, 181, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Aladjidi, N.; Jutand, M.A.; Beaubois, C.; Fernandes, H.; Jeanpetit, J.; Coureau, G.; Gilleron, V.; Kostrzewa, A.; Lauroua, P.; Jeanne, M.; et al. Reliable assessment of the incidence of childhood autoimmune hemolytic anemia. Pediatr. Blood Cancer 2017, 64. [Google Scholar] [CrossRef] [PubMed]
- Keeling, D.M.; Isenberg, D.A. Haematological manifestations of systemic lupus erythematosus. Blood Rev. 1993, 7, 199–207. [Google Scholar] [CrossRef]
- Newman, K.; Owlia, M.B.; El-Hemaidi, I.; Akhtari, M. Management of immune cytopenias in patients with systemic lupus erythematosus—Old and new. Autoimmun. Rev. 2013, 12, 784–791. [Google Scholar] [CrossRef]
- Hodgson, K.; Ferrer, G.; Pereira, A.; Moreno, C.; Montserrat, E. Autoimmune cytopenia in chronic lymphocytic leukaemia: Diagnosis and treatment. Br. J. Haematol. 2011, 154, 14–22. [Google Scholar] [CrossRef]
- Borthakur, G.; O’Brien, S.; Wierda, W.G.; Thomas, D.A.; Cortes, J.E.; Giles, F.J.; Kantarjian, H.M.; Lerner, S.; Keating, M.J. Immune anaemias in patients with chronic lymphocytic leukaemia treated with fludarabine, cyclophosphamide and rituximab–incidence and predictors. Br. J. Haematol. 2007, 136, 800–805. [Google Scholar] [CrossRef]
- Barcellini, W.; Fattizzo, B. Clinical applications of hemolytic markers in the differential diagnosis and management of hemolytic anemia. Dis. Markers 2015, 2015, 635670. [Google Scholar] [CrossRef]
- Petz, L.D.; Garratty, G. Immune Hemolytic Anemias, 2nd ed.; Churchill Livingstone: Philadelphia, PA, USA, 2004. [Google Scholar]
- Barcellini, W.; Fattizzo, B.; Zaninoni, A.; Radice, T.; Nichele, I.; Di Bona, E.; Lunghi, M.; Tassinari, C.; Alfinito, F.; Ferrari, A.; et al. Clinical heterogeneity and predictors of outcome in primary autoimmune hemolytic anemia: A GIMEMA study of 308 patients. Blood 2014, 124, 2930–2936. [Google Scholar] [CrossRef]
- Barcellini, W.; Zaninoni, A.; Fattizzo, B.; Giannotta, J.A.; Lunghi, M.; Ferrari, A.; Leporace, A.P.; Maschio, N.; Scaramucci, L.; Cantoni, S.; et al. Predictors of refractoriness to therapy and healthcare resource utilization in 378 patients with primary autoimmune hemolytic anemia from eight Italian reference centers. Am. J. Hematol. 2018, 93, E243–E246. [Google Scholar] [CrossRef] [PubMed]
- Jäger, U.; Barcellini, W.; Broome, C.M.; Gertz, M.A.; Hill, A.; Hill, Q.A.; Jilma, B.; Kuter, D.J.; Michel, M.; Montillo, M.; et al. Diagnosis and treatment of autoimmune hemolytic anemia in adults: Recommendations from the First International Consensus Meeting. Blood Rev. 2019, 5, 100648. [Google Scholar] [CrossRef] [PubMed]
- Barcellini, W. Immune hemolysis: Diagnosis and treatment recommendations. Semin. Hematol. 2015, 52, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Hill, Q.A.; Stamps, R.; Massey, E.; Grainger, J.D.; Provan, D.; Hill, A. Guidelines on the management of drug-induced immune and secondary autoimmune, haemolytic anaemia. Br. J. Haematol. 2017, 177, 208–220. [Google Scholar] [CrossRef]
- Feuille, E.J.; Anooshiravani, N.; Sullivan, K.E.; Fuleihan, R.L.; Cunningham-Rundles, C. Autoimmune cytopenias and associated conditions in CVID: A report from the USIDNET Registry. J. Clin. Immunol. 2018, 38, 28–34. [Google Scholar] [CrossRef]
- Bariş, H.E.; Kiykim, A.; Nain, E.; Özen, A.O.; Karakoç-Aydiner, E.; Bariş, S. The plethora, clinical manifestations and treatment options of autoimmunity in patients with primary immunodeficiency. Turk. Pediatri. ARS 2016, 51, 186–192. [Google Scholar] [CrossRef]
- Barcellini, W.; Giannotta, J.; Fattizzo, B. Autoimmune hemolytic anemia in adults: Primary risk factors and diagnostic procedures. Expert Rev. Hematol. 2020, 13, 585–597. [Google Scholar] [CrossRef]
- Fattizzo, B.; Barcellini, W. Autoimmune cytopenias in chronic lymphocytic leukemia: Focus on molecular aspects. Front. Oncol. 2020, 9, 1435. [Google Scholar] [CrossRef]
- Abdel-Khalik, A.; Paton, L.; White, A.G.; Urbaniak, S.J. Human leucocyte antigens A, B, C, and DRW in idiopathic “warm” autoimmune haemolytic anaemia. Br. Med. J. 1980, 280, 760–761. [Google Scholar] [CrossRef]
- Wang-Rodriguez, J.; Rearden, A. Reduced frequency of HLA-DQ6 in individuals with a positive direct antiglobulin test. Transfusion 1996, 3, 979–984. [Google Scholar] [CrossRef]
- Visco, C.; Barcellini, W.; Maura, F.; Neri, A.; Cortelezzi, A.; Rodeghiero, F. Autoimmune cytopenias in chronic lymphocytic leukemia. Am. J. Hematol. 2014, 89, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Maura, F.; Cutrona, G.; Fabris, S.; Colombo, M.; Tuana, G.; Agnelli, L.; Matis, S.; Lionetti, M.; Gentile, M.; Recchia, A.G.; et al. Relevance of stereotyped B-cell receptors in the context of the molecular, cytogenetic and clinical features of chronic lymphocytic leukemia. PLoS ONE 2011, 6, e24313. [Google Scholar] [CrossRef][Green Version]
- Maura, F.; Visco, C.; Falisi, E.; Reda, G.; Fabris, S.; Agnelli, L.; Tuana, G.; Lionetti, M.; Guercini, N.; Novella, E.; et al. B-cell receptor configuration and adverse cytogenetics are associated with autoimmune hemolytic anemia in chronic lymphocytic leukemia. Am. J. Hematol. 2013, 88, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Berentsen, S.; Bø, K.; Shammas, F.V.; Myking, A.O.; Ulvestad, E. Chronic cold agglutinin disease of the “idiopathic” type is a premalignant or low- grade malignant lymphoproliferative disease. APMIS 1997, 105, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Lafarge, A.; Bertinchamp, R.; Pichereau, C.; Valade, S.; Chermak, A.; Theodose, I.; Canet, E.; Lemiale, V.; Schlemmer, B.; Galicier, L.; et al. Prognosis of autoimmune hemolytic anemia in critically ill patients. Ann. Hematol. 2019, 98, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Fattizzo, B.; Zaninoni, A.; Nesa, F.; Sciumbata, V.M.; Zanella, A.; Cortelezzi, A.; Barcellini, W. Lessons from very severe, refractory, and fatal primary autoimmune hemolytic anemias. Am. J. Hematol. 2015, 90, E149–E151. [Google Scholar] [CrossRef]
- Fattizzo, B.; Zaninoni, A.; Gianelli, U.; Zanella, A.; Cortelezzi, A.; Kulasekararaj, A.G.; Barcellini, W. Prognostic impact of bone marrow fibrosis and dyserythropoiesis in autoimmune hemolytic anemia. Am. J. Hematol. 2018, 93, E88–E91. [Google Scholar] [CrossRef]
- Fattizzo, B.; Michel, M.; Giannotta, J.; Guillet, S.; Frederiksen, H.; Vos, J.M.I.; Mauro, F.R.; Jilma, B.; Patriarca, A.; Zaja, F.; et al. Efficacy of recombinant erythropoietin in autoimmune hemolytic anemia: A multicenter international study. Haematologica 2020. [Google Scholar] [CrossRef]
- Sokol, R.J.; Hewitt, S.; Stamps, B.K. Erythrocyte autoantibodies, autoimmune haemolysis and pregnancy. Vox Sang 1982, 43, 169–176. [Google Scholar] [CrossRef]
- Ng, S.C.; Wong, K.K.; Raman, S.; Bosco, J. Autoimmune haemolytic anaemia in pregnancy: A case report. Eur. J. Obstet. Gynecol. Reprod. Biol. 1990, 37, 83–85. [Google Scholar] [CrossRef]
- Agapidou, A.; Vlachaki, E.; Theodoridis, T.; Economou, M.; Perifanis, V. Cyclosporine therapy during pregnancy in a patient with β-thalassemia major and autoimmune haemolytic anemia: A case report and review of the literature. Hippokratia 2013, 17, 85–87. [Google Scholar] [PubMed]
- Dhingra, S.; Wiener, J.I.; Jackson, H. Management of cold agglutinin immune hemolytic anemia in pregnancy. Obstet. Gynecol. 2007, 110 Pt 2, 485–486. [Google Scholar] [CrossRef]
- Batalias, L.; Trakakis, E.; Loghis, C.; Salabasis, C.; Simeonidis, G.; Karanikolopoulos, P.; Kassanos, D.; Salamalekis, E. Autoimmune hemolytic anemia caused by cold agglutinins in a young pregnant woman. J. Matern. Fetal. Neonatal. Med. 2006, 19, 251–253. [Google Scholar] [CrossRef] [PubMed]
- Lefkou, E.; Nelson-Piercy, C.; Hunt, B.J. Evans’ syndrome in pregnancy: A systematic literature review and two new cases. Eur. J. Obstet. Gynecol. Reprod. Biol. 2010, 149, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Grigoriadis, C.; Tympa, A.; Liapis, A.; Hassiakos, D.; Bakas, P. Alpha-methyldopa-induced autoimmune hemolytic anemia in the third trimester of pregnancy. Case Rep. Obstet. Gynecol. 2013, 2013, 150278. [Google Scholar] [CrossRef] [PubMed]
- Benraad, C.E.; Scheerder, H.A.; Overbeeke, M.A. Autoimmune haemolytic anaemia during pregnancy. Eur. J. Obstet. Gynecol. Reprod. Biol. 1994, 55, 209–211. [Google Scholar] [CrossRef]
- Kumar, R.; Advani, A.R.; Sharan, J.; Basharutallah, M.S.; Al-Lumai, A.S. Pregnancy induced hemolytic anemia: An unexplained entity. Ann. Hematol. 2001, 80, 623–626. [Google Scholar] [CrossRef]
- Katsuragi, S.; Sameshima, H.; Omine, M.; Ikenoue, T. Pregnancy-induced hemolytic anemia with a possible immune-related mechanism. Obstet. Gynecol. 2008, 111, 528–529. [Google Scholar] [CrossRef]
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune-related adverse events associated with immune checkpoint blockade. N. Engl. J. Med. 2018, 378, 158–168. [Google Scholar] [CrossRef]
- Zhuang, J.; Du, J.; Guo, X.; Zhou, J.; Duan, L.; Qiu, W.; Si, X.; Zhang, L.; Li, Y.; Liu, X.; et al. Clinical diagnosis and treatment recommendations for immune checkpoint inhibitor- related hematological adverse events. Thorac. Cancer 2020, 11, 799–804. [Google Scholar] [CrossRef]
- Petrelli, F.; Ardito, R.; Borgonovo, K.; Lonati, V.; Cabiddu, M.; Ghilardi, M.; Barni, S. Haematological toxicities with immunotherapy in patients with cancer: A systematic review and meta-analysis. Eur. J. Cancer 2018, 103, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Tanios, G.E.; Doley, P.B.; Munker, R. Autoimmune hemolytic anemia associated with the use of immune checkpoint inhibitors for cancer: 68 cases from the Food and Drug Administration database and review. Eur. J. Haematol. 2019, 102, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Leaf, R.K.; Ferreri, C.; Rangachari, D.; Mier, J.; Witteles, W.; Ansstas, G.; Anagnostou, T.; Zubiri, L.; Piotrowska, Z.; Oo, T.H.; et al. Clinical and laboratory features of autoimmune hemolytic anemia associated with immune checkpoint inhibitors. Am. J. Hematol. 2019, 94, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Barcellini, W.; Fattizzo, B.; Zaninoni, A. Management of refractory autoimmune hemolytic anemia after allogeneic hematopoietic stem cell transplantation: Current perspectives. J. Blood Med. 2019, 10, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Skeate, R.; Singh, C.; Cooley, S.; Geller, M.; Northouse, J.; Welbig, J.; Slungaard, A.; Miller, J.; McKenna, D. Hemolytic anemia due to passenger lymphocyte syndrome in solid malignancy patients treated with allogeneic natural killer cell products. Transfusion 2013, 53, 419–423. [Google Scholar] [CrossRef]
- Kruizinga, M.D.; van Tol, M.J.D.; Bekker, V.; Netelenbos, T.; Smiers, F.J.; Bresters, D.; Jansen-Hoogendijk, A.M.; van Ostaijen-Ten Dam, M.M.; Kollen, W.J.W.; Zwaginga, J.J.; et al. Risk factors, treatment, and immune dysregulation in autoimmune cytopenia after allogeneic hematopoietic stem cell transplantation in pediatric patients. Biol. Blood Marrow Transpl. 2018, 24, 772–778. [Google Scholar] [CrossRef]
- González-Vicent, M.; Sanz, J.; Fuster, J.L.; Cid, J.; de Heredia, C.D.; Morillo, D.; Fernández, J.M.; Pascual, A.; Badell, I.; Serrano, D.; et al. Autoimmune hemolytic anemia (AIHA) following allogeneic hematopoietic stem cell transplantation (HSCT): A retrospective analysis and a proposal of treatment on behalf of the Grupo Español De Trasplante de Medula Osea en Niños (GETMON) and the Grupo Español de Trasplante Hematopoyetico (GETH). Transfus. Med. Rev. 2018, 32, 179–185. [Google Scholar] [CrossRef]
- Barcellini, W.; Fattizzo, B. The changing landscape of autoimmune hemolytic anemia. Front. Immunol. 2020, 11, 946. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Exogenous Predisposing Factors | ||
|---|---|---|
| Infections | Frequency | Comments |
| Parvovirus B19; HCV; HAV; HBV; HIV Mycoplasma spp.; Tuberculosis; Babesiosis; Brucellosis; Syphilis; EBV; Respiratory Syncytial Virus | 0.02% to 20% | ParvoB19 infection and HCV and its treatment correlate with AIHA development; case reports of association with AIHA are available for the other infectious agents. |
| Drugs | ||
| Antibiotics (penicillins, cephalosporins, etc.), cytotoxic drugs (oxaliplatin, etc.), antidiabetics (metformin), anti-inflammatory drugs (diclofenac, etc.), neurologic drugs (α-methyldopa, L-dopa, chlorpromazine, etc.), cardiologic drugs (procainamide, etc.) | Case reports and reviews | Various mechanisms are demonstrated: hapten and drug absorption mechanisms; Immune/ternary complex mechanisms; autoantibody mechanism; non-immunologic protein formation; unknown mechanisms. |
| CLL therapy: fludarabine and Tyrosine kinase inhibitors | 6–21% | Fludarabine induced AIHA may be avoided by rituximab association. Ibrutinib was associated to low risk of AIHA development in registrative trials in CLL |
| Vaccines | ||
| Vaccines | 0.8/100,000 person-years | AIHA was the rarest autoimmune complication in a population study |
| Endogenous predisposing factors | ||
| Congenital Syndromes and Immunodeficiencies | ||
| Kabuki syndrome and Hemoglobinopathies | 4–6% | AIHA and ITP are the most frequent autoimmune complications of Kabuki Syndrome; DAT positivity is frequent, but clinically overt AIHA is rarer in thalassemia (particularly beta intermedia, alloimmunized and transfused pts) |
| ALPS; CVID; IgA deficiency | 2–70% | AIHA is the most frequent autoimmune complication together with ITP and ES |
| Genes involved in PIDsTNFRSF6, CTLA4, STAT3, PIK3CD, CBL, ADAR1, LRBA, RAG1, and KRAS | 40% of pediatric ES | Majority of pediatric ES display somatic mutations found in immunodeficiencies |
| Genetic Findings | ||
| HLA I and II | Case series | HLA-B8 and BW6 are strongly associated to wAIHA. |
| IGHV and IGKV region | >60% cAIHA | Specific IGVH and IGKV regions are related to AIHA development |
| TCRG and TCRB | 50% | Pathogenic T-cells are clonally restricted in AIHA |
| CTLA-4 exon 1 | 73% | CTLA-4 signaling is defective in AIHA, particularly in CLL cases |
| Cytokine polymorphisms | 41% | AIHA shows higher frequency of LT-α (+252) AG phenotype |
| KMT2D and CARD11 | 69% and 31% of cAIHA tested | Autoreactive B-cells display somatic mutations favoring proliferation |
| Mixed (host and environmental) predisposing factors | ||
| Autoimmune Diseases | ||
| SLE, Systemic sclerosis; autoimmune thyroiditis; Sjogren Syndrome; IBDs; Autoimmune hepatitis/Primary biliary cirrhosis | 1.4–14% | AIHA frequency is higher in pediatric than in adult patients with SLE. AIHA may be rarely associated to systemic sclerosis or Sjogren syndrome, Hashimoto thyroiditis and Graves’ disease, ulcerative colitis, and autoimmune hepatitis. |
| Lymphoproliferative Disorders | Frequency | Results |
| Chronic lymphoid leukemia and NHL | 5–20% | Autoimmune cytopenias may frequently complicate chronic lymphoproliferative disorders and usually correlate with advanced disease and high biologic risk (unmutated IGHV status, stereotyped IGHV frames, and chromosome 17p and/or 11q deletions) |
| Solid Cancers | ||
| Thymoma; Ovarian/Prostate | 1.29–30% autoimmune phenomena | Thymoma and prostate and ovarian carcinomas have the highest association with autoimmunity |
| Setting | Clinical Presentation | Diagnostic Issues | Therapeutic Hints | Reference |
|---|---|---|---|---|
| AIHA in the intensive care unit 0.05% of primary AIHA cases | Very severe anemia Massive hemolysis Unresponsiveness to transfusion Multi-organ failure | Factors associated with severity are concomitant immune thrombocytopenia, severe infections, and thrombosis | Intensive support with transfusion, along with steroid boli, iv Ig, rituximab, erythropoietin, and plasma-exchange may be required | [11,12,26,27] |
| AIHA with reticulocytopenia 20% of AIHA cases | DAT positive AIHA with inadequate reticulocytosis typical of patients with severe or very severe AIHA | BMRI < 121 s [(absolute reticulocyte count) × (patient’ Hb/normal Hb)] Bone marrow mandatory to exclude underlying lymphoid or myeloid neoplasm MS-DAT may show anti-erythroblasts autoantibodies Endogenous EPO levels generally inadequate | Along with AIHA treatment recombinant EPO, i.e., alpha-epoetin 40,000 UI/week subcutaneously may be effective in up to 70% of cases | [13,28,29] |
| AIHA in pregnancy 1 in 50,000 pregnancies | Acute Hb drop during pregnancy or after delivery with alteration of hemolytic markers | Exclude HELLP and thrombotic microangiopathies DAT negative cases are also described | Typically responds to conventional treatment (steroids and iv Ig) and often resolves after delivery Evaluate thrombotic riskConsider that the newborn may be DAT+ in wAIHA since IgG cross the placenta | [30,31,32,33,34,35,36,37,38,39] |
| Novel anti-cancer drugs | Mono- or bilineage cytopenia | History of therapy with anti-PD1, anti-PDL1, or anti-CTLA4 MoAb Most of cases IgG wAIHA DAT may be negative in up to 38% of cases | Prednisone 1.5 mg to 2 mg/kg per day along with CPIs discontinuation is recommended | [40,41,42,43,44] |
| Post-transplant AIHA Passenger lymphocyte syndrome 9–70% of solid organ transplant | 3 to 24 days post-transplant transient hemolysis | Mainly group O donors, though few cases have been described in AB recipients with non-AB donors risk of hemolysis ranging from 9 to 70% (kidney < liver < heart-lung transplants) | The syndrome is self-limiting and usually requires supportive treatment only | [15] |
| Hematopoietic stem cell transplant (HSCT) 2–4% of HSCTs | Severe immune hemolysis 3–10 months post-HSCT | Warm forms develop later (6–18 months) than cold forms (2–8 months) | Steroids are effective only in 20%, early rituximab is advised, case reports of novel drugs have been described | [45,46,47] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fattizzo, B.; Giannotta, J.A.; Serpenti, F.; Barcellini, W. Difficult Cases of Autoimmune Hemolytic Anemia: A Challenge for the Internal Medicine Specialist. J. Clin. Med. 2020, 9, 3858. https://doi.org/10.3390/jcm9123858
Fattizzo B, Giannotta JA, Serpenti F, Barcellini W. Difficult Cases of Autoimmune Hemolytic Anemia: A Challenge for the Internal Medicine Specialist. Journal of Clinical Medicine. 2020; 9(12):3858. https://doi.org/10.3390/jcm9123858
Chicago/Turabian StyleFattizzo, Bruno, Juri Alessandro Giannotta, Fabio Serpenti, and Wilma Barcellini. 2020. "Difficult Cases of Autoimmune Hemolytic Anemia: A Challenge for the Internal Medicine Specialist" Journal of Clinical Medicine 9, no. 12: 3858. https://doi.org/10.3390/jcm9123858
APA StyleFattizzo, B., Giannotta, J. A., Serpenti, F., & Barcellini, W. (2020). Difficult Cases of Autoimmune Hemolytic Anemia: A Challenge for the Internal Medicine Specialist. Journal of Clinical Medicine, 9(12), 3858. https://doi.org/10.3390/jcm9123858