Potential Second-Hits in Hereditary Hemorrhagic Telangiectasia



Abstract
{kind=link}
{kind=link}
{kind=link}
1. Clinical Characteristics of HHT
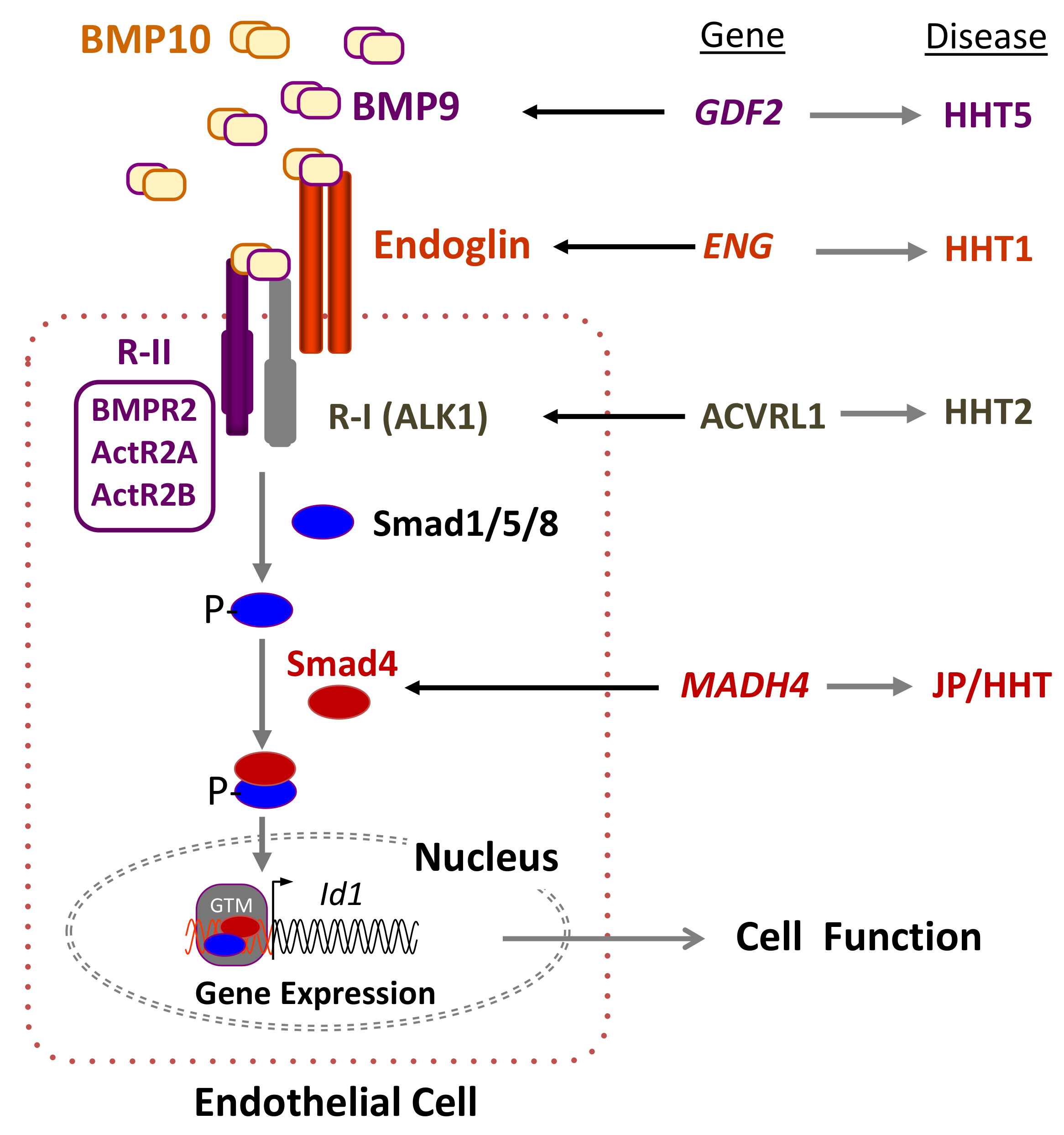
2. Genetics of HHT: The Germline Mutation
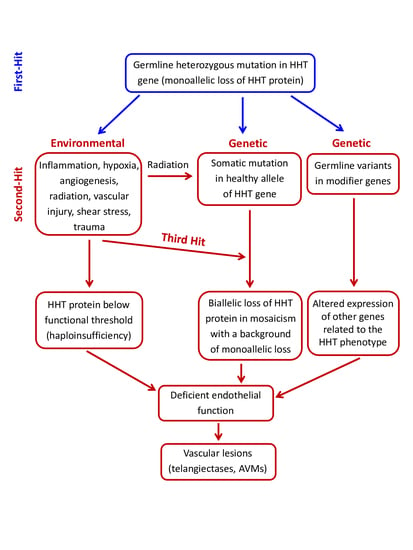
3. Pathogenic Mechanisms in HHT: The Second-Hit Hypothesis
3.1. Environmental Second-Hits
3.1.1. Mechanical and Light-Induced Triggers
3.1.2. Modulators of Endothelial Function
VEGF-Dependent Angiogenic Stimuli
ALK1 Signaling and the Notch Pathway
Proliferation and Apoptosis Stimuli
Inflammation and Endothelial Cell Adhesion and Nitric Oxide Regulation
3.2. Genetic Second-Hits
3.2.1. Germline Modifier Variants/Genes
3.2.2. Somatic Mutations in the Second Allele of the HHT Genes
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Plauchu, H.; de Chadarevian, J.P.; Bideau, A.; Robert, J.M. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am. J. Med. Genet. 1989, 32, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Faughnan, M.E.; Palda, V.A.; Garcia-Tsao, G.; Geisthoff, U.W.; McDonald, J.; Proctor, D.D.; Spears, J.; Brown, D.H.; Buscarini, E.; Chesnutt, M.S.; et al. International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. J. Med. Genet. 2011, 48, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Faughnan, M.E.; Mager, J.J.; Hetts, S.W.; Palda, V.A.; Lang-Robertson, K.; Buscarini, E.; Deslandres, E.; Kasthuri, R.S.; Lausman, A.; Poetker, D.; et al. Second International Guidelines for the Diagnosis and Management of Hereditary Hemorrhagic Telangiectasia. Ann. Intern. Med. 2020. Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Cottin, V.; Chinet, T.; Lavolé, A.; Corre, R.; Marchand, E.; Reynaud-Gaubert, M.; Plauchu, H.; Cordier, J.F.; Groupe d’Etudes et de Recherche sur les Maladies “Orphelines” Pulmonaires (GERM”O”P). Pulmonary arteriovenous malformations in hereditary hemorrhagic telangiectasia: A series of 126 patients. Medicine 2007, 86, 1–17. [Google Scholar] [CrossRef]
- Dupuis-Girod, S.; Cottin, V.; Shovlin, C.L. The Lung in Hereditary Hemorrhagic Telangiectasia. Respiration 2017, 94, 315–330. [Google Scholar] [CrossRef]
- Majumdar, S.; McWilliams, J.P. Approach to Pulmonary Arteriovenous Malformations: A Comprehensive Update. J. Clin. Med. 2020, 9, 1927. [Google Scholar] [CrossRef]
- Ianora, A.A.; Memeo, M.; Sabba, C.; Cirulli, A.; Rotondo, A.; Angelelli, G. Hereditary hemorrhagic telangiectasia: Multi-detector row helical CT assessment of hepatic involvement. Radiology 2004, 230, 250–259. [Google Scholar] [CrossRef]
- Memeo, M.; Stabile Ianora, A.A.; Scardapane, A.; Suppressa, P.; Cirulli, A.; Sabbà, C.; Rotondo, A.; Angelelli, G. Hereditary haemorrhagic telangiectasia: Study of hepatic vascular alterations with multi-detector row helical CT and reconstruction programs. Radiol. Med. 2005, 109, 125–138. [Google Scholar] [PubMed]
- Buscarini, E.; Danesino, C.; Olivieri, C.; Lupinacci, G.; De Grazia, F.; Reduzzi, L.; Blotta, P.; Gazzaniga, P.; Pagella, F.; Grosso, M.; et al. Doppler ultrasonographic grading of hepatic vascular malformations in hereditary hemorrhagic telangiectasia—Results of extensive screening. Ultraschall Med. 2004, 25, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Tsao, G.; Korzenik, J.R.; Young, L.; Henderson, K.J.; Jain, D.; Byrd, B.; Pollak, J.S.; White, R.I., Jr. Liver disease in patients with hereditary hemorrhagic telangiectasia. N. Engl. J. Med. 2000, 343, 931–936. [Google Scholar] [CrossRef]
- Kim, H.; Nelson, J.; Krings, T.; terBrugge, K.G.; McCulloch, C.E.; Lawton, M.T.; Young, W.L.; Faughnan, M.E.; Brain Vascular Malformation Consortium HHT Investigator Group. Hemorrhage rates from brain arteriovenous malformation in patients with hereditary hemorrhagic telangiectasia. Stroke 2015, 46, 1362–1364. [Google Scholar] [CrossRef] [PubMed]
- Brinjikji, W.; Iyer, V.N.; Yamaki, V.; Lanzino, G.; Cloft, H.J.; Thielen, K.R.; Swanson, K.L.; Wood, C.P. Neurovascular Manifestations of Hereditary Hemorrhagic Telangiectasia: A Consecutive Series of 376 Patients during 15 Years. AJNR Am. J. Neuroradiol. 2016, 37, 1479–1486. [Google Scholar] [CrossRef]
- Shovlin, C.L.; Guttmacher, A.E.; Buscarini, E.; Faughnan, M.E.; Hyland, R.H.; Westermann, C.J.; Kjeldsen, A.D.; Plauchu, H. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am. J. Med. Genet. 2000, 91, 66–67. [Google Scholar] [CrossRef]
- McAllister, K.A.; Grogg, K.M.; Johnson, D.W.; Gallione, C.J.; Baldwin, M.A.; Jackson, C.E.; Helmbold, E.A.; Markel, D.S.; McKinnon, W.C.; Murrel, J.; et al. Endoglin, a TGF-β binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat. Genet. 1994, 8, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.W.; Berg, J.N.; Baldwin, M.A.; Gallione, C.J.; Marondel, I.; Yoon, S.J.; Stenzel, T.T.; Speer, M.; Pericak-Vance, M.A.; Diamond, A.; et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type. Nat. Genet. 1996, 13, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Gallione, C.J.; Repetto, G.M.; Legius, E.; Rustgi, A.K.; Schelley, S.L.; Tejpar, S.; Mitchell, G.; Drouin, E.; Westermann, C.J.; Marchuk, D.A. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet 2004, 363, 852–859. [Google Scholar] [CrossRef]
- Wooderchak-Donahue, W.L.; McDonald, J.; O’Fallon, B.; Upton, P.D.; Li, W.; Roman, B.L.; Young, S.; Plant, P.; Fülöp, G.T.; Langa, C.; et al. BMP9 mutations cause a vascular-anomaly syndrome with phenotypic overlap with hereditary hemorrhagic telangiectasia. Am. J. Hum. Genet. 2013, 93, 530–537. [Google Scholar] [CrossRef]
- Cole, S.G.; Begbie, M.E.; Wallace, G.M.; Shovlin, C.L. A new locus for hereditary haemorrhagic telangiectasia (HHT3) maps to chromosome 5. J. Med. Genet. 2005, 42, 577–582. [Google Scholar] [CrossRef]
- Bayrak-Toydemir, P.; McDonald, J.; Akarsu, N.; Toydemir, R.M.; Calderon, F.; Tuncali, T.; Tang, W.; Miller, F.; Mao, R. A fourth locus for hereditary hemorrhagic telangiectasia maps to chromosome 7. Am. J. Med. Genet. A 2006, 140, 2155–2162. [Google Scholar] [CrossRef]
- Abdalla, S.A.; Letarte, M. Hereditary haemorrhagic telangiectasia: Current views on genetics and mechanisms of disease. J. Med. Genet. 2006, 43, 97–110. [Google Scholar] [CrossRef]
- McDonald, J.; Bayrak-Toydemir, P.; DeMille, D.; Wooderchak-Donahue, W.; Whitehead, K. Curaçao diagnostic criteria for hereditary hemorrhagic telangiectasia is highly predictive of a pathogenic variant in ENG or ACVRL1 (HHT1 and HHT2). Genet. Med. 2020, 22, 1201–1205. [Google Scholar] [CrossRef]
- Bossler, A.D.; Richards, J.; George, C.; Godmilow, L.; Ganguly, A. Novel mutations in ENG and ACVRL1 identified in a series of 200 individuals undergoing clinical genetic testing for hereditary hemorrhagic telangiectasia (HHT): Correlation of genotype with phenotype. Hum. Mutat. 2006, 27, 667–675. [Google Scholar] [CrossRef]
- Bayrak-Toydemir, P.; McDonald, J.; Markewitz, B.; Lewin, S.; Miller, F.; Chou, L.S.; Gedge, F.; Tang, W.; Coon, H.; Mao, R. Genotype-phenotype correlation in hereditary hemorrhagic telangiectasia: Mutations and manifestations. Am. J. Med. Genet. A 2006, 140, 463–470. [Google Scholar] [CrossRef]
- Jelsig, A.M.; Tørring, P.M.; Kjeldsen, A.D.; Qvist, N.; Bojesen, A.; Jensen, U.B.; Andersen, M.K.; Gerdes, A.M.; Brusgaard, K.; Ousager, L.B. JP-HHT phenotype in Danish patients with SMAD4 mutations. Clin. Genet. 2016, 90, 55–62. [Google Scholar] [CrossRef]
- McDonald, N.M.; Ramos, G.P.; Sweetser, S. SMAD4 mutation and the combined juvenile polyposis and hereditary hemorrhage telangiectasia syndrome: A single center experience. Int. J. Colorectal Dis. 2020, 35, 1963–1965. [Google Scholar] [CrossRef]
- Gedge, F.; McDonald, J.; Phansalkar, A.; Chou, L.S.; Calderon, F.; Mao, R.; Lyon, E.; Bayrak-Toydemir, P. Clinical and analytical sensitivities in hereditary hemorrhagic telangiectasia testing and a report of de novo mutations. J. Mol. Diagn. 2007, 9, 258–265. [Google Scholar] [CrossRef]
- Scharpfenecker, M.; van Dinther, M.; Liu, Z.; van Bezooijen, R.L.; Zhao, Q.; Pukac, L.; Löwik, C.W.; ten Dijke, P. BMP-9 signals via ALK1 and inhibits bFGF-induced endothelial cell proliferation and VEGF-stimulated angiogenesis. J. Cell Sci. 2007, 120, 964–972. [Google Scholar] [CrossRef]
- Castonguay, R.; Werner, E.D.; Matthews, R.G.; Presman, E.; Mulivor, A.W.; Solban, N.; Sako, D.; Pearsall, R.S.; Underwood, K.W.; Seehra, J.; et al. Soluble endoglin specifically binds bone morphogenetic proteins 9 and 10 via its orphan domain, inhibits blood vessel formation, and suppresses tumor growth. J. Biol. Chem. 2011, 286, 30034–30046. [Google Scholar] [CrossRef]
- Alt, A.; Miguel-Romero, L.; Donderis, J.; Aristorena, M.; Blanco, F.J.; Round, A.; Rubio, V.; Bernabeu, C.; Marina, A. Structural and functional insights into endoglin ligand recognition and binding. PLoS ONE 2012, 7, e29948. [Google Scholar] [CrossRef] [PubMed]
- Tillet, E.; Bailly, S. Emerging roles of BMP9 and BMP10 in hereditary hemorrhagic telangiectasia. Front. Genet. 2015, 5, 456. [Google Scholar] [CrossRef] [PubMed]
- Tillet, E.; Ouarné, M.; Desroches-Castan, A.; Mallet, C.; Subileau, M.; Didier, R.; Lioutsko, A.; Belthier, G.; Feige, J.J.; Bailly, S. A heterodimer formed by bone morphogenetic protein 9 (BMP9) and BMP10 provides most BMP biological activity in plasma. J. Biol. Chem. 2018, 293, 10963–10974. [Google Scholar] [CrossRef]
- Santibañez, J.F.; Quintanilla, M.; Bernabeu, C. TGF-β/TGF-β receptor system and its role in physiological and pathological conditions. Clin. Sci. 2011, 121, 233–251. [Google Scholar] [CrossRef] [PubMed]
- Roman, B.L.; Hinck, A.P. ALK1 signaling in development and disease: New paradigms. Cell. Mol. Life Sci. 2017, 74, 4539–4560. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Llorente, L.; Gallardo-Vara, E.; Rossi, E.; Smadja, D.M.; Botella, L.M.; Bernabeu, C. Endoglin and alk1 as therapeutic targets for hereditary hemorrhagic telangiectasia. Expert. Opin. Ther. Targets 2017, 21, 933–947. [Google Scholar] [CrossRef]
- Wood, J.H.; Guo, J.; Morrell, N.W.; Li, W. Advances in the molecular regulation of endothelial BMP9 signalling complexes and implications for cardiovascular disease. Biochem. Soc. Trans. 2019, 47, 779–791. [Google Scholar] [CrossRef]
- Barbara, N.P.; Wrana, J.L.; Letarte, M. Endoglin is an accessory protein that interacts with the signaling receptor complex of multiple members of the transforming growth factor-beta superfamily. J. Biol. Chem. 1999, 274, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Esteo, M.; Sanchez-Elsner, T.; Letamendia, A.; Bernabeu, C. Extracellular and cytoplasmic domains of endoglin interact with the transforming growth factor-beta receptors I and II. J. Biol. Chem. 2002, 277, 29197–29209. [Google Scholar] [CrossRef]
- Bernabeu, C.; Conley, B.A.; Vary, C.P. Novel biochemical pathways of endoglin in vascular cell physiology. J. Cell Biochem. 2007, 102, 1375–1388. [Google Scholar] [CrossRef]
- Xu, G.; Barrios-Rodiles, M.; Jerkic, M.; Turinsky, A.L.; Nadon, R.; Vera, S.; Voulgaraki, D.; Wrana, J.L.; Toporsian, M.; Letarte, M. Novel protein interactions with endoglin and activin receptor-like kinase 1: Potential role in vascular networks. Mol. Cell. Proteom. 2014, 13, 489–502. [Google Scholar] [CrossRef]
- Conley, B.A.; Koleva, R.; Smith, J.D.; Kacer, D.; Zhang, D.; Bernabéu, C.; Vary, C.P. Endoglin controls cell migration and composition of focal adhesions: Function of the cytosolic domain. J. Biol. Chem. 2004, 279, 27440–27449. [Google Scholar] [CrossRef]
- Sanz-Rodriguez, F.; Guerrero-Esteo, M.; Botella, L.M.; Banville, D.; Vary, C.P.; Bernabéu, C. Endoglin regulates cytoskeletal organization through binding to ZRP-1, a member of the Lim family of proteins. J. Biol. Chem. 2004, 279, 32858–32868. [Google Scholar] [CrossRef]
- Fernandez-L, A.; Sanz-Rodriguez, F.; Zarrabeitia, R.; Pérez-Molino, A.; Hebbel, R.P.; Nguyen, J.; Bernabéu, C.; Botella, L.M. Blood outgrowth endothelial cells from Hereditary Haemorrhagic Telangiectasia patients reveal abnormalities compatible with vascular lesions. Cardiovasc. Res. 2005, 68, 235–248. [Google Scholar] [CrossRef]
- Gallardo-Vara, E.; Ruiz-Llorente, L.; Casado-Vela, J.; Ruiz-Rodríguez, M.J.; López-Andrés, N.; Pattnaik, A.K.; Quintanilla, M.; Bernabeu, C. Endoglin Protein Interactome Profiling Identifies TRIM21 and Galectin-3 as New Binding Partners. Cells 2019, 8, 1082. [Google Scholar] [CrossRef]
- Bourdeau, A.; Faughnan, M.E.; Letarte, M. Endoglin-deficient mice, a unique model to study hereditary hemorrhagic telangiectasia. Trends Cardiovasc. Med. 2000, 10, 279–285. [Google Scholar] [CrossRef]
- Tual-Chalot, S.; Oh, S.P.; Arthur, H.M. Mouse models of hereditary hemorrhagic telangiectasia: Recent advances and future challenges. Front. Genet. 2015, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Bourdeau, A.; Dumont, D.J.; Letarte, M. A murine model of hereditary hemorrhagic telangiectasia. J. Clin. Investig. 1999, 104, 1343–1351. [Google Scholar] [CrossRef]
- Li, D.Y.; Sorensen, L.K.; Brooke, B.S.; Urness, L.D.; Davis, E.C.; Taylor, D.G.; Boak, B.B.; Wendel, D.P. Defective angiogenesis in mice lacking endoglin. Science 1999, 284, 1534–1537. [Google Scholar] [CrossRef] [PubMed]
- Arthur, H.M.; Ure, J.; Smith, A.J.; Renforth, G.; Wilson, D.I.; Torsney, E.; Charlton, R.; Parums, D.V.; Jowett, T.; Marchuk, D.A.; et al. Endoglin, an ancillary TGFbeta receptor, is required for extraembryonic angiogenesis and plays a key role in heart development. Dev. Biol. 2000, 217, 42–53. [Google Scholar] [CrossRef]
- Oh, S.P.; Seki, T.; Goss, K.A.; Imamura, T.; Yi, Y.; Donahoe, P.K.; Li, L.; Miyazono, K.; ten Dijke, P.; Kim, S.; et al. Activin receptor-like kinase 1 modulates transforming growth factor-b1 signaling in the regulation of angiogenesis. Proc. Natl. Acad. Sci. USA 2000, 97, 2626–2631. [Google Scholar] [CrossRef] [PubMed]
- Urness, L.D.; Sorensen, L.K.; Li, D.Y. Arteriovenous malformations in mice lacking activin receptor-like kinase-1. Nat. Genet. 2000, 26, 328–331. [Google Scholar] [CrossRef]
- Maris, J.M.; Knudson, A.G. Revisiting tissue specificity of germline cancer predisposing mutations. Nat. Rev. Cancer 2015, 15, 65–66. [Google Scholar] [CrossRef]
- Snellings, D.A.; Gallione, C.J.; Clark, D.S.; Vozoris, N.T.; Faughnan, M.E.; Marchuk, D.A. Somatic Mutations in Vascular Malformations of Hereditary Hemorrhagic Telangiectasia Result in Bi-allelic Loss of ENG or ACVRL1. Am. J. Hum. Genet. 2019, 105, 894–906. [Google Scholar] [CrossRef]
- Shovlin, C.L.; Hughes, J.M.; Scott, J.; Seidman, C.E.; Seidman, J.G. Characterization of endoglin and identification of novel mutations in hereditary hemorrhagic telangiectasia. Am. J. Hum. Genet. 1997, 61, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Lux, A.; Attisano, L.; Marchuk, D.A. Assignment of transforming growth factor beta1 and beta3 and a third new ligand to the type I receptor ALK-1. J. Biol. Chem. 1999, 274, 9984–9992. [Google Scholar] [CrossRef]
- Gu, Y.; Jin, P.; Zhang, L.; Zhao, X.; Gao, X.; Ning, Y.; Meng, A.; Chen, Y.G. Functional analysis of mutations in the kinase domain of the TGF-beta receptor ALK1 reveals different mechanisms for induction of hereditary hemorrhagic telangiectasia. Blood 2006, 107, 1951–1954. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Förg, T.; Hafner, M.; Lux, A. Investigation of endoglin wild-type and missense mutant protein heterodimerisation using fluorescence microscopy based IF, BiFC and FRET analyses. PLoS ONE 2014, 9, e102998. [Google Scholar] [CrossRef]
- Mallet, C.; Lamribet, K.; Giraud, S.; Dupuis-Girod, S.; Feige, J.J.; Bailly, S.; Tillet, E. Functional analysis of endoglin mutations from hereditary hemorrhagic telangiectasia type 1 patients reveals different mechanisms for endoglin loss of function. Hum. Mol. Genet. 2015, 24, 1142–1154. [Google Scholar] [CrossRef]
- López-Novoa, J.M.; Bernabeu, C. The physiological role of endoglin in the cardiovascular system. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H959–H974. [Google Scholar] [CrossRef]
- Gonzalez, C.D.; Cipriano, S.D.; Topham, C.A.; Stevenson, D.A.; Whitehead, K.J.; Vanderhooft, S.; Presson, A.P.; McDonald, J. Localization and age distribution of telangiectases in children and adolescents with hereditary hemorrhagic telangiectasia: A retrospective cohort study. J. Am. Acad. Dermatol. 2019, 81, 950–955. [Google Scholar] [CrossRef] [PubMed]
- Geisthoff, U.; Nguyen, H.L.; Lefering, R.; Maune, S.; Thangavelu, K.; Droege, F. Trauma Can Induce Telangiectases in Hereditary Hemorrhagic Telangiectasia. J. Clin. Med. 2020, 9, 1507. [Google Scholar] [CrossRef]
- Park, S.O.; Wankhede, M.; Lee, Y.J.; Choi, E.J.; Fliess, N.; Choe, S.W.; Oh, S.H.; Walter, G.; Raizada, M.K.; Sorg, B.S.; et al. Real-time imaging of de novo arteriovenous malformation in a mouse model of hereditary hemorrhagic telangiectasia. J. Clin. Investig. 2009, 119, 3487–3496. [Google Scholar] [CrossRef]
- Han, C.; Choe, S.W.; Kim, Y.H.; Acharya, A.P.; Keselowsky, B.G.; Sorg, B.S.; Lee, Y.J.; Oh, S.P. VEGF neutralization can prevent and normalize arteriovenous malformations in an animal model for hereditary hemorrhagic telangiectasia 2. Angiogenesis 2014, 17, 823–830. [Google Scholar] [CrossRef]
- Kim, Y.H.; Choe, S.W.; Chae, M.Y.; Hong, S.; Oh, S.P. SMAD4 Deficiency Leads to Development of Arteriovenous Malformations in Neonatal and Adult Mice. J. Am. Heart Assoc. 2018, 7, e009514. [Google Scholar] [CrossRef]
- Corti, P.; Young, S.; Chen, C.Y.; Patrick, M.J.; Rochon, E.R.; Pekkan, K.; Roman, B.L. Interaction between alk1 and blood flow in the development of arteriovenous malformations. Development 2011, 138, 1573–1582. [Google Scholar] [CrossRef]
- Baeyens, N.; Larrivée, B.; Ola, R.; Hayward-Piatkowskyi, B.; Dubrac, A.; Huang, B.; Ross, T.D.; Coon, B.G.; Min, E.; Tsarfati, M.; et al. Defective fluid shear stress mechanotransduction mediates hereditary hemorrhagic telangiectasia. J. Cell Biol. 2016, 214, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Hiepen, C.; Mendez, P.L.; Knaus, P. It Takes Two to Tango: Endothelial TGFβ/BMP Signaling Crosstalk with Mechanobiology. Cells 2020, 9, 1965. [Google Scholar] [CrossRef]
- Botella, L.M.; Sánchez-Elsner, T.; Sanz-Rodriguez, F.; Kojima, S.; Shimada, J.; Guerrero-Esteo, M.; Cooreman, M.P.; Ratziu, V.; Langa, C.; Vary, C.P.; et al. Transcriptional activation of endoglin and transforming growth factor-beta signaling components by cooperative interaction between Sp1 and KLF6: Their potential role in the response to vascular injury. Blood 2002, 100, 4001–4010. [Google Scholar] [CrossRef]
- Garrido-Martín, E.M.; Blanco, F.J.; Roque, M.; Novensa, L.; Tarocchi, M.; Lang, U.E.; Suzuki, T.; Friedman, S.L.; Botella, L.M.; Bernabeu, C. Vascular injury triggers Krüppel-like factor 6 mobilization and cooperation with specificity protein 1 to promote endothelial activation through upregulation of the activin receptor-like kinase 1 gene. Circ. Res. 2013, 112, 113–127. [Google Scholar] [CrossRef]
- Seghers, L.; de Vries, M.R.; Pardali, E.; Hoefer, I.E.; Hierck, B.P.; ten Dijke, P.; Goumans, M.J.; Quax, P.H. Shear induced collateral artery growth modulated by endoglin but not by ALK1. J. Cell. Mol. Med. 2012, 16, 2440–2450. [Google Scholar] [CrossRef]
- Jin, Y.; Muhl, L.; Burmakin, M.; Wang, Y.; Duchez, A.C.; Betsholtz, C.; Arthur, H.M.; Jakobsson, L. Endoglin prevents vascular malformation by regulating flow-induced cell migration and specification through VEGFR2 signalling. Nat. Cell Biol. 2017, 19, 639–652. [Google Scholar] [CrossRef]
- Ola, R.; Künzel, S.H.; Zhang, F.; Genet, G.; Chakraborty, R.; Pibouin-Fragner, L.; Martin, K.; Sessa, W.; Dubrac, A.; Eichmann, A. SMAD4 Prevents Flow Induced Arteriovenous Malformations by Inhibiting Casein Kinase 2. Circulation 2018, 138, 2379–2394. [Google Scholar] [CrossRef]
- Sugden, W.W.; Meissner, R.; Aegerter-Wilmsen, T.; Tsaryk, R.; Leonard, E.V.; Bussmann, J.; Hamm, M.J.; Herzog, W.; Jin, Y.; Jakobsson, L.; et al. Endoglin controls blood vessel diameter through endothelial cell shape changes in response to haemodynamic cues. Nat. Cell Biol. 2017, 19, 653–665. [Google Scholar] [CrossRef] [PubMed]
- Sugden, W.W.; Siekmann, A.F. Endothelial cell biology of Endoglin in hereditary hemorrhagic telangiectasia. Curr. Opin. Hematol. 2018, 25, 237–244. [Google Scholar] [CrossRef]
- Klostranec, J.M.; Chen, L.; Mathur, S.; McDonald, J.; Faughnan, M.E.; Ratjen, F.; Krings, T. A theory for polymicrogyria and brain arteriovenous malformations in HHT. Neurology 2019, 92, 34–42. [Google Scholar] [CrossRef]
- Scharpfenecker, M.; Floot, B.; Russell, N.S.; Ten Dijke, P.; Stewart, F.A. Endoglin haploinsufficiency reduces radiation-induced fibrosis and telangiectasia formation in mouse kidneys. Radiother. Oncol. 2009, 92, 484–491. [Google Scholar] [CrossRef]
- Sadick, H.; Naim, R.; Sadick, M.; Hörmann, K.; Riedel, F. Plasma level and tissue expression of angiogenic factors in patients with hereditary hemorrhagic telangiectasia. Int. J. Mol. Med. 2005, 15, 591–596. [Google Scholar] [CrossRef]
- Sadick, H.; Naim, R.; Gossler, U.; Hormann, K.; Riedel, F. Angiogenesis in hereditary hemorrhagic telangiectasia: VEGF165 plasma concentration in correlation to the VEGF expression and microvessel density. Int. J. Mol. Med. 2005, 15, 15–19. [Google Scholar] [CrossRef]
- Botella, L.M.; Albiñana, V.; Ojeda-Fernandez, L.; Recio-Poveda, L.; Bernabéu, C. Research on potential biomarkers in hereditary hemorrhagic telangiectasia. Front. Genet. 2015, 6, 115. [Google Scholar] [CrossRef]
- Xu, B.; Wu, Y.Q.; Huey, M.; Arthur, H.M.; Marchuk, D.A.; Hashimoto, T.; Young, W.L.; Yang, G.Y. Vascular endothelial growth factor induces abnormal microvasculature in the endoglin heterozygous mouse brain. J. Cereb. Blood Flow Metab. 2004, 24, 237–244. [Google Scholar] [CrossRef]
- Choi, E.J.; Chen, W.; Jun, K.; Arthur, H.M.; Young, W.L.; Su, H. Novel brain arteriovenous malformation mouse models for type 1 hereditary hemorrhagic telangiectasia. PLoS ONE 2014, 9, e88511. [Google Scholar] [CrossRef] [PubMed]
- Hao, Q.; Zhu, Y.; Su, H.; Shen, F.; Yang, G.Y.; Kim, H.; Young, W.L. VEGF Induces More Severe Cerebrovascular Dysplasia in Endoglin than in Alk1 Mice. Version 2. Transl. Stroke Res. 2010, 1, 197–201. [Google Scholar] [CrossRef]
- Ardelean, D.S.; Jerkic, M.; Yin, M.; Peter, M.; Ngan, B.; Kerbel, R.S.; Foster, F.S.; Letarte, M. Endoglin and activin receptor-like kinase 1 heterozygous mice have a distinct pulmonary and hepatic angiogenic profile and response to anti-VEGF treatment. Angiogenesis 2014, 17, 129–146. [Google Scholar] [CrossRef]
- Buscarini, E.; Botella, L.M.; Geisthoff, U.; Kjeldsen, A.D.; Mager, H.J.; Pagella, F.; Suppressa, P.; Zarrabeitia, R.; Dupuis-Girod, S.; Shovlin, C.L.; et al. Safety of thalidomide and bevacizumab in patients with hereditary hemorrhagic telangiectasia. Orphanet J. Rare Dis. 2019, 14, 28. [Google Scholar] [CrossRef] [PubMed]
- Larrivée, B.; Prahst, C.; Gordon, E.; del Toro, R.; Mathivet, T.; Duarte, A.; Simons, M.; Eichmann, A. ALK1 signaling inhibits angiogenesis by cooperating with the Notch pathway. Dev. Cell 2012, 22, 489–500. [Google Scholar] [CrossRef]
- Peacock, H.M.; Caolo, V.; Jones, E.A. Arteriovenous malformations in hereditary haemorrhagic telangiectasia: Looking beyond ALK1-NOTCH interactions. Cardiovasc Res. 2016, 109, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Schumacher, N.; Maier, M.; Sendtner, M.; Gessler, M. The Notch target genes Hey1 and Hey2 are required for embryonic vascular development. Genes Dev. 2004, 18, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Lawson, N.D.; Scheer, N.; Pham, V.N.; Kim, C.H.; Chitnis, A.B.; Campos-Ortega, J.A.; Weinstein, B.M. Notch signaling is required for arterial-venous differentiation during embryonic vascular development. Development 2001, 128, 3675–3683. [Google Scholar]
- Krebs, L.T.; Shutter, J.R.; Tanigaki, K.; Honjo, T.; Stark, K.L.; Gridley, T. Haploinsufficient lethality and formation of arteriovenous malformations in Notch pathway mutants. Genes Dev. 2004, 18, 2469–2473. [Google Scholar] [CrossRef] [PubMed]
- Lebrin, F.; Goumans, M.J.; Jonker, L.; Carvalho, R.L.; Valdimarsdottir, G.; Thorikay, M.; Mummery, C.; Arthur, H.M.; ten Dijke, P. Endoglin promotes endothelial cell proliferation and TGF-beta/ALK1 signal transduction. EMBO J. 2004, 23, 4018–4028. [Google Scholar] [CrossRef]
- Li, C.; Hampson, I.N.; Hampson, L.; Kumar, P.; Bernabeu, C.; Kumar, S. CD105 antagonizes the inhibitory signaling of transforming growth factor beta1 on human vascular endothelial cells. FASEB J. 2000, 14, 55–64. [Google Scholar] [CrossRef]
- She, X.; Matsuno, F.; Harada, N.; Tsai, H.; Seon, B.K. Synergy between anti-endoglin (CD105) monoclonal antibodies and TGF-beta in suppression of growth of human endothelial cells. Int. J. Cancer 2004, 108, 251–257. [Google Scholar] [CrossRef]
- Li, C.; Issa, R.; Kumar, P.; Hampson, I.N.; Lopez-Novoa, J.M.; Bernabeu, C.; Kumar, S. CD105 prevents apoptosis in hypoxic endothelial cells. J. Cell Sci. 2003, 116, 2677–2685. [Google Scholar] [CrossRef]
- Nivet, A.; Schlienger, M.; Clavère, P.; Huguet, F. Effects of high-dose irradiation on vascularization: Physiopathology and clinical consequences. Cancer Radiother. 2019, 23, 161–167. [Google Scholar] [CrossRef]
- David, L.; Mallet, C.; Keramidas, M.; Lamandé, N.; Gasc, J.M.; Dupuis-Girod, S.; Plauchu, H.; Feige, J.J.; Bailly, S. Bone morphogenetic protein-9 is a circulating vascular quiescence factor. Circ. Res. 2008, 102, 914–922. [Google Scholar] [CrossRef]
- Suzuki, Y.; Ohga, N.; Morishita, Y.; Hida, K.; Miyazono, K.; Watabe, T. BMP-9 induces proliferation of multiple types of endothelial cells in vitro and in vivo. J. Cell Sci. 2010, 123, 1684–1692. [Google Scholar] [CrossRef]
- Valdimarsdottir, G.; Goumans, M.J.; Rosendahl, A.; Brugman, M.; Itoh, S.; Lebrin, F.; Sideras, P.; ten Dijke, P. Stimulation of Id1 expression by bone morphogenetic protein is sufficient and necessary for bone morphogenetic protein-induced activation of endothelial cells. Circulation 2002, 106, 2263–2270. [Google Scholar] [CrossRef]
- David, L.; Mallet, C.; Mazerbourg, S.; Feige, J.J.; Bailly, S. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood 2007, 109, 1953–1961. [Google Scholar] [CrossRef]
- Torsney, E.; Charlton, R.; Parums, D.; Collis, M.; Arthur, H.M. Inducible expression of human endoglin during inflammation and wound healing in vivo. Inflamm. Res. 2002, 51, 464–470. [Google Scholar] [CrossRef]
- Braverman, I.M.; Keh, A.; Jacobson, B.S. Ultrastructure and three-dimensional organization of the telangiectases of hereditary hemorrhagic telangiectasia. J. Investig. Dermatol. 1990, 95, 422–427. [Google Scholar] [CrossRef]
- Zhang, R.; Hanc, Z.; Degos, V.; Shen, F.; Choi, E.J.; Sun, Z.; Kang, S.; Wong, M.; Zhu, W.; Zhan, L.; et al. Persistent infiltration and pro-inflammatory differentiation of monocytes cause unresolved inflammation in brain arteriovenous malformation. Angiogenesis 2016, 19, 451–461. [Google Scholar] [CrossRef]
- Van Laake, L.W.; van den Driesche, S.; Post, S.; Feijen, A.; Jansen, M.A.; Driessens, M.H.; Mager, J.J.; Snijder, R.J.; Westermann, C.J.; Doevendans, P.A.; et al. Endoglin has a crucial role in blood cell-mediated vascular repair. Circulation 2006, 114, 2288–2297. [Google Scholar] [CrossRef] [PubMed]
- Jaipersad, A.S.; Lip, G.Y.; Silverman, S.; Shantsila, E. The role of monocytes in angiogenesis and atherosclerosis. J. Am. Coll. Cardiol. 2014, 63, 1–11. [Google Scholar] [CrossRef]
- Jerkic, M.; Peter, M.; Ardelean, D.; Fine, M.; Konerding, M.A.; Letarte, M. Dextran sulfate sodium leads to chronic colitis and pathological angiogenesis in Endoglin heterozygous mice. Inflamm. Bowel Dis. 2010, 16, 1859–1870. [Google Scholar] [CrossRef]
- Rossi, E.; Sanz-Rodriguez, F.; Eleno, N.; Düwell, A.; Blanco, F.J.; Langa, C.; Botella, L.M.; Cabañas, C.; Lopez-Novoa, J.M.; Bernabeu, C. Endothelial endoglin is involved in inflammation: Role in leukocyte adhesion and transmigration. Blood 2013, 121, 403–415. [Google Scholar] [CrossRef]
- Peter, M.R.; Jerkic, M.; Sotov, V.; Douda, D.N.; Ardelean, D.S.; Ghamami, N.; Lakschevitz, F.; Khan, M.A.; Robertson, S.J.; Glogauer, M.; et al. Impaired resolution of inflammation in the Endoglin heterozygous mouse model of chronic colitis. Mediat. Inflamm. 2014, 2014, 767185. [Google Scholar] [CrossRef] [PubMed]
- Shen, F.; Degos, V.; Chu, P.L.; Han, Z.; Westbroek, E.M.; Choi, E.J.; Marchuk, D.; Kim, H.; Lawton, M.T.; Maze, M.; et al. Endoglin deficiency impairs stroke recovery. Stroke 2014, 45, 2101–2106. [Google Scholar] [CrossRef][Green Version]
- Han, Z.; Shaligram, S.; Faughnan, M.E.; Clark, D.; Sun, Z.; Su, H. Reduction of endoglin receptor impairs mononuclear cell-migration. Explor. Med. 2020, 1, 136–148. [Google Scholar] [CrossRef]
- Rossi, E.; Smadja, D.M.; Boscolo, E.; Langa, C.; Arevalo, M.A.; Pericacho, M.; Gamella-Pozuelo, L.; Kauskot, A.; Botella, L.M.; Gaussem, P.; et al. Endoglin regulates mural cell adhesion in the circulatory system. Cell. Mol. Life Sci. 2016, 73, 1715–1739. [Google Scholar] [CrossRef]
- Rossi, E.; Pericacho, M.; Bachelot-Loza, C.; Pidard, D.; Gaussem, P.; Poirault-Chassac, S.; Blanco, F.J.; Langa, C.; González-Manchón, C.; Lopez-Novoa, J.M.; et al. Human endoglin as a potential new partner involved in platelet-endothelium interactions. Cell. Mol. Life Sci. 2018, 75, 1269–1284. [Google Scholar] [CrossRef]
- Rossi, E.; Lopez-Novoa, J.M.; Bernabeu, C. Endoglin involvement in integrin-mediated cell adhesion as a putative pathogenic mechanism in hereditary hemorrhagic telangiectasia type 1 (HHT1). Front. Genet. 2015, 5, 457. [Google Scholar] [CrossRef]
- Koleva, R.I.; Conley, B.A.; Romero, D.; Riley, K.S.; Marto, J.A.; Lux, A.; Vary, C.P. Endoglin structure and function: Determinants of endoglin phosphorylation by transforming growth factor-beta receptors. J. Biol. Chem. 2006, 281, 25110–25123. [Google Scholar] [CrossRef]
- Blanco, F.J.; Santibanez, J.F.; Guerrero-Esteo, M.; Langa, C.; Vary, C.P.; Bernabeu, C. Interaction and functional interplay between endoglin and ALK-1, two components of the endothelial transforming growth factor-beta receptor complex. J. Cell. Physiol. 2005, 204, 574–584. [Google Scholar] [CrossRef]
- Ma, L.; Shen, F.; Jun, K.; Bao, C.; Kuo, R.; Young, W.L.; Nishimura, S.L.; Su, H. Integrin β8 Deletion Enhances Vascular Dysplasia and Hemorrhage in the Brain of Adult Alk1 Heterozygous Mice. Transl. Stroke Res. 2016, 7, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef]
- Jerkic, M.; Rivas-Elena, J.V.; Prieto, M.; Carrón, R.; Sanz-Rodríguez, F.; Pérez-Barriocanal, F.; Rodríguez-Barbero, A.; Bernabéu, C.; López-Novoa, J.M. Endoglin regulates nitric oxide-dependent vasodilatation. FASEB J. 2004, 18, 609–611. [Google Scholar] [CrossRef]
- Toporsian, M.; Gros, R.; Kabir, M.G.; Vera, S.; Govindaraju, K.; Eidelman, D.H.; Husain, M.; Letarte, M. A role for endoglin in coupling eNOS activity and regulating vascular tone revealed in hereditary hemorrhagic telangiectasia. Circ. Res. 2005, 96, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Jerkic, M.; Letarte, M. Contribution of oxidative stress to endothelial dysfunction in hereditary hemorrhagic telangiectasia. Front. Genet. 2015, 6, 34. [Google Scholar] [CrossRef]
- Jerkic, M.; Sotov, V.; Letarte, M. Oxidative stress contributes to endothelial dysfunction in mouse models of hereditary hemorrhagic telangiectasia. Oxid. Med. Cell Longev. 2012, 2012, 686972. [Google Scholar] [CrossRef]
- De Gussem, E.M.; Snijder, R.J.; Disch, F.J.; Zanen, P.; Westermann, C.J.; Mager, J.J. The effect of N-acetylcysteine on epistaxis and quality of life in patients with HHT: A pilot study. Rhinology 2009, 47, 85–88. [Google Scholar]
- Albiñana, V.; Cuesta, A.M.; Rojas-P., I.; Gallardo-Vara, E.; Recio-Poveda, L.; Bernabéu, C.; Botella, L.M. Review of Pharmacological Strategies with Repurposed Drugs for Hereditary Hemorrhagic Telangiectasia Related Bleeding. J. Clin. Med. 2020, 9, 1766. [Google Scholar] [CrossRef]
- Bourdeau, A.; Faughnan, M.E.; McDonald, M.L.; Paterson, A.D.; Wanless, I.R.; Letarte, M. Potential role of modifier genes influencing transforming growth factor-beta1 levels in the development of vascular defects in endoglin heterozygous mice with hereditary hemorrhagic telangiectasia. Am. J. Pathol. 2001, 158, 2011–2020. [Google Scholar] [CrossRef]
- Satomi, J.; Mount, R.J.; Toporsian, M.; Paterson, A.D.; Wallace, M.C.; Harrison, R.V.; Letarte, M. Cerebral vascular abnormalities in a murine model of hereditary hemorrhagic telangiectasia. Stroke 2003, 34, 783–789. [Google Scholar] [CrossRef]
- Coulson, P.S.; Wilson, R.A. Portal shunting and resistance to Schistosoma mansoni in 129 strain mice. Parasitology 1989, 99, 383–389. [Google Scholar] [CrossRef]
- Elsaghier, A.A.; McLaren, D.J. Schistosoma mansoni: Evidence that vascular abnormalities correlate with the ‘non-permissive’ trait in 129/Ola mice. Parasitology 1989, 99, 377–381. [Google Scholar] [CrossRef]
- Srinivasan, S.; Hanes, M.A.; Dickens, T.; Porteous, M.E.; Oh, S.P.; Hale, L.P.; Marchuk, D.A. A mouse model for hereditary hemorrhagic telangiectasia (HHT) type 2. Hum. Mol. Genet. 2003, 12, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, L.K.; Brooke, B.S.; Li, D.Y.; Urness, L.D. Loss of distinct arterial and venous boundaries in mice lacking endoglin, a vascular-specific TGFbeta coreceptor. Dev. Biol. 2003, 261, 235–250. [Google Scholar] [CrossRef]
- Mahmoud, M.; Allinson, K.R.; Zhai, Z.; Oakenfull, R.; Ghandi, P.; Adams, R.H.; Fruttiger, M.; Arthur, H.M. Pathogenesis of arteriovenous malformations in the absence of endoglin. Circ. Res. 2010, 106, 1425–1433. [Google Scholar] [CrossRef]
- Garrido-Martin, E.M.; Nguyen, H.L.; Cunningham, T.A.; Choe, S.W.; Jiang, Z.; Arthur, H.M.; Lee, Y.J.; Oh, S.P. Common and distinctive pathogenetic features of arteriovenous malformations in hereditary hemorrhagic telangiectasia 1 and hereditary hemorrhagic telangiectasia 2 animal models--brief report. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2232–2236. [Google Scholar] [CrossRef] [PubMed]
- Benzinou, M.; Clermont, F.F.; Letteboer, T.G.; Kim, J.H.; Espejel, S.; Harradine, K.A.; Arbelaez, J.; Luu, M.T.; Roy, R.; Quigley, D.; et al. Mouse and human strategies identify PTPN14 as a modifier of angiogenesis and hereditary haemorrhagic telangiectasia. Nat. Commun. 2012, 3, 616. [Google Scholar] [CrossRef]
- Letteboer, T.G.; Benzinou, M.; Merrick, C.B.; Quigley, D.A.; Zhau, K.; Kim, I.J.; To, M.D.; Jablons, D.M.; van Amstel, J.K.; Westermann, C.J.; et al. Genetic variation in the functional ENG allele inherited from the non-affected parent associates with presence of pulmonary arteriovenous malformation in hereditary hemorrhagic telangiectasia 1 (HHT1) and may influence expression of PTPN14. Front. Genet. 2015, 6, 67. [Google Scholar] [CrossRef]
- Kawasaki, K.; Freimuth, J.; Meyer, D.S.; Lee, M.M.; Tochimoto-Okamoto, A.; Benzinou, M.; Clermont, F.F.; Wu, G.; Roy, R.; Letteboer, T.G.; et al. Genetic variants of Adam17 differentially regulate TGFβ signaling to modify vascular pathology in mice and humans. Proc. Natl. Acad. Sci. USA 2014, 111, 7723–7728. [Google Scholar] [CrossRef]
- Pawlikowska, L.; Tran, M.N.; Achrol, A.S.; Ha, C.; Burchard, E.; Choudhry, S.; Zaroff, J.; Lawton, M.T.; Castro, R.; McCulloch, C.E.; et al. Polymorphisms in transforming growth factor-beta-related genes ALK1 and ENG are associated with sporadic brain arteriovenous malformations. Stroke 2005, 36, 2278–2280. [Google Scholar] [CrossRef]
- Simon, M.; Franke, D.; Ludwig, M.; Aliashkevich, A.F.; Köster, G.; Oldenburg, J.; Boström, A.; Ziegler, A.; Schramm, J. Association of a polymorphism of the ACVRL1 gene with sporadic arteriovenous malformations of the central nervous system. J. Neurosurg. 2006, 104, 945–949. [Google Scholar] [CrossRef]
- Pawlikowska, L.; Nelson, J.; Guo, D.E.; McCulloch, C.E.; Lawton, M.T.; Young, W.L.; Kim, H.; Faughnan, M.E.; Brain Vascular Malformation Consortium HHT Investigator Group. The ACVRL1 c.314-35A>G polymorphism is associated with organ vascular malformations in hereditary hemorrhagic telangiectasia patients with ENG mutations, but not in patients with ACVRL1 mutations. Am. J. Med. Genet. A 2015, 167, 1262–1267. [Google Scholar] [CrossRef]
- Pawlikowska, L.; Nelson, J.; Guo, D.E.; McCulloch, C.E.; Lawton, M.T.; Kim, H.; Faughnan, M.E.; Brain Vascular Malformation Consortium HHT Investigator Group. Association of common candidate variants with vascular malformations and intracranial hemorrhage in hereditary hemorrhagic telangiectasia. Mol. Genet. Genom. Med. 2018, 6, 350–356. [Google Scholar] [CrossRef]
- Davies, J.C.; Griesenbach, U.; Alton, E. Modifier genes in cystic fibrosis. Pediatr. Pulmonol. 2005, 39, 383–391. [Google Scholar] [CrossRef]
- Kormann, M.S.D.; Dewerth, A.; Eichner, F.; Baskaran, P.; Hector, A.; Regamey, N.; Hartl, D.; Handgretinger, R.; Antony, J.S. Transcriptomic profile of cystic fibrosis patients identifies type I interferon response and ribosomal stalk proteins as potential modifiers of disease severity. PLoS ONE 2017, 12, e0183526. [Google Scholar] [CrossRef]
- Rigamonti, D.; Hadley, M.N.; Drayer, B.P.; Johnson, P.C.; Hoenig-Rigamonti, K.; Knight, J.T.; Spetzler, R.F. Cerebral cavernous malformations. Incidence and familial occurrence. N. Engl. J. Med. 1988, 319, 343–347. [Google Scholar] [CrossRef]
- Akers, A.L.; Johnson, E.; Steinberg, G.K.; Zabramski, J.M.; Marchuk, D.A. Biallelic somatic and germline mutations in cerebral cavernous malformations (CCMs): Evidence for a two-hit mechanism of CCM pathogenesis. Hum. Mol. Genet. 2009, 18, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.C.; Drakos, S.G.; Ruiz, O.E.; Smith, A.C.; Gibson, C.C.; Ling, J.; Passi, S.F.; Stratman, A.N.; Sacharidou, A.; Revelo, M.P.; et al. Mutations in 2 distinct genetic pathways result in cerebral cavernous malformations in mice. J. Clin. Investig. 2011, 121, 1871–1881. [Google Scholar] [CrossRef] [PubMed]
- Limaye, N.; Wouters, V.; Uebelhoer, M.; Tuominen, M.; Wirkkala, R.; Mulliken, J.B.; Eklund, L.; Boon, L.M.; Vikkula, M. Somatic mutations in angiopoietin receptor gene TEK cause solitary and multiple sporadic venous malformations. Nat. Genet. 2009, 41, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Macmurdo, C.F.; Wooderchak-Donahue, W.; Bayrak-Toydemir, P.; Le, J.; Wallenstein, M.B.; Milla, C.; Teng, J.M.; Bernstein, J.A.; Stevenson, D.A. RASA1 somatic mutation and variable expressivity in capillary malformation/arteriovenous malformation (CM/AVM) syndrome. Am. J. Med. Genet. A 2016, 170, 1450–1454. [Google Scholar] [CrossRef]
- Cai, R.; Liu, F.; Liu, Y.; Chen, H.; Lin, X. RASA-1 somatic “second hit” mutation in capillary malformation-arteriovenous malformation. J. Dermatol. 2018, 45, 1478–1480. [Google Scholar] [CrossRef]
- Choi, E.J.; Walker, E.J.; Shen, F.; Oh, S.P.; Arthur, H.M.; Young, W.L.; Su, H. Minimal homozygous endothelial deletion of Eng with VEGF stimulation is sufficient to cause cerebrovascular dysplasia in the adult mouse. Cerebrovasc. Dis. 2012, 33, 540–547. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernabeu, C.; Bayrak-Toydemir, P.; McDonald, J.; Letarte, M. Potential Second-Hits in Hereditary Hemorrhagic Telangiectasia. J. Clin. Med. 2020, 9, 3571. https://doi.org/10.3390/jcm9113571
Bernabeu C, Bayrak-Toydemir P, McDonald J, Letarte M. Potential Second-Hits in Hereditary Hemorrhagic Telangiectasia. Journal of Clinical Medicine. 2020; 9(11):3571. https://doi.org/10.3390/jcm9113571
Chicago/Turabian StyleBernabeu, Carmelo, Pinar Bayrak-Toydemir, Jamie McDonald, and Michelle Letarte. 2020. "Potential Second-Hits in Hereditary Hemorrhagic Telangiectasia" Journal of Clinical Medicine 9, no. 11: 3571. https://doi.org/10.3390/jcm9113571
APA StyleBernabeu, C., Bayrak-Toydemir, P., McDonald, J., & Letarte, M. (2020). Potential Second-Hits in Hereditary Hemorrhagic Telangiectasia. Journal of Clinical Medicine, 9(11), 3571. https://doi.org/10.3390/jcm9113571