Atrial Fibrillation Progression Is Associated with Cell Senescence Burden as Determined by p53 and p16 Expression

 , , ,
, , ,
Abstract
1. Introduction
2. Material and Methods
2.1. Patients
2.2. Sample Preparation
2.3. Western Blot Analysis (WB)
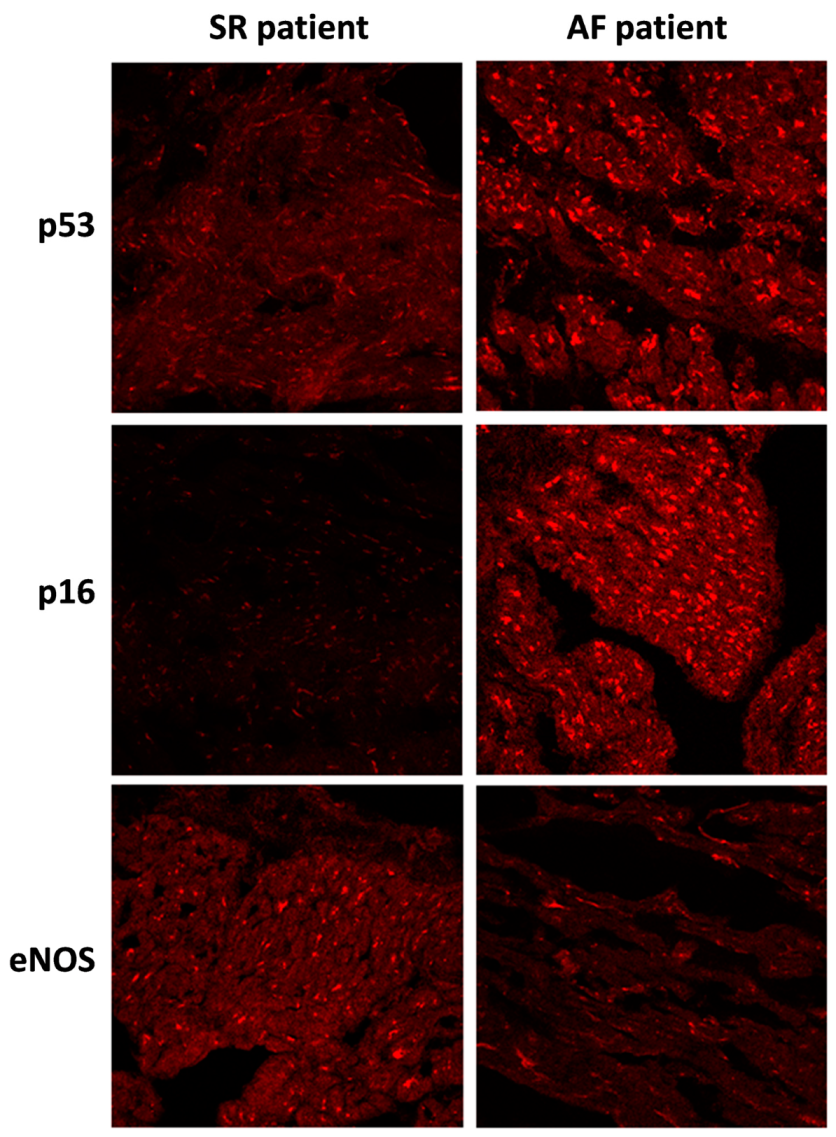
2.4. Immunofluorescence Studies
2.5. Statistical Analysis
3. Results
3.1. Patients Characteristics
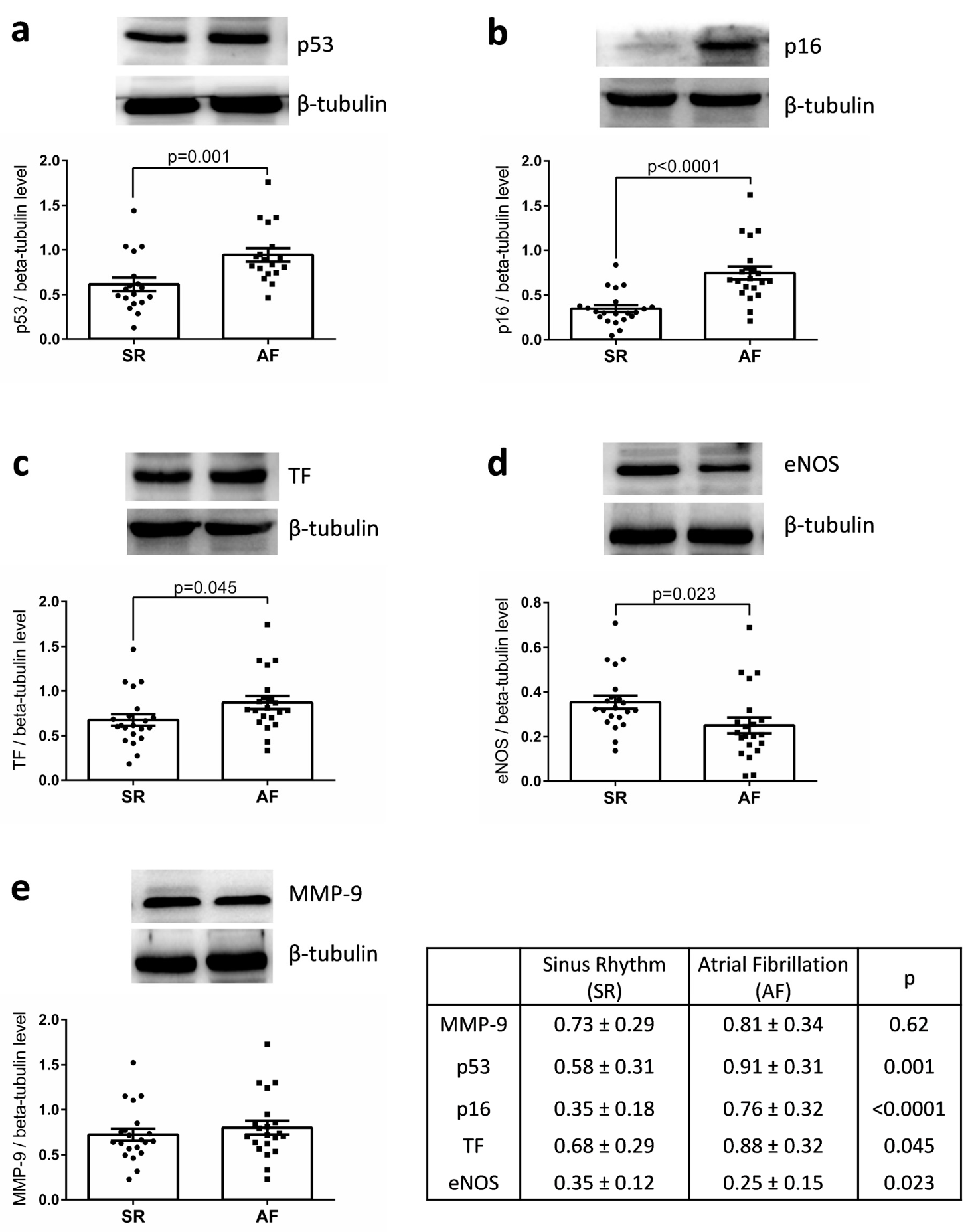
3.2. p53, p16, TF, and MMP-9 are Upregulated and eNOS Down-Regulated in the Right Atrial Appendages of Patients with AF
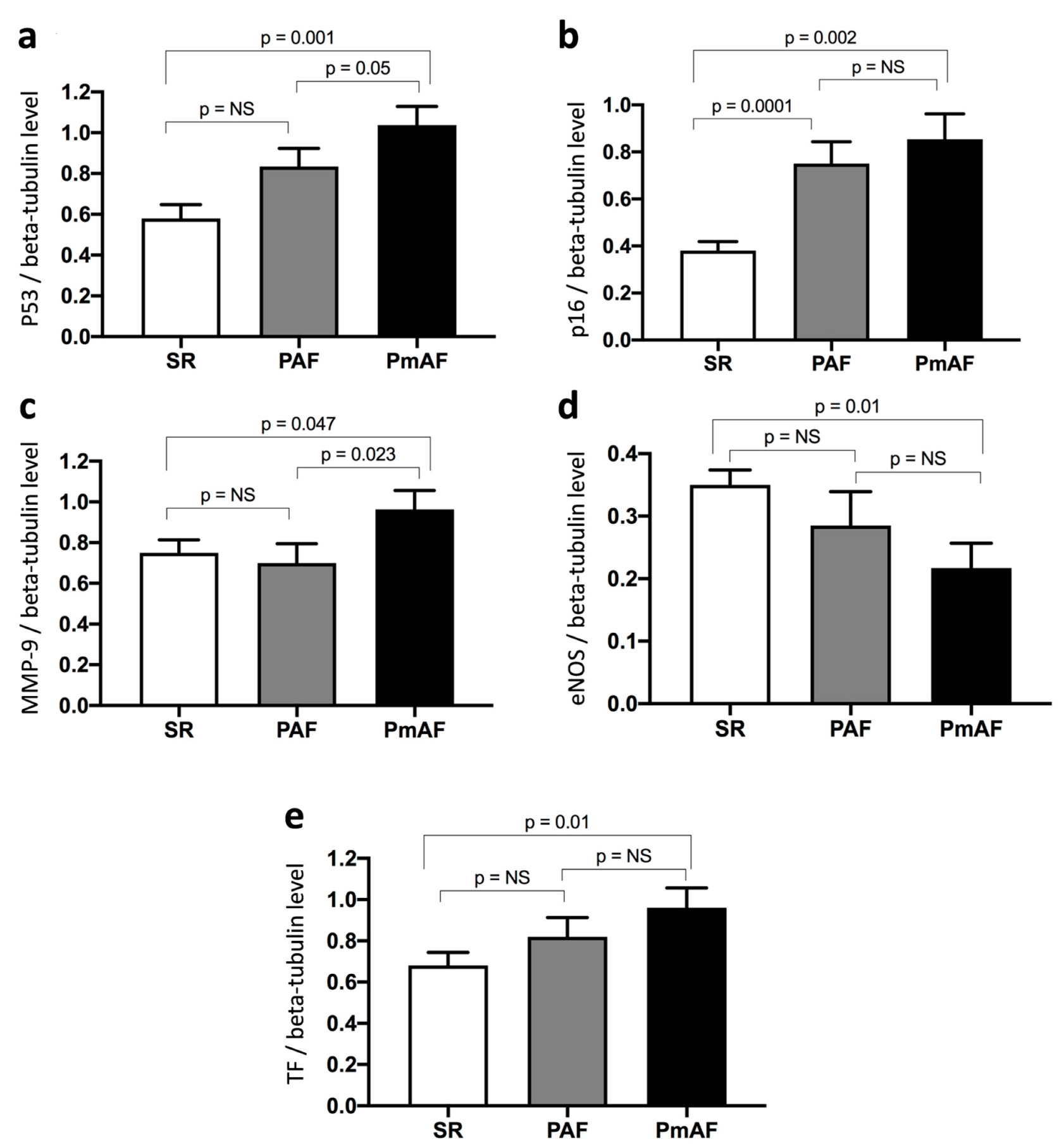
3.3. Changes in the Protein Expression Level of p53, p16, MMP-9, eNOS, and TF are Related to the Progression of AF
3.4. Predictive Factors of p53 and p16 Elevation
3.5. Predictive Factors of TF, eNOS, and MMP-9 Elevation
4. Discussion
4.1. Senescence Burden and Atrial Fibrillation
4.2. Senescence is Associated with Endothelial Dysfunction and Enhanced Tissue Factor Expression
4.3. MMP-9 Expression as a Surrogate Marker of Atrial Remodeling and AF
5. Study Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Watson, T.; Shantsila, E.; Lip, G.Y. Mechanisms of thrombogenesis in atrial fibrillation: Virchow’s triad revisited. Lancet 2009, 373, 155–166. [Google Scholar] [CrossRef]
- Vanhoutte, P.M. Regenerated Endothelium and Its Senescent Response to Aggregating Platelets. Circ. J. 2016, 80, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, Y.; Lip, G.Y.; Lane, D.A. Recent developments in understanding epidemiology and risk determinants of atrial fibrillation as a cause of stroke. Can. J. Cardiol. 2013, 29, S4–S13. [Google Scholar] [CrossRef] [PubMed]
- Burton, D.G.; Krizhanovsky, V. Physiological and pathological consequences of cellular senescence. Cell Mol. Life Sci. 2014, 71, 4373–4386. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Pickering, J.G. Cellular Senescence and Vascular Disease: Novel Routes to Better Understanding and Therapy. Can. J. Cardiol. 2016, 32, 612–623. [Google Scholar] [CrossRef]
- Abbas, M.; Jesel, L.; Auger, C.; Amoura, L.; Messas, N.; Manin, G.; Rumig, C.; Leon-Gonzalez, A.J.; Ribeiro, T.P.; Silva, G.C.; et al. Endothelial Microparticles From Acute Coronary Syndrome Patients Induce Premature Coronary Artery Endothelial Cell Aging and Thrombogenicity: Role of the Ang II/AT1 Receptor/NADPH Oxidase-Mediated Activation of MAPKs and PI3-Kinase Pathways. Circulation 2017, 135, 280–296. [Google Scholar] [CrossRef]
- Yoshida, Y.; Shimizu, I.; Katsuumi, G.; Jiao, S.; Suda, M.; Hayashi, Y.; Minamino, T. p53-Induced inflammation exacerbates cardiac dysfunction during pressure overload. J. Mol. Cell Cardiol. 2015, 85, 183–198. [Google Scholar] [CrossRef]
- Meyer, K.; Hodwin, B.; Ramanujam, D.; Engelhardt, S.; Sarikas, A. Essential Role for Premature Senescence of Myofibroblasts in Myocardial Fibrosis. J. Am. Coll. Cardiol. 2016, 67, 2018–2028. [Google Scholar] [CrossRef]
- Anselm, E.; Chataigneau, M.; Ndiaye, M.; Chataigneau, T.; Schini-Kerth, V.B. Grape juice causes endothelium-dependent relaxation via a redox-sensitive Src- and Akt-dependent activation of eNOS. Cardiovasc. Res. 2007, 73, 404–413. [Google Scholar] [CrossRef]
- Xie, J.; Chen, Y.; Hu, C.; Pan, Q.; Wang, B.; Li, X.; Geng, J.; Xu, B. Premature senescence of cardiac fibroblasts and atrial fibrosis in patients with atrial fibrillation. Oncotarget 2017, 8, 57981–57990. [Google Scholar] [CrossRef]
- Kim, N.H.; Ahn, Y.; Oh, S.K.; Cho, J.K.; Park, H.W.; Kim, Y.S.; Hong, M.H.; Nam, K.I.; Park, W.J.; Jeong, M.H.; et al. Altered patterns of gene expression in response to chronic atrial fibrillation. Int. Heart J. 2005, 46, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Hasan, H.; Park, S.H.; Auger, C.; Belcastro, E.; Matsushita, K.; Marchandot, B.; Lee, H.H.; Qureshi, A.W.; Kauffenstein, G.; Ohlmann, P.; et al. Thrombin Induces Angiotensin II-Mediated Senescence in Atrial Endothelial Cells: Impact on Pro-Remodeling Patterns. J. Clin. Med. 2019, 8, 1570. [Google Scholar] [CrossRef] [PubMed]
- Krtolica, A.; Parrinello, S.; Lockett, S.; Desprez, P.Y.; Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc. Natl. Acad. Sci. USA 2001, 98, 12072–12077. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Correia-Melo, C.; Hewitt, G.; Passos, J.F. Telomeres, oxidative stress and inflammatory factors: Partners in cellular senescence? Longev. Healthspan. 2014, 3, 1. [Google Scholar] [CrossRef]
- Akar, J.G.; Jeske, W.; Wilber, D.J. Acute onset human atrial fibrillation is associated with local cardiac platelet activation and endothelial dysfunction. J. Am. Coll. Cardiol. 2008, 51, 1790–1793. [Google Scholar] [CrossRef]
- Shin, S.Y.; Na, J.O.; Lim, H.E.; Choi, C.U.; Choi, J.I.; Kim, S.H.; Kim, E.J.; Park, S.W.; Rha, S.W.; Park, C.G.; et al. Improved endothelial function in patients with atrial fibrillation through maintenance of sinus rhythm by successful catheter ablation. J. Cardiovasc. Electrophysiol. 2011, 22, 376–382. [Google Scholar] [CrossRef]
- Kim, Y.M.; Guzik, T.J.; Zhang, Y.H.; Zhang, M.H.; Kattach, H.; Ratnatunga, C.; Pillai, R.; Channon, K.M.; Casadei, B. A myocardial Nox2 containing NAD(P)H oxidase contributes to oxidative stress in human atrial fibrillation. Circ. Res. 2005, 97, 629–636. [Google Scholar] [CrossRef]
- Hecker, L.; Vittal, R.; Jones, T.; Jagirdar, R.; Luckhardt, T.R.; Horowitz, J.C.; Pennathur, S.; Martinez, F.J.; Thannickal, V.J. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 2009, 15, 1077–1081. [Google Scholar] [CrossRef]
- Simmers, M.B.; Cole, B.K.; Ogletree, M.L.; Chen, Z.; Xu, Y.; Kong, L.J.; Mackman, N.; Blackman, B.R.; Wamhoff, B.R. Hemodynamics associated with atrial fibrillation directly alters thrombotic potential of endothelial cells. Thromb. Res. 2016, 143, 34–39. [Google Scholar] [CrossRef]
- Wakula, P.; Neumann, B.; Kienemund, J.; Thon-Gutschi, E.; Stojakovic, T.; Manninger, M.; Scherr, D.; Scharnagl, H.; Kapl, M.; Pieske, B.; et al. CHA2DS2-VASc score and blood biomarkers to identify patients with atrial high-rate episodes and paroxysmal atrial fibrillation. Europace 2017, 19, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Hoit, B.D. Matrix metalloproteinases and atrial structural remodeling. J. Am. Coll. Cardiol. 2003, 42, 345–347. [Google Scholar] [CrossRef][Green Version]
- Gramley, F.; Lorenzen, J.; Plisiene, J.; Rakauskas, M.; Benetis, R.; Schmid, M.; Autschbach, R.; Knackstedt, C.; Schimpf, T.; Mischke, K.; et al. Decreased plasminogen activator inhibitor and tissue metalloproteinase inhibitor expression may promote increased metalloproteinase activity with increasing duration of human atrial fibrillation. J. Cardiovasc. Electrophysiol. 2007, 18, 1076–1082. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Fujimaki, T.; Yoshida, T.; Oguri, M.; Yajima, K.; Hibino, T.; Murohara, T. Impact of matrix metalloproteinase-2 levels on long-term outcome following pharmacological or electrical cardioversion in patients with atrial fibrillation. Europace 2009, 11, 332–337. [Google Scholar] [CrossRef]
- Chiao, Y.A.; Ramirez, T.A.; Zamilpa, R.; Okoronkwo, S.M.; Dai, Q.; Zhang, J.; Jin, Y.F.; Lindsey, M.L. Matrix metalloproteinase-9 deletion attenuates myocardial fibrosis and diastolic dysfunction in ageing mice. Cardiovasc. Res. 2012, 96, 444–455. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Variable | Sinus Rhythm | Atrial Fibrillation | p |
|---|---|---|---|
| Age (years) | 68 ± 11 | 70 ± 12 | 0.63 |
| Sex (male) | 13 | 13 | 1 |
| Hypertension | 14 | 13 | 0.75 |
| Diabetes | 7 | 8 | 0.75 |
| Smoking | 7 | 7 | 1 |
| LVEF < 40% | 1 | 1 | |
| Vascular Disease | 6 | 4 | 0.47 |
| Body Mass Index | 28.4 ± 5.7 | 30.5 ± 6.9 | 0.30 |
| Euroscore I | 4.2 ± 3.1 | 6.8 ± 5.1 | 0.06 |
| Euroscore II | 1.9 ± 1.8 | 3.5 ± 3.2 | 0.06 |
| CHA2DS2-VASc | |||
| 0–1 | 6 | 5 | |
| 2–3 | 8 | 7 | |
| ≥4 | 7 | 9 | |
| Drugs | |||
| Beta-Blockers | 12 | 15 | 0.33 |
| ACE inhibitor/AT1 blocker | 14 | 11 | 0.20 |
| Aspirin | 14 | 2 | <0.001 |
| VKA | 2 | 14 | <0.001 |
| Statin | 12 | 11 | |
| Echographic Data | |||
| LA Area (cm²) | 23 ± 9 | 36 ± 19 | 0.02 |
| RA Area (cm²) | 18 ± 7 | 23 ± 9 | 0.15 |
| LVEF (%) | 62 ± 8 | 60 ± 8 | 0.41 |
| EDLVD (mm) | 51 ± 8 | 53 ± 10 | 0.50 |
| LV Mass (g) | 133 ± 41 | 127 ± 40 | 0.65 |
| Biology | |||
| Hb (g/L) | 13.5 ± 1.1 | 13.5 ± 1.6 | 0.99 |
| Leukocytes | 6677 ± 2131 | 7191 ± 2004 | 0.43 |
| Fibrinogen (g/L) | 3.4 ± 0.5 | 3.6 ± 0.9 | 0.58 |
| Creatinin Clearance (mL/min) | 87 ± 30 | 78 ± 33 | 0.45 |
| Variable | Univariate Analysis | Multivariate Analysis | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | p | HR | 95% CI | p | |
| Age | 1.011 | 0.959–1.066 | 0.68 | |||
| AF | 10.240 | 2.475–42.370 | 0.001 | 7.849 | 1.330–46.333 | 0.023 |
| Permanent AF | 15.000 | 1.685–133.551 | 0.015 | |||
| Paroxysmal AF | 1.600 | 0.413–6.193 | 0.49 | |||
| AF at the Time of Surgery | 8.000 | 2.012–31.803 | 0.003 | |||
| Hypertension | 1.231 | 0.38–4.358 | 0.75 | |||
| Diabetes Mellitus | 0.533 | 0.148–1.922 | 0.34 | |||
| Smoking | 0.538 | 0.422–5.606 | 0.51 | |||
| Female Sex | 1.083 | 0.288–4.081 | 0.91 | |||
| LVEF <40% | 2.105 | 0.176–25.170 | 0.56 | |||
| CHADS2-VASc | 1.085 | 0.760–1.549 | 0.65 | |||
| Statins | 0.699 | 0.433–1.129 | 0.14 | |||
| ACE inhibitor/AT1 blocker | 0.667 | 0.190–2.334 | 0.53 | |||
| Coronary Artery Disease | 0.300 | 0.083–1.081 | 0.07 | |||
| Euroscore I | 1.089 | 0.937–1.266 | 0.27 | |||
| Euroscore II | 1.063 | 0.844–1.338 | 0.60 | |||
| LA Area | 1.032 | 0.973–1.094 | 0.30 | |||
| p16 | 16.121 | 1.461–177.851 | 0.023 | 1.986 | 0.109–36.312 | 0.643 |
| eNOS | 0.041 | 0.001-4.086 | 0.174 | |||
| Variable | Univariate Analysis | Multivariate Analysis | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | p | HR | 95% CI | p | |
| Age | 1.010 | 0.959–1.065 | 0.70 | |||
| AF | 18.062 | 0.871–84.283 | <0.001 | 15.741 | 2.863–86.556 | 0.002 |
| Permanent AF | 3.000 | 0.655–13.747 | 0.16 | |||
| Paroxysmal AF | 8.636 | 1.593–46.807 | 0.012 | |||
| AF at the Time of Surgery | 13.600 | 3.091–59.831 | 0.001 | |||
| Hypertension | 0.533 | 0.148–1.922 | 0.34 | |||
| Diabetes Mellitus | 0.813 | 0.229–2.877 | 0.75 | |||
| Smoking | 1.538 | 0.422–5.606 | 0.51 | |||
| Female Sex | 0.686 | 0.182–2.589 | 0.58 | |||
| LVEF <40% | 1.90 | 0.180–19.015 | 0.56 | |||
| CHADS2-VASc | 1.050 | 0.737–1.498 | 0.79 | |||
| Statins | 1.250 | 0.274–5.705 | 0.77 | |||
| ACE inhibitor/AT1blocker | 0.284 | 0.076–1.063 | 0.06 | |||
| Coronary artery disease | 0.677 | 0.198–2.312 | 0.53 | |||
| Euroscore I | 1.036 | 0.899–1.194 | 0.62 | |||
| Euroscore II | 1.091 | 0.861–1.383 | 0.47 | |||
| LA Area | 1.028 | 0.972–1.087 | 0.33 | |||
| p53 | 7.842 | 1.167–52.711 | 0.034 | 1.532 | 0.133–17.610 | 0.73 |
| eNOS | 0.05 | 0.01–4.834 | 0.199 | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jesel, L.; Abbas, M.; Park, S.-H.; Matsushita, K.; Kindo, M.; Hasan, H.; Auger, C.; Sato, C.; Ohlmann, P.; Mazzucotelli, J.-P.; et al. Atrial Fibrillation Progression Is Associated with Cell Senescence Burden as Determined by p53 and p16 Expression. J. Clin. Med. 2020, 9, 36. https://doi.org/10.3390/jcm9010036
Jesel L, Abbas M, Park S-H, Matsushita K, Kindo M, Hasan H, Auger C, Sato C, Ohlmann P, Mazzucotelli J-P, et al. Atrial Fibrillation Progression Is Associated with Cell Senescence Burden as Determined by p53 and p16 Expression. Journal of Clinical Medicine. 2020; 9(1):36. https://doi.org/10.3390/jcm9010036
Chicago/Turabian StyleJesel, Laurence, Malak Abbas, Sin-Hee Park, Kensuke Matsushita, Michel Kindo, Hira Hasan, Cyril Auger, Chisato Sato, Patrick Ohlmann, Jean-Philippe Mazzucotelli, and et al. 2020. "Atrial Fibrillation Progression Is Associated with Cell Senescence Burden as Determined by p53 and p16 Expression" Journal of Clinical Medicine 9, no. 1: 36. https://doi.org/10.3390/jcm9010036
APA StyleJesel, L., Abbas, M., Park, S.-H., Matsushita, K., Kindo, M., Hasan, H., Auger, C., Sato, C., Ohlmann, P., Mazzucotelli, J.-P., Toti, F., Kauffenstein, G., Schini-Kerth, V., & Morel, O. (2020). Atrial Fibrillation Progression Is Associated with Cell Senescence Burden as Determined by p53 and p16 Expression. Journal of Clinical Medicine, 9(1), 36. https://doi.org/10.3390/jcm9010036