Bioinformatics and Computational Tools for Next-Generation Sequencing Analysis in Clinical Genetics



Abstract
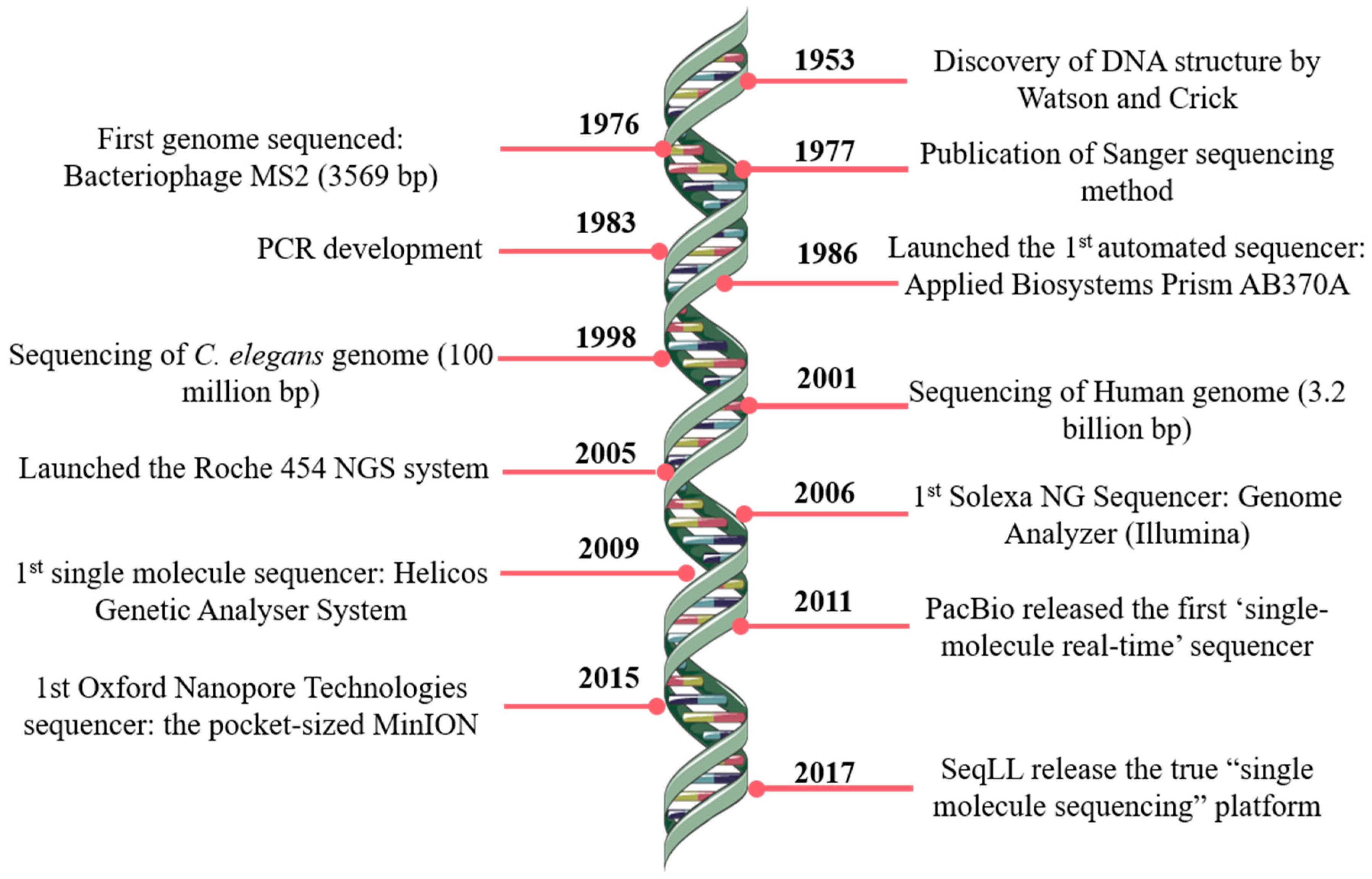
1. Introduction
2. NGS Library
3. NGS Platforms
3.1. Second-Generation Sequencing Platforms
3.2. Third-Generation Sequencers
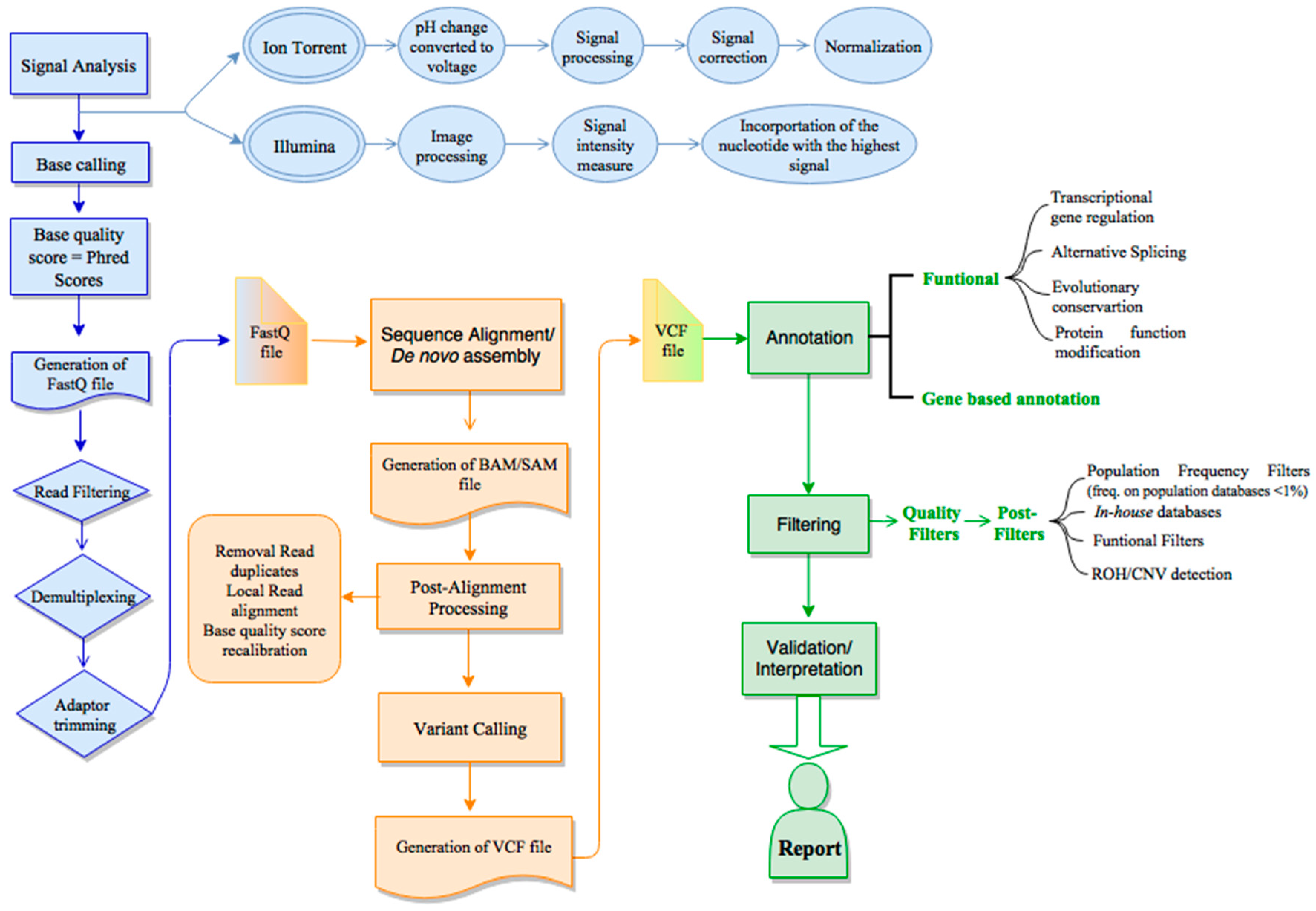
4. NGS Bioinformatics
4.1. Primary Analysis
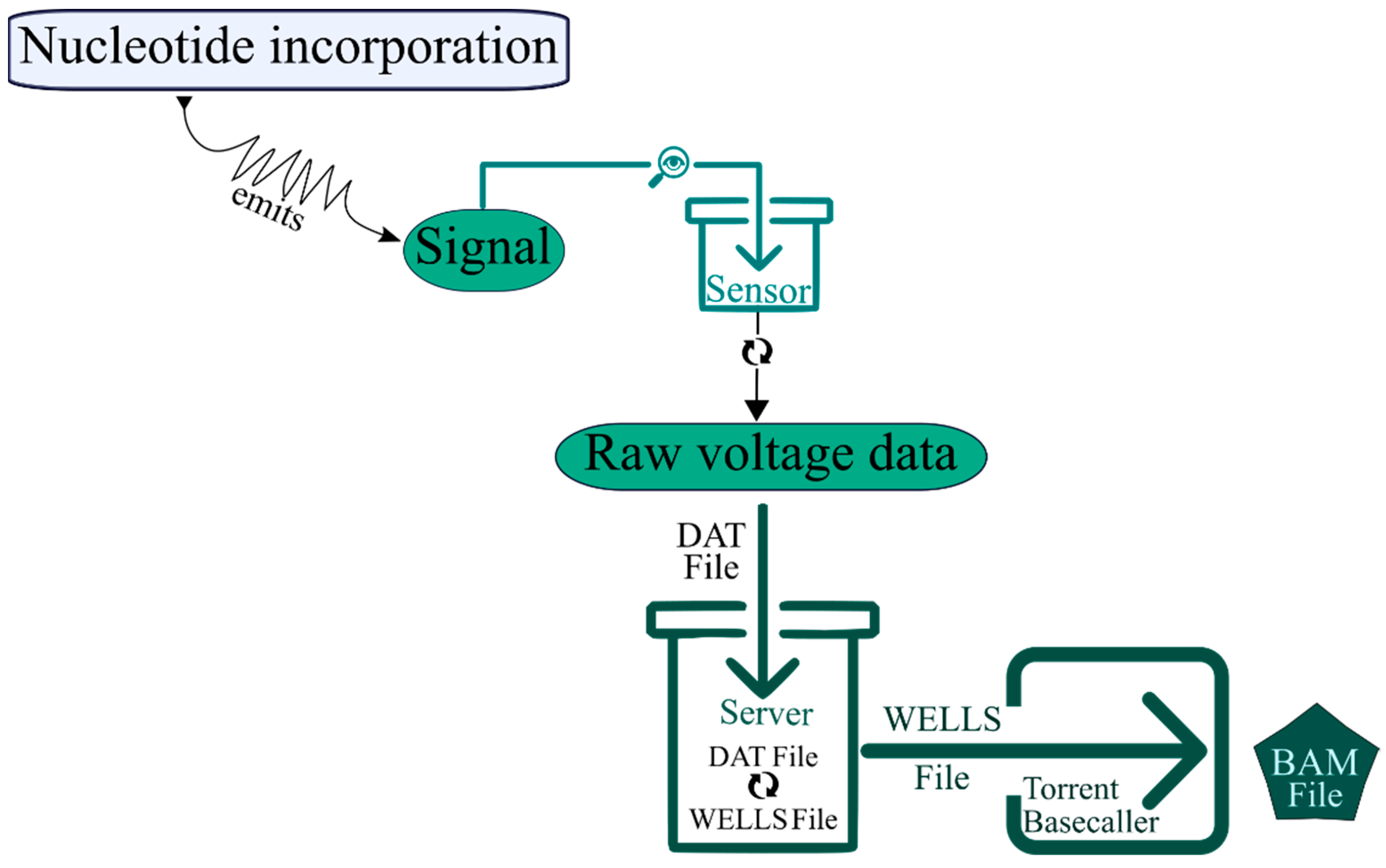
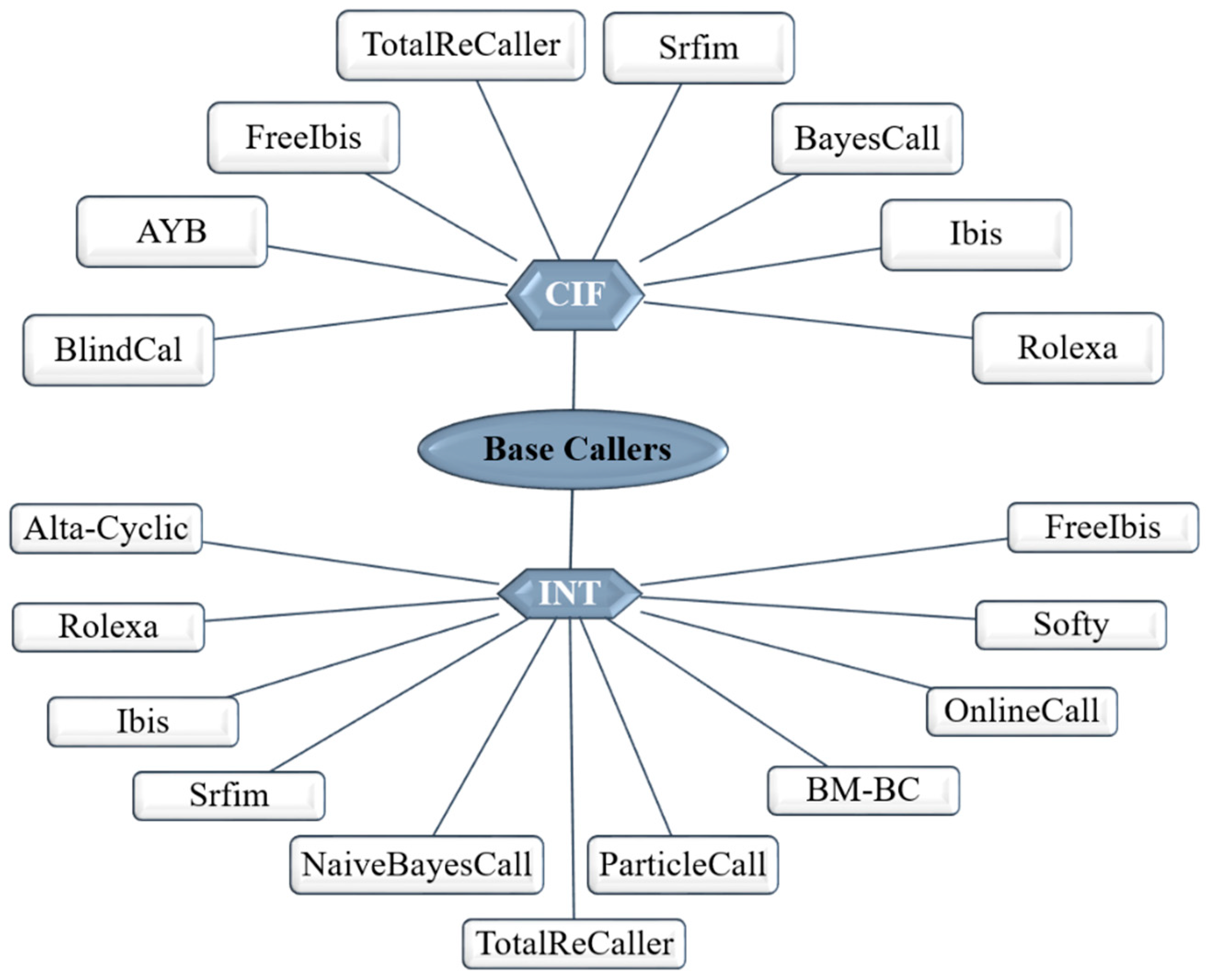
4.1.1. Ion Torrent
4.1.2. Illumina
4.1.3. Quality Control: Read Filtering and Trimming
4.2. Secondary Analysis
4.2.1. Sequence Alignment
De novo Assembly
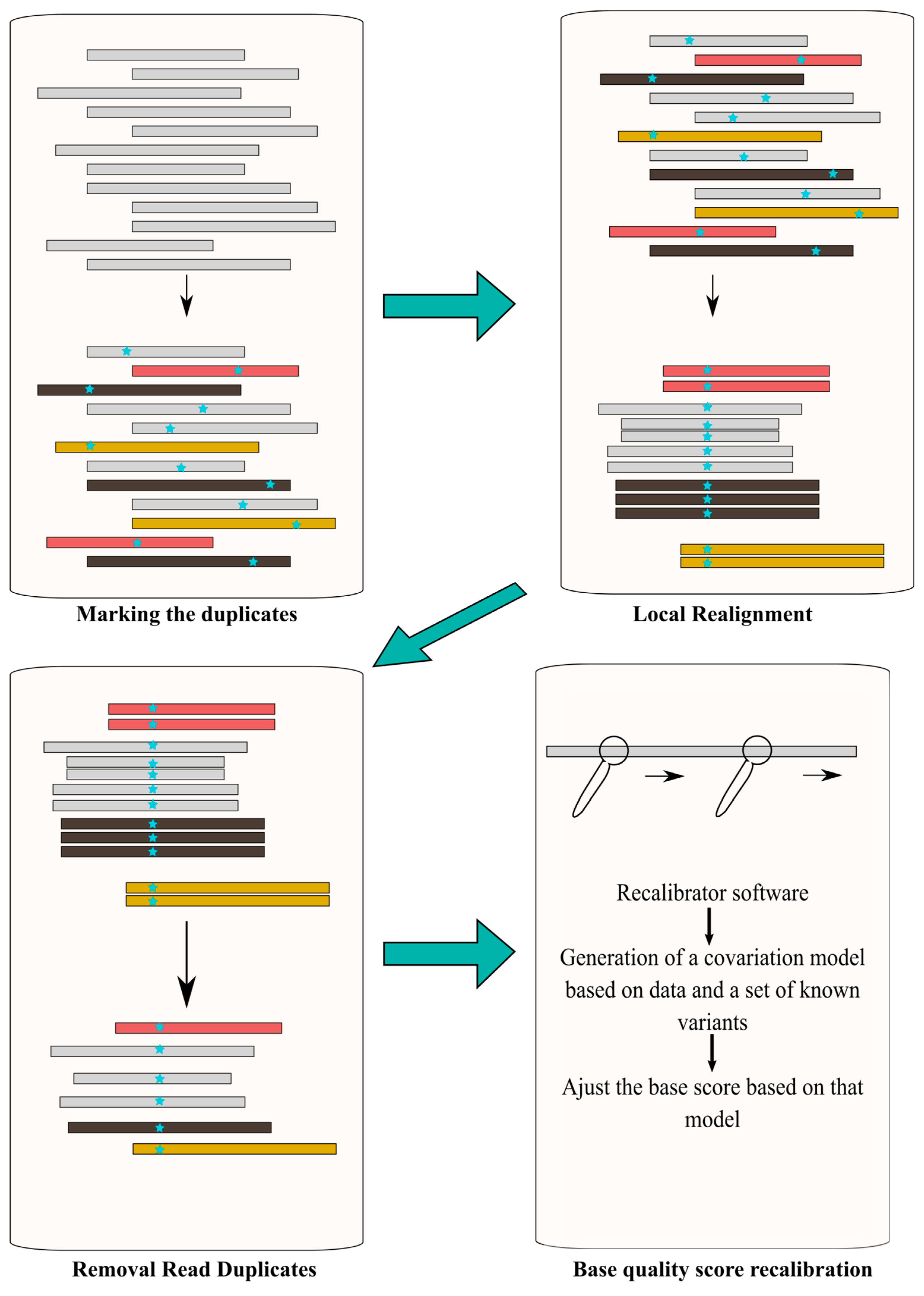
4.2.2. Post-Alignment Processing
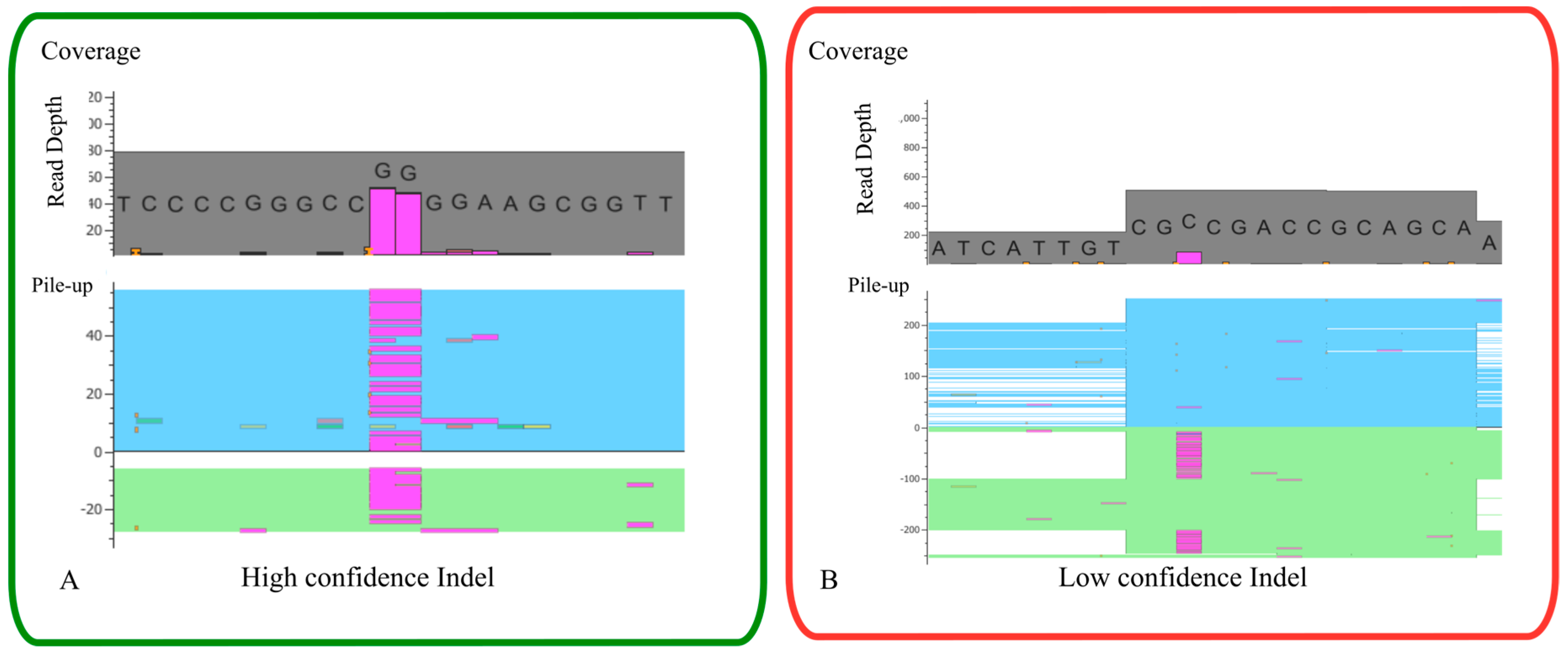
4.2.3. Variant Calling
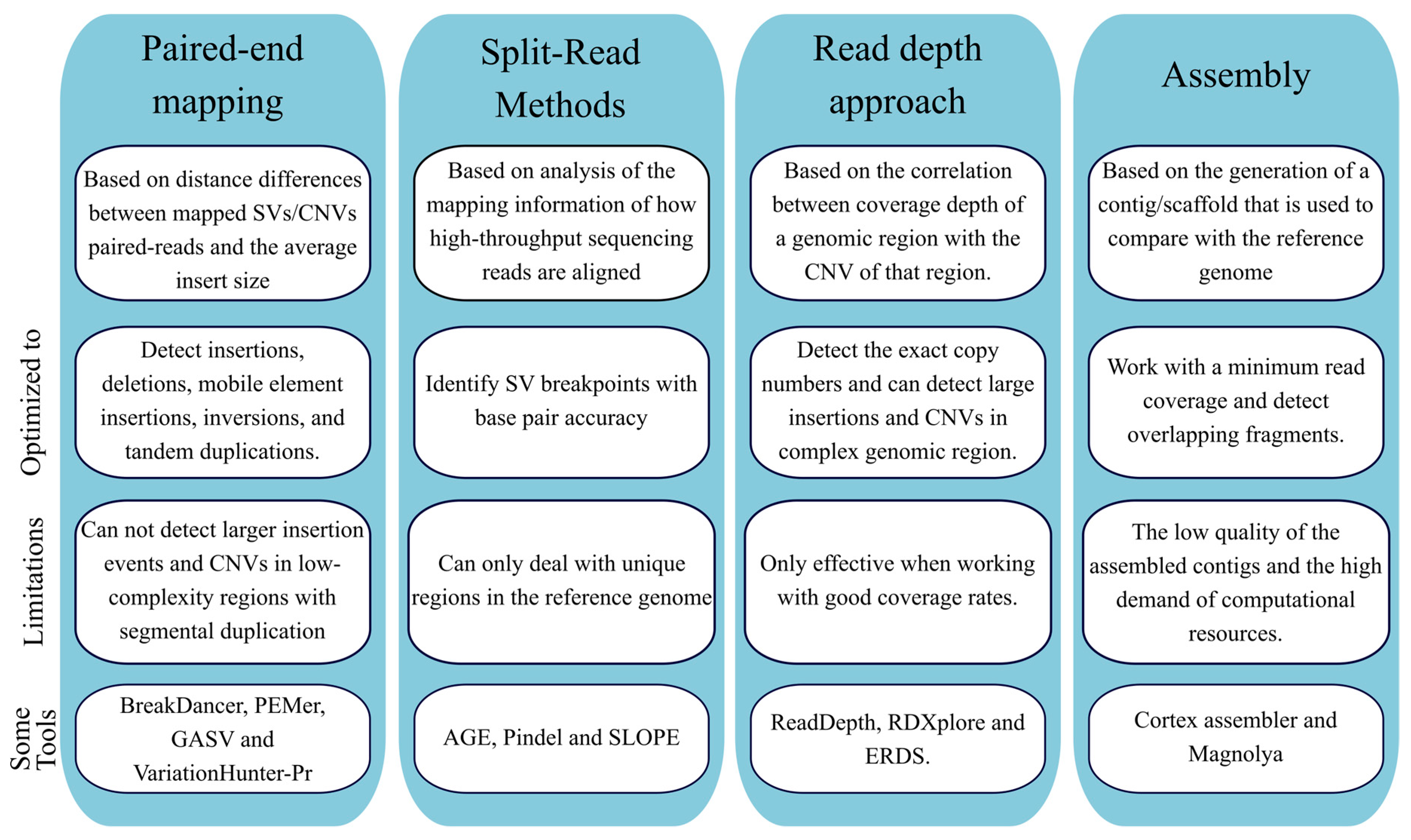
Structural Variants Calling
4.3. Tertiary Analysis
4.3.1. Variant Annotation
4.3.2. Variant Filtering, Prioritization and Visualization
5. NGS Pitfalls
6. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Jackson, D.A.; Symonst, R.H.; Berg, P. Biochemical Method for Inserting New Genetic Information into DNA of Simian Virus 40: Circular SV40 DNA Molecules Containing Lambda Phage Genes and the Galactose Operon of Escherichia coli. Proc. Natl. Acad. Sci. USA 1972, 69, 2904–2909. [Google Scholar] [CrossRef]
- Sanger, F.; Coulson, A.R. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J. Mol. Biol. 1975, 94, 441–448. [Google Scholar] [CrossRef]
- Maxam, A.M.; Gilbert, W. A new method for sequencing DNA. Proc. Natl. Acad. Sci. USA 1977, 74, 560–564. [Google Scholar] [CrossRef]
- Sanger, F.; Nicklen, S.; Coulson, A.R. Biochemistry DNA sequencing with chain-terminating inhibitors (DNA polymerase/nucleotide sequences/bacteriophage 4X174). Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [PubMed]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [PubMed]
- Shendure, J.; Ji, H. Next-generation DNA sequencing. Natl. Biotechnol. 2008, 26, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Schadt, E.E.; Turner, S.; Kasarskis, A. A window into third-generation sequencing. Hum. Mol. Genet. 2010, 19, R227–R240. [Google Scholar] [CrossRef] [PubMed]
- Ameur, A.; Kloosterman, W.P.; Hestand, M.S. Single-Molecule Sequencing: Towards Clinical Applications. Trends Biotechnol. 2019, 37, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, E.L.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The Third Revolution in Sequencing Technology. Trends Genet. 2018, 34, 666–681. [Google Scholar] [CrossRef]
- Aird, D.; Ross, M.G.; Chen, W.-S.; Danielsson, M.; Fennell, T.; Russ, C.; Jaffe, D.B.; Nusbaum, C.; Gnirke, A. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. 2011, 12, R18. [Google Scholar] [CrossRef]
- Head, S.R.; Komori, H.K.; LaMere, S.A.; Whisenant, T.; Van Nieuwerburgh, F.; Salomon, D.R.; Ordoukhanian, P. Library construction for next-generation sequencing: Overviews and challenges. Biotechniques 2014, 56, 61. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, E.L.; Jaszczyszyn, Y.; Thermes, C. Library preparation methods for next-generation sequencing: Tone down the bias. Exp. Cell Res. 2014, 322, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Knierim, E.; Lucke, B.; Schwarz, J.M.; Schuelke, M.; Seelow, D. Systematic Comparison of Three Methods for Fragmentation of Long-Range PCR Products for Next Generation Sequencing. PLoS ONE 2011, 6, e28240. [Google Scholar] [CrossRef] [PubMed]
- Illumina. Nextera XT Library Prep: Tips and Troubleshooting. Available online: https://www.illumina.com/content/dam/illumina-marketing/documents/products/technotes/nextera-xt-troubleshooting-technical-note.pdf (accessed on 1 November 2019).
- Ion Torrent. APPLICATION NOTE Ion PGM™ Small Genome Sequencing. Available online: https://tools.thermofisher.com/content/sfs/brochures/Small-Genome-Ecoli-De-Novo-App-Note.pdf (accessed on 13 December 2019).
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef]
- Sakharkar, M.K.; Chow, V.T.K.; Kangueane, P. Distributions of Exons and Introns in the Human Genome. Silico Biol. 2004, 4, 387–393. [Google Scholar]
- Samorodnitsky, E.; Jewell, B.M.; Hagopian, R.; Miya, J.; Wing, M.R.; Lyon, E.; Damodaran, S.; Bhatt, D.; Reeser, J.W.; Datta, J.; et al. Evaluation of Hybridization Capture Versus Amplicon-Based Methods for Whole-Exome Sequencing. Hum. Mutat. 2015, 36, 903–914. [Google Scholar] [CrossRef]
- Hung, S.S.; Meissner, B.; Chavez, E.A.; Ben-Neriah, S.; Ennishi, D.; Jones, M.R.; Shulha, H.P.; Chan, F.C.; Boyle, M.; Kridel, R.; et al. Assessment of Capture and Amplicon-Based Approaches for the Development of a Targeted Next-Generation Sequencing Pipeline to Personalize Lymphoma Management. J. Mol. Diagn. 2018, 20, 203–214. [Google Scholar] [CrossRef]
- Horn, S. Target Enrichment via DNA Hybridization Capture. In Ancient DNA: Methods and Protocols; Shapiro, B., Hofreiter, M., Eds.; Humana Press: Totowa, NJ, USA, 2012; pp. 177–188. [Google Scholar] [CrossRef]
- Kanagawa, T. Bias and artifacts in multitemplate polymerase chain reactions (PCR). J. Biosci. Bioeng. 2003, 96, 317–323. [Google Scholar] [CrossRef]
- Sloan, D.B.; Broz, A.K.; Sharbrough, J.; Wu, Z. Detecting Rare Mutations and DNA Damage with Sequencing-Based Methods. Trends Biotechnol. 2018, 36, 729–740. [Google Scholar] [CrossRef]
- Fu, Y.; Wu, P.-H.; Beane, T.; Zamore, P.D.; Weng, Z. Elimination of PCR duplicates in RNA-seq and small RNA-seq using unique molecular identifiers. BMC Genom. 2018, 19, 531. [Google Scholar] [CrossRef]
- Hong, J.; Gresham, D. Incorporation of unique molecular identifiers in TruSeq adapters improves the accuracy of quantitative sequencing. Biotechniques 2017, 63, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Hall, K.P.; Evers, D.J.; Barnes, C.L.; Bignell, H.R.; et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008, 456, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Fuller, C.W.; Middendorf, L.R.; Benner, S.A.; Church, G.M.; Harris, T.; Huang, X.; Jovanovich, S.B.; Nelson, J.R.; Schloss, J.A.; Schwartz, D.C.; et al. The challenges of sequencing by synthesis. Nat. Biotechnol. 2009, 27, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Heyn, P.; Kelso, J. Addressing challenges in the production and analysis of illumina sequencing data. BMC Genom. 2011, 12, 382. [Google Scholar] [CrossRef]
- Rothberg, J.M.; Hinz, W.; Rearick, T.M.; Schultz, J.; Mileski, W.; Davey, M.; Leamon, J.H.; Johnson, K.; Milgrew, M.J.; Edwards, M. An integrated semiconductor device enabling non-optical genome sequencing. Nature 2011, 475, 348. [Google Scholar] [CrossRef]
- Merriman, B.; Ion Torrent R&D Team; Rothberg, J.M. Progress in Ion Torrent semiconductor chip based sequencing. Electrophoresis 2012, 33, 3397–3417. [Google Scholar] [CrossRef]
- Quail, M.A.; Smith, M.; Coupland, P.; Otto, T.D.; Harris, S.R.; Connor, T.R.; Bertoni, A.; Swerdlow, H.P.; Gu, Y. A tale of three next generation sequencing platforms: Comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genom. 2012, 13, 1. [Google Scholar] [CrossRef]
- Thompson, J.F.; Steinmann, K.E. Single molecule sequencing with a HeliScope genetic analysis system. Curr. Protoc. Mol. Biol. 2010, 7, 10. [Google Scholar] [CrossRef]
- Pushkarev, D.; Neff, N.F.; Quake, S.R. Single-molecule sequencing of an individual human genome. Nat. Biotechnol. 2009, 27, 847–850. [Google Scholar] [CrossRef]
- McCarthy, A. Third Generation DNA Sequencing: Pacific Biosciences’ Single Molecule Real Time Technology. Chem. Biol. 2010, 17, 675–676. [Google Scholar] [CrossRef]
- Nakano, K.; Shiroma, A.; Shimoji, M.; Tamotsu, H.; Ashimine, N.; Ohki, S.; Shinzato, M.; Minami, M.; Nakanishi, T.; Teruya, K.; et al. Advantages of genome sequencing by long-read sequencer using SMRT technology in medical area. Hum. Cell 2017, 30, 149–161. [Google Scholar] [CrossRef] [PubMed]
- PacBio. SMRT Sequencing—Delivering Highly Accurate Long Reads to Drive Discovery in Life Science. Available online: https://www.pacb.com/wp-content/uploads/SMRT-Sequencing-Brochure-Delivering-highly-accurate-long-reads-to-drive-discovery-in-life-science.pdf (accessed on 9 December 2019).
- Merker, J.D.; Wenger, A.M.; Sneddon, T.; Grove, M.; Zappala, Z.; Fresard, L.; Waggott, D.; Utiramerur, S.; Hou, Y.; Smith, K.S.; et al. Long-read genome sequencing identifies causal structural variation in a Mendelian disease. Genet. Med. 2018, 20, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Technologies, O.N. Company History. Available online: https://nanoporetech.com/about-us/history (accessed on 13 December 2019).
- Deamer, D.; Akeson, M.; Branton, D. Three decades of nanopore sequencing. Nat. Biotechnol. 2016, 34, 518. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Fiddes, I.T.; Miga, K.H.; Olsen, H.E.; Paten, B.; Akeson, M. Improved data analysis for the MinION nanopore sequencer. Nat. Methods 2015, 12, 351–356. [Google Scholar] [CrossRef]
- Nanopore, O. High-Throughput, Real-Time and On-Demand Sequencing for Your Lab. Available online: https://nanoporetech.com/sites/default/files/s3/literature/GridION-Brochure-14Mar2019.pdf (accessed on 9 December 2019).
- Goodwin, S.; Gurtowski, J.; Ethe-Sayers, S.; Deshpande, P.; Schatz, M.C.; McCombie, W.R. Oxford Nanopore sequencing, hybrid error correction, and de novo assembly of a eukaryotic genome. Genome Res. 2015, 25, 1750–1756. [Google Scholar] [CrossRef]
- De Coster, W.; De Rijk, P.; De Roeck, A.; De Pooter, T.; D’Hert, S.; Strazisar, M.; Sleegers, K.; Van Broeckhoven, C. Structural variants identified by Oxford Nanopore PromethION sequencing of the human genome. Genome Res. 2019, 29, 1178–1187. [Google Scholar] [CrossRef]
- Xu, L.; Seki, M. Recent advances in the detection of base modifications using the Nanopore sequencer. J. Hum. Genet. 2020, 65, 25–33. [Google Scholar] [CrossRef]
- Genomics, X. The Power of Massively Parallel Partitioning. Available online: https://pages.10xgenomics.com/rs/446-PBO-704/images/10x_BR025_Chromium-Brochure_Letter_Digital.pdf (accessed on 13 December 2019).
- Zheng, G.X.Y.; Lau, B.T.; Schnall-Levin, M.; Jarosz, M.; Bell, J.M.; Hindson, C.M.; Kyriazopoulou-Panagiotopoulou, S.; Masquelier, D.A.; Merrill, L.; Terry, J.M.; et al. Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nat. Biotechnol. 2016, 34, 303–311. [Google Scholar] [CrossRef]
- Zheng, G.X.Y.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef]
- Granja, J.M.; Klemm, S.; McGinnis, L.M.; Kathiria, A.S.; Mezger, A.; Corces, M.R.; Parks, B.; Gars, E.; Liedtke, M.; Zheng, G.X.Y.; et al. Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia. Nat. Biotechnol. 2019, 37, 1458–1465. [Google Scholar] [CrossRef]
- Zeng, Y.; Liu, C.; Gong, Y.; Bai, Z.; Hou, S.; He, J.; Bian, Z.; Li, Z.; Ni, Y.; Yan, J.; et al. Single-Cell RNA Sequencing Resolves Spatiotemporal Development of Pre-thymic Lymphoid Progenitors and Thymus Organogenesis in Human Embryos. Immunity 2019, 51, 930–948. [Google Scholar] [CrossRef] [PubMed]
- Laurentino, S.; Heckmann, L.; Di Persio, S.; Li, X.; Meyer zu Hörste, G.; Wistuba, J.; Cremers, J.-F.; Gromoll, J.; Kliesch, S.; Schlatt, S.; et al. High-resolution analysis of germ cells from men with sex chromosomal aneuploidies reveals normal transcriptome but impaired imprinting. Clin. Epigenet. 2019, 11, 127. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xiong, X.; Cao, W.; Zhang, C.; Werren, J.H.; Wang, X. Genome Assembly of the A-Group Wolbachia in Nasonia oneida Using Linked-Reads Technology. Genome Biol. Evol. 2019, 11, 3008–3013. [Google Scholar] [CrossRef] [PubMed]
- Delaneau, O.; Zagury, J.-F.; Robinson, M.R.; Marchini, J.L.; Dermitzakis, E.T. Accurate, scalable and integrative haplotype estimation. Nat. Commun. 2019, 10, 5436. [Google Scholar] [CrossRef]
- Nanopore, O. Terabases of Long-Read Sequence Data, Analysed in Real Time. Available online: https://nanoporetech.com/sites/default/files/s3/literature/PromethION-Brochure-14Mar2019.pdf (accessed on 9 December 2019).
- Ion Torrent. Torrent Suite-Signal Processing and Base Calling Application Note Torrent Suite Software Analysis Pipeline. Technical Note. Available online: http://coolgenes.cahe.wsu.edu/ion-docs/Technical-Note---Analysis-Pipeline_6455567.html (accessed on 26 February 2019).
- McKinnon, K.I. Convergence of the Nelder-Mead Simplex Method to a Nonstationary Point. SIAM J. Optim. 1998, 9, 148–158. [Google Scholar] [CrossRef]
- Mehlhorn, K.; Sanders, P. Generic Approaches to Optimization. In Algorithms and Data Structures: The Basic Toolbox; Springer Science & Business Media: Heidelberg, Germany, 2008. [Google Scholar] [CrossRef]
- Illumina. Illumina Sequencing Technology: Technology Spotlight: Illumina® Sequencing. Available online: https://www.illumina.com/documents/products/techspotlights/techspotlight_sequencing.pdf (accessed on 13 December 2019).
- Erlich, Y.; Mitra, P.P.; delaBastide, M.; McCombie, W.R.; Hannon, G.J. Alta-Cyclic: A self-optimizing base caller for next-generation sequencing. Nat. Methods 2008, 5, 679–682. [Google Scholar] [CrossRef]
- Kao, W.-C.; Stevens, K.; Song, Y.S. BayesCall: A model-based base-calling algorithm for high-throughput short-read sequencing. Genome Res. 2009, 19, 1884–1895. [Google Scholar] [CrossRef]
- Ledergerber, C.; Dessimoz, C. Base-calling for next-generation sequencing platforms. Brief. Bioinform. 2011, 12, 489–497. [Google Scholar] [CrossRef]
- Cacho, A.; Smirnova, E.; Huzurbazar, S.; Cui, X. A Comparison of Base-calling Algorithms for Illumina Sequencing Technology. Brief. Bioinform. 2015, 17, 786–795. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, B.; Wan, L.; Li, L.M. Estimating Phred scores of Illumina base calls by logistic regression and sparse modeling. BMC Bioinform. 2017, 18, 335. [Google Scholar] [CrossRef]
- Ewing, B.; Hillier, L.; Wendl, M.C.; Green, P. Base-calling of automated sequencer traces usingPhred. I. Accuracy assessment. Genome Res. 1998, 8, 175–185. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 11, 11.10.11–11.10.33. [Google Scholar] [CrossRef]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A Toolkit for Quality Control of Next Generation Sequencing Data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Su, X.; Wang, A.; Xu, J.; Ning, K. QC-Chain: Fast and Holistic Quality Control Method for Next-Generation Sequencing Data. PLoS ONE 2013, 8, e60234. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC A Quality Control tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 October 2019).
- Kong, Y. Btrim: A fast, lightweight adapter and quality trimming program for next-generation sequencing technologies. Genomics 2011, 98, 152–153. [Google Scholar] [CrossRef]
- Renaud, G.; Stenzel, U.; Kelso, J. leeHom: Adaptor trimming and merging for Illumina sequencing reads. Nucleic Acids Res. 2014, 42, e141. [Google Scholar] [CrossRef]
- Lindgreen, S. AdapterRemoval: Easy Cleaning of Next Generation Sequencing Reads. BMC Res. Notes 2012, 5, 337. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Del Fabbro, C.; Scalabrin, S.; Morgante, M.; Giorgi, F.M.; Binkley, G. An Extensive Evaluation of Read Trimming Effects on Illumina NGS Data Analysis. PLoS ONE 2013, 8, e85024. [Google Scholar] [CrossRef]
- Gargis, A.S.; Kalman, L.; Lubin, I.M. Assay Validation. In Clinical Genomics; Kulkarni, S., Pfeifer, J., Eds.; Academic Press: Boston, MA, USA, 2015; pp. 363–376. [Google Scholar]
- Flicek, P.; Birney, E. Sense from sequence reads: Methods for alignment and assembly. Nat. Methods 2009, 6, S6–S12. [Google Scholar] [CrossRef]
- Ameur, A.; Che, H.; Martin, M.; Bunikis, I.; Dahlberg, J.; Höijer, I.; Häggqvist, S.; Vezzi, F.; Nordlund, J.; Olason, P.; et al. De Novo Assembly of Two Swedish Genomes Reveals Missing Segments from the Human GRCh38 Reference and Improves Variant Calling of Population-Scale Sequencing Data. Genes 2018, 9, 486. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2012, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, N.A.; Rung, J.; Brazma, A.; Marioni, J.C. Tools for mapping high-throughput sequencing data. Bioinformatics 2012, 28, 3169–3177. [Google Scholar] [CrossRef]
- Miller, J.R.; Koren, S.; Sutton, G. Assembly algorithms for next-generation sequencing data. Genomics 2010, 95, 315–327. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows—Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Ruffalo, M.; LaFramboise, T.; Koyutürk, M. Comparative analysis of algorithms for next-generation sequencing read alignment. Bioinformatics 2011, 27, 2790–2796. [Google Scholar] [CrossRef]
- Homer, N. TMAP: The Torrent Mapping Program. Available online: https://github.com/iontorrent/TMAP/blob/master/doc/tmap-book.pdf (accessed on 1 October 2019).
- Li, Z.; Chen, Y.; Mu, D.; Yuan, J.; Shi, Y.; Zhang, H.; Gan, J.; Li, N.; Hu, X.; Liu, B.; et al. Comparison of the two major classes of assembly algorithms: Overlap–layout–consensus and de-bruijn-graph. Brief. Funct. Genom. 2012, 11, 25–37. [Google Scholar] [CrossRef]
- Pop, M.; Phillippy, A.; Delcher, A.L.; Salzberg, S.L. Comparative genome assembly. Brief. Bioinform. 2004, 5, 237–248. [Google Scholar] [CrossRef]
- Compeau, P.E.C.; Pevzner, P.A.; Tesler, G. How to apply de Bruijn graphs to genome assembly. Nat. Biotechnol. 2011, 29, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Sedlazeck, F.J.; Lee, H.; Darby, C.A.; Schatz, M.C. Piercing the dark matter: Bioinformatics of long-range sequencing and mapping. Nat. Rev. Genet. 2018, 19, 329–346. [Google Scholar] [CrossRef]
- Tian, S.; Yan, H.; Kalmbach, M.; Slager, S.L. Impact of post-alignment processing in variant discovery from whole exome data. BMC Bioinform. 2016, 17, 403. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P.N.; Piro, R.M.; Jager, M. Postprocessing the Alignment. In Computational Exome and Genome Analysis, 1st ed.; Chapman and Hall/CRC: New York, NY, USA, 2017. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Nielsen, R.; Paul, J.S.; Albrechtsen, A.; Song, Y.S. Genotype and SNP calling from next-generation sequencing data. Nat. Rev. Genet. 2011, 12, 443. [Google Scholar] [CrossRef] [PubMed]
- Libbrecht, M.W.; Noble, W.S. Machine learning applications in genetics and genomics. Nat. Rev. Genet. 2015, 16, 321. [Google Scholar] [CrossRef]
- Kuhn, M.; Stange, T.; Herold, S.; Thiede, C.; Roeder, I. Finding small somatic structural variants in exome sequencing data: A machine learning approach. Comput. Stat. 2018, 33, 1145–1158. [Google Scholar] [CrossRef]
- Hwang, S.; Kim, E.; Lee, I.; Marcotte, E.M. Systematic comparison of variant calling pipelines using gold standard personal exome variants. Sci. Rep. 2015, 5, 17875. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- The Variant Call Format (VCF) Version 4.2 Specification. Available online: https://samtools.github.io/hts-specs/VCFv4.2.pdf (accessed on 1 October 2019).
- Feuk, L.; Carson, A.R.; Scherer, S.W. Structural variation in the human genome. Nat. Rev. Genet. 2006, 7, 85–97. [Google Scholar] [CrossRef]
- Stankiewicz, P.; Lupski, J.R. Structural Variation in the Human Genome and its Role in Disease. Annu. Rev. Med. 2010, 61, 437–455. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhashi, S.; Matsumoto, N. Long-read sequencing for rare human genetic diseases. J. Hum. Genet. 2020, 65, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Kraft, F.; Kurth, I. Long-read sequencing in human genetics. Med. Genet. 2019, 31, 198–204. [Google Scholar] [CrossRef]
- Zhao, M.; Wang, Q.; Wang, Q.; Jia, P.; Zhao, Z. Computational tools for copy number variation (CNV) detection using next-generation sequencing data: Features and perspectives. BMC Bioinform. 2013, 14, S1. [Google Scholar] [CrossRef] [PubMed]
- Pirooznia, M.; Goes, F.S.; Zandi, P.P. Whole-genome CNV analysis: Advances in computational approaches. Front. Genet. 2015, 6, 138. [Google Scholar] [CrossRef] [PubMed]
- Korbel, J.O.; Urban, A.E.; Affourtit, J.P.; Godwin, B.; Grubert, F.; Simons, J.F.; Kim, P.M.; Palejev, D.; Carriero, N.J.; Du, L.; et al. Paired-End Mapping Reveals Extensive Structural Variation in the Human Genome. Science 2007, 318, 420–426. [Google Scholar] [CrossRef]
- Chen, K.; Wallis, J.W.; McLellan, M.D.; Larson, D.E.; Kalicki, J.M.; Pohl, C.S.; McGrath, S.D.; Wendl, M.C.; Zhang, Q.; Locke, D.P.; et al. BreakDancer: An algorithm for high-resolution mapping of genomic structural variation. Nat. Methods 2009, 6, 677. [Google Scholar] [CrossRef]
- Ye, K.; Guo, L.; Yang, X.; Lamijer, E.-W.; Raine, K.; Ning, Z. Split-Read Indel and Structural Variant Calling Using PINDEL. In Copy Number Variants: Methods and Protocols; Bickhart, D.M., Ed.; Springer: New York, NY, USA, 2018; pp. 95–105. [Google Scholar] [CrossRef]
- Duncavage, E.J.; Abel, H.J.; Pfeifer, J.D.; Armstrong, J.R.; Becker, N.; Magrini, V.J. SLOPE: A quick and accurate method for locating non-SNP structural variation from targeted next-generation sequence data. Bioinformatics 2010, 26, 2684–2688. [Google Scholar] [CrossRef]
- Park, H.; Chun, S.-M.; Shim, J.; Oh, J.-H.; Cho, E.J.; Hwang, H.S.; Lee, J.-Y.; Kim, D.; Jang, S.J.; Nam, S.J.; et al. Detection of chromosome structural variation by targeted next-generation sequencing and a deep learning application. Sci. Rep. 2019, 9, 3644. [Google Scholar] [CrossRef]
- Russnes, H.G.; Navin, N.; Hicks, J.; Borresen-Dale, A.-L. Insight into the heterogeneity of breast cancer through next-generation sequencing. J. Clin. Investig. 2011, 121, 3810–3818. [Google Scholar] [CrossRef]
- Magi, A.; Tattini, L.; Palombo, F.; Benelli, M.; Gialluisi, A.; Giusti, B.; Abbate, R.; Seri, M.; Gensini, G.F.; Romeo, G.; et al. H3M2: Detection of runs of homozygosity from whole-exome sequencing data. Bioinformatics 2014, 30, 2852–2859. [Google Scholar] [CrossRef] [PubMed]
- Zarrei, M.; MacDonald, J.R.; Merico, D.; Scherer, S.W. A copy number variation map of the human genome. Nat. Rev. Genet. 2015, 16, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Ion Torrent. CNV Detection by Ion Semiconductor Sequencing. Available online: https://assets.thermofisher.com/TFS-Assets/LSG/brochures/CNV-Detection-by-Ion.pdf (accessed on 1 November 2019).
- Scherer, S.W.; Lee, C.; Birney, E.; Altshuler, D.M.; Eichler, E.E.; Carter, N.P.; Hurles, M.E.; Feuk, L. Challenges and standards in integrating surveys of structural variation. Nat. Genet. 2007, 39, S7–S15. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef]
- González-Pérez, A.; López-Bigas, N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am. J. Hum. Genet. 2011, 88, 440–449. [Google Scholar] [CrossRef]
- Yang, H.; Wang, K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat. Protoc. 2015, 10, 1556–1566. [Google Scholar] [CrossRef]
- McLaren, W.; Pritchard, B.; Rios, D.; Chen, Y.; Flicek, P.; Cunningham, F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 2010, 26, 2069–2070. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Ng, S.B.; Turner, E.H.; Robertson, P.D.; Flygare, S.D.; Bigham, A.W.; Lee, C.; Shaffer, T.; Wong, M.; Bhattacharjee, A.; Eichler, E.E.; et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature 2009, 461, 272. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Keren, H.; Lev-Maor, G.; Ast, G. Alternative splicing and evolution: Diversification, exon definition and function. Nat. Rev. Genet. 2010, 11, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.D.; Harrow, J.; Harte, R.A.; Wallin, C.; Diekhans, M.; Maglott, D.R.; Searle, S.; Farrell, C.M.; Loveland, J.E.; Ruef, B.J.; et al. The consensus coding sequence (CCDS) project: Identifying a common protein-coding gene set for the human and mouse genomes. Genome Res. 2009, 19, 1316–1323. [Google Scholar] [CrossRef]
- McCarthy, D.J.; Humburg, P.; Kanapin, A.; Rivas, M.A.; Gaulton, K.; Cazier, J.-B.; Donnelly, P. Choice of transcripts and software has a large effect on variant annotation. Genome Med. 2014, 31, 26. [Google Scholar] [CrossRef]
- The International HapMap 3 Consortium. Integrating common and rare genetic variation in diverse human populations. Nature 2010, 467, 52–58. [Google Scholar] [CrossRef]
- Stoneking, M.; Krause, J. Learning about human population history from ancient and modern genomes. Nat. Rev. Genet. 2011, 12, 603–614. [Google Scholar] [CrossRef]
- Siva, N. 1000 Genomes project. Nat. Biotechnol. 2008, 26, 256. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef]
- Genomics, A. Practical Guidelines. Available online: https://www.acmg.net/ACMG/Medical-Genetics-Practice-Resources/Practice-Guidelines.aspx (accessed on 13 December 2019).
- Harper, P.S. The European Society of Human Genetics: Beginnings, early history and development over its first 25 years. Eur. J. Hum. Genet. 2017, 2017, 1–8. [Google Scholar] [CrossRef][Green Version]
- Gilissen, C.; Hoischen, A.; Brunner, H.G.; Veltman, J.A. Disease gene identification strategies for exome sequencing. Eur. J. Hum. Genet. 2012, 20, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Sauna, Z.E.; Kimchi-Sarfaty, C. Understanding the contribution of synonymous mutations to human disease. Nat. Rev. Genet. 2011, 12, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Cartegni, L.; Chew, S.L.; Krainer, A.R. Listening to silence and understanding nonsense: Exonic mutations that affect splicing. Nat. Rev. Genet. 2002, 3, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Desmet, F.-O.; Hamroun, D.; Lalande, M.; Collod-Béroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [PubMed]
- Desvignes, J.-P.; Bartoli, M.; Miltgen, M.; Delague, V.; Salgado, D.; Krahn, M.; Béroud, C. VarAFT: A variant annotation and filtration system for human next generation sequencing data. Nucleic Acids Res. 2018, 46, W545–W553. [Google Scholar] [CrossRef]
- MacArthur, D.G.; Balasubramanian, S.; Frankish, A.; Huang, N.; Morris, J.; Walter, K.; Jostins, L.; Habegger, L.; Pickrell, J.K.; Montgomery, S.B.; et al. A Systematic Survey of Loss-of-Function Variants in Human Protein-Coding Genes. Science 2012, 335, 823–828. [Google Scholar] [CrossRef]
- Lelieveld, S.H.; Veltman, J.A.; Gilissen, C. Novel bioinformatic developments for exome sequencing. Hum. Genet. 2016, 135, 603–614. [Google Scholar] [CrossRef]
- Robinson, P.N.; Köhler, S.; Oellrich, A.; Sanger Mouse Genetics, P.; Wang, K.; Mungall, C.J.; Lewis, S.E.; Washington, N.; Bauer, S.; Seelow, D.; et al. Improved exome prioritization of disease genes through cross-species phenotype comparison. Genome Res. 2014, 24, 340–348. [Google Scholar] [CrossRef]
- Eilbeck, K.; Quinlan, A.; Yandell, M. Settling the score: Variant prioritization and Mendelian disease. Nat. Rev. Genet. 2017, 18, 599. [Google Scholar] [CrossRef]
- Khurana, E.; Fu, Y.; Chen, J.; Gerstein, M. Interpretation of Genomic Variants Using a Unified Biological Network Approach. PLoS Comp. Biol. 2013, 9, e1002886. [Google Scholar] [CrossRef]
- Petrovski, S.; Wang, Q.; Heinzen, E.L.; Allen, A.S.; Goldstein, D.B. Genic Intolerance to Functional Variation and the Interpretation of Personal Genomes. PLoS Genet. 2013, 9, e1003709. [Google Scholar] [CrossRef]
- Zemojtel, T.; Köhler, S.; Mackenroth, L.; Jäger, M.; Hecht, J.; Krawitz, P.; Graul-Neumann, L.; Doelken, S.; Ehmke, N.; Spielmann, M.; et al. Effective diagnosis of genetic disease by computational phenotype analysis of the disease-associated genome. Sci. Transl. Med. 2014, 6, 252ra123. [Google Scholar] [CrossRef] [PubMed]
- Singleton, M.V.; Guthery, S.L.; Voelkerding, K.V.; Chen, K.; Kennedy, B.; Margraf, R.L.; Durtschi, J.; Eilbeck, K.; Reese, M.G.; Jorde, L.B.; et al. Phevor Combines Multiple Biomedical Ontologies for Accurate Identification of Disease-Causing Alleles in Single Individuals and Small Nuclear Families. Am. J. Hum. Genet. 2014, 94, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Pereira, R.; Oliveira, M.E.; Santos, R.; Oliveira, E.; Barbosa, T.; Santos, T.; Gonçalves, P.; Ferraz, L.; Pinto, S.; Barros, A.; et al. Characterization of CCDC103 expression profiles: Further insights in primary ciliary dyskinesia and in human reproduction. J. Assist. Reprod. Genet. 2019, 36, 1683–1700. [Google Scholar] [CrossRef]
- Pereira, R.; Barbosa, T.; Gales, L.; Oliveira, E.; Santos, R.; Oliveira, J.; Sousa, M. Clinical and Genetic Analysis of Children with Kartagener Syndrome. Cells 2019, 8, 900. [Google Scholar] [CrossRef]
- Stelzer, G.; Plaschkes, I.; Oz-Levi, D.; Alkelai, A.; Olender, T.; Zimmerman, S.; Twik, M.; Belinky, F.; Fishilevich, S.; Nudel, R.; et al. VarElect: The phenotype-based variation prioritizer of the GeneCards Suite. BMC Genom. 2016, 17, 444. [Google Scholar] [CrossRef]
- Pollard, K.S.; Hubisz, M.J.; Rosenbloom, K.R.; Siepel, A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010, 20, 110–121. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Rödelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef]
- Bao, L.; Zhou, M.; Cui, Y. nsSNPAnalyzer: Identifying disease-associated nonsynonymous single nucleotide polymorphisms. Nucleic Acids Res. 2005, 33, W480–W482. [Google Scholar] [CrossRef]
- Stitziel, N.O.; Binkowski, T.A.; Tseng, Y.Y.; Kasif, S.; Liang, J. topoSNP: A topographic database of non-synonymous single nucleotide polymorphisms with and without known disease association. Nucleic Acids Res. 2004, 32, 520–522. [Google Scholar] [CrossRef]
- Esposito, A.; Colantuono, C.; Ruggieri, V.; Chiusano, M.L. Bioinformatics for agriculture in the Next-Generation sequencing era. Chem. Biol. Technol. Agric. 2016, 3, 9. [Google Scholar] [CrossRef]
- Bamshad, M.J.; Ng, S.B.; Bigham, A.W.; Tabor, H.K.; Emond, M.J.; Nickerson, D.A.; Shendure, J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 2011, 12, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Ku, C.S.; Cooper, D.N.; Polychronakos, C.; Naidoo, N.; Wu, M.; Soong, R. Exome sequencing: Dual role as a discovery and diagnostic tool. Ann. Neurol. 2012, 71, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Sboner, A.; Mu, X.; Greenbaum, D.; Auerbach, R.K.; Gerstein, M.B. The real cost of sequencing: Higher than you think! Genome Biol. 2011, 12, 125. [Google Scholar] [CrossRef]
- Mardis, E.R.; Mu, X.J.; Greenbaum, D.; Auerbach, R.K.; Gerstein, M.B.; Nisbett, J.; Guigo, R.; Dermitzakis, E.; Gilad, Y.; Pritchard, J.; et al. The $1000 genome, the $100,000 analysis? Genome Med. 2010, 2, 84. [Google Scholar] [CrossRef]
- Moorthie, S.; Hall, A.; Wright, C.F. Informatics and clinical genome sequencing: Opening the black box. Genet. Med. 2013, 15, 165–171. [Google Scholar] [CrossRef]
- Daber, R.; Sukhadia, S.; Morrissette, J.J.D. Understanding the limitations of next generation sequencing informatics, an approach to clinical pipeline validation using artificial data sets. Cancer Genet. 2013, 206, 441–448. [Google Scholar] [CrossRef]
- Biesecker, L.G.; Green, R.C. Diagnostic clinical genome and exome sequencing. N. Engl. J. Med. 2014, 370, 2418–2425. [Google Scholar] [CrossRef]
- Gagan, J.; Van Allen, E.M. Next-generation sequencing to guide cancer therapy. Genome Med. 2015, 7, 80. [Google Scholar] [CrossRef]
- Oliveira, J.; Martins, M.; Pinto Leite, R.; Sousa, M.; Santos, R. The new neuromuscular disease related with defects in the ASC-1 complex: Report of a second case confirms ASCC1 involvement. Clin. Genet. 2017, 92, 434–439. [Google Scholar] [CrossRef]
- Tebani, A.; Afonso, C.; Marret, S.; Bekri, S. Omics-Based Strategies in Precision Medicine: Toward a Paradigm Shift in Inborn Errors of Metabolism Investigations. Int. J. Mol. Sci. 2016, 17, 1555. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, H.; Hasegawa, M.; Wakimoto, K.; Miyamoto-Sato, E. Next-Generation Technologies for Multiomics Approaches Including Interactome Sequencing. BioMed Res. Int. 2015, 2015, 1–9. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Software | Short Description | Ref. |
|---|---|---|
| PhyloP Phylogenetic p-values | Based on a model of neutral evolution, the patterns of conservation (positive scores)/acceleration (negative scores) are analyzed for various annotation classes and clades of interest. | [146] |
| SIFT Sorting Intolerant from Tolerant | Predicts based on sequence homology, if an AA substitution will affect protein function and potentially alter the phenotype. Scores less than 0.05 indicating a variant as deleterious. | [112] |
| PolyPhen-2 Polymorphism Phenotyping v2 | Predicts the functional impact of an AA replacement from its individual features using a naive Bayes classifier. Includes two tools HumDiv (designed to be applied in complex phenotypes) and HumVar (designed to diagnostic of Mendelian diseases). Higher scores (>0.85) predicts, more confidently, damaging variants. | [113] |
| CADD Combined Annotation Dependent Depletion | Integrates diverse genome annotations and scores all human SNV and Indel. It prioritizes functional, deleterious, and disease causal variants according to functional categories, effect sizes and genetic architectures. Scores above 10 should be applied as a cut-off for identifying pathogenic variants. | [114] |
| MutationTaster | Analyses evolutionary conservation, splice-site changes, loss of protein features and changes that might affect the amount of mRNA. Variants are classified, as polymorphism or disease-causing | [147] |
| Human Splice Finder | Predict the effects of mutations on splicing signals or to identify splicing motifs in any human sequence. | [133] |
| nsSNPAnalyzer | Extracts structural and evolutionary information from a query nsSNP and uses a machine learning method (Random Forest) to predict its phenotypic effect. Classifies the variant as neutral and disease. | [148] |
| TopoSNP Topographic mapping of SNP | Analyze SNP based on its geometric location and conservation information, produces an interactive visualization of disease and non-disease associated with each SNP. | [149] |
| Condel Consensus Deleteriousness | Condel integrates the output of different methods to predict the impact of nsSNP on protein function. The algorithm based on the weighted average of the normalized scores classifies the variants as neutral or deleterious. | [115] |
| ANNOVAR * Annotate Variation | Annotates the variants based on several parameters, such as identification whether SNPs or CNVs affect the protein (gene-based), identification of variants in specific genomic regions outside protein-coding regions (region-based) and identification of known variants documented in public and licensed database (filter-based) | [116] |
| VEP * Variant Effect Predictor | Determines the effect of multiple variants (SNPs, insertions, deletions, CNVs or structural variants) on genes, transcripts and protein sequence, as well as regulatory regions. | [117] |
| snpEff * | Annotation and classification of SNV based on their effects on annotated genes, such as synonymous/nsSNP, start or stop codon gains or losses, their genomic locations, among others. Considered as a structural based tool for annotation. | [118] |
| SeattleSeq * | Provides annotation of SNVs and small indels, by providing to each the dbSNP rs IDs, gene names and accession numbers, variation functions, protein positions and AA changes, conservation scores, HapMap frequencies, PolyPhen predictions and clinical association. | [119] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pereira, R.; Oliveira, J.; Sousa, M. Bioinformatics and Computational Tools for Next-Generation Sequencing Analysis in Clinical Genetics. J. Clin. Med. 2020, 9, 132. https://doi.org/10.3390/jcm9010132
Pereira R, Oliveira J, Sousa M. Bioinformatics and Computational Tools for Next-Generation Sequencing Analysis in Clinical Genetics. Journal of Clinical Medicine. 2020; 9(1):132. https://doi.org/10.3390/jcm9010132
Chicago/Turabian StylePereira, Rute, Jorge Oliveira, and Mário Sousa. 2020. "Bioinformatics and Computational Tools for Next-Generation Sequencing Analysis in Clinical Genetics" Journal of Clinical Medicine 9, no. 1: 132. https://doi.org/10.3390/jcm9010132
APA StylePereira, R., Oliveira, J., & Sousa, M. (2020). Bioinformatics and Computational Tools for Next-Generation Sequencing Analysis in Clinical Genetics. Journal of Clinical Medicine, 9(1), 132. https://doi.org/10.3390/jcm9010132