1. Introduction
Most of the cancer-related mortality is caused due to metastasis—the spread of cancer to secondary vital organs [
1,
2]. This is a complex phenomenon involving dissemination of cells from the primary site, intravasation into the circulatory system followed by extravasation, and finally successful colonization in secondary tumor sites such as liver, lung, bone and brain [
1,
2]. Cancer that is diagnosed at the primary site is relatively easier to manage compared to those with metastatic lesions. Circulating tumor cells (CTCs) derived from either the primary or metastatic tumors serve as precursors of metastasis [
3]. To begin to understand the biology of CTCs and their role in the metastatic process, it is important to culture CTCs in a suitable microenvironment that recapitulates their physiological features.
CTCs represent an extraordinarily rare population in the milieu of billions of blood cells, and hence, their identification and isolation pose critical impediments to their characterization [
4]. In the past decade or so, several technologies have come up to isolate and detect CTCs. Broadly CTC detection is done by two methods, one involving pre-enrichment with markers, and the other without enrichment, which is also known as direct detection of CTCs. The reported technologies for direct detection include (1) line-confocal microscopy and (2) surface-enhanced Raman scattering nanoparticles (SERS) [
5,
6]. On the other hand, marker-based pre-enrichment methods include several techniques, for example, Cell Search™, Dynabeads
® CD45, EPISPOT (EPithelial ImmunoSPOT), ClearCell
® FX1 System, Herringbone CTC-Chip, CTC-iChip, DEPArray™ System, etc. [
4,
7,
8,
9]. However, pre-enrichment results in the loss of CTCs that do not express the chosen markers. Further, several detection techniques involve cell-fixation which does not allow subsequent CTC expansion for biological characterization including stemness, drug resistance, etc. In addition, most of these methods detect only low numbers of CTCs (<20%) [
10]. Hence, there is an unmet need to establish a robust method with improved efficiency for CTC enrichment to enable a better understanding of their biology.
Conventional two-dimensional (2D) culture system suffers from major limitations in terms of altered cellular morphology, motility, polarity and other functional aspects. When grown on a 2D substrate, cells lose their in vivo morphology and importantly their cell–cell and cell–matrix interactions. Further, stiffness of materials typically used for 2D cell cultures, such as tissue culture polystyrene (TCPS) and glass, are several orders of magnitude (GigaPascals) higher than the stiffness of human tissues (kiloPascals) [
11]. In addition, studies have also demonstrated altered signaling in cancer cells when cultured in 2D platforms [
12]. Therefore, we sought to develop a three-dimensional (3D) culture system to enrich and expand the rare population of CTCs.
Three-dimensional culture systems have been widely explored to study breast cancer over the past three decades. A variety of 3D gel-based matrices like collagen, Matrigel, laminin-rich extracellular matrix (lrECM), fibrin, etc., have been routinely used to mimic cell-basement membrane interactions. However, the abundance of ECM already present in these matrices reduces the secretion of native ECM molecules by cancer cells [
12,
13]. To overcome these limitations, we recently developed a 3D porous scaffold synthesized using a synthetic biomaterial, poly(ε-caprolactone) (PCL) that better mimics the architecture and stiffness of breast tumors, and enables deposition of native ECM [
13]. Our 3D culture system showed improved cell–cell and cell–matrix interactions. Furthermore, global microarray analysis revealed that cancer cells cultured in this 3D scaffold are able to maintain increased stemness and epithelial–mesenchymal transition (EMT) properties, and show closer association to in vivo tumor growth than conventional 2D cultures on TCPS [
13]. Thus, this 3D porous PCL scaffold system offers a superior model system to mimic the native tumor microenvironment.
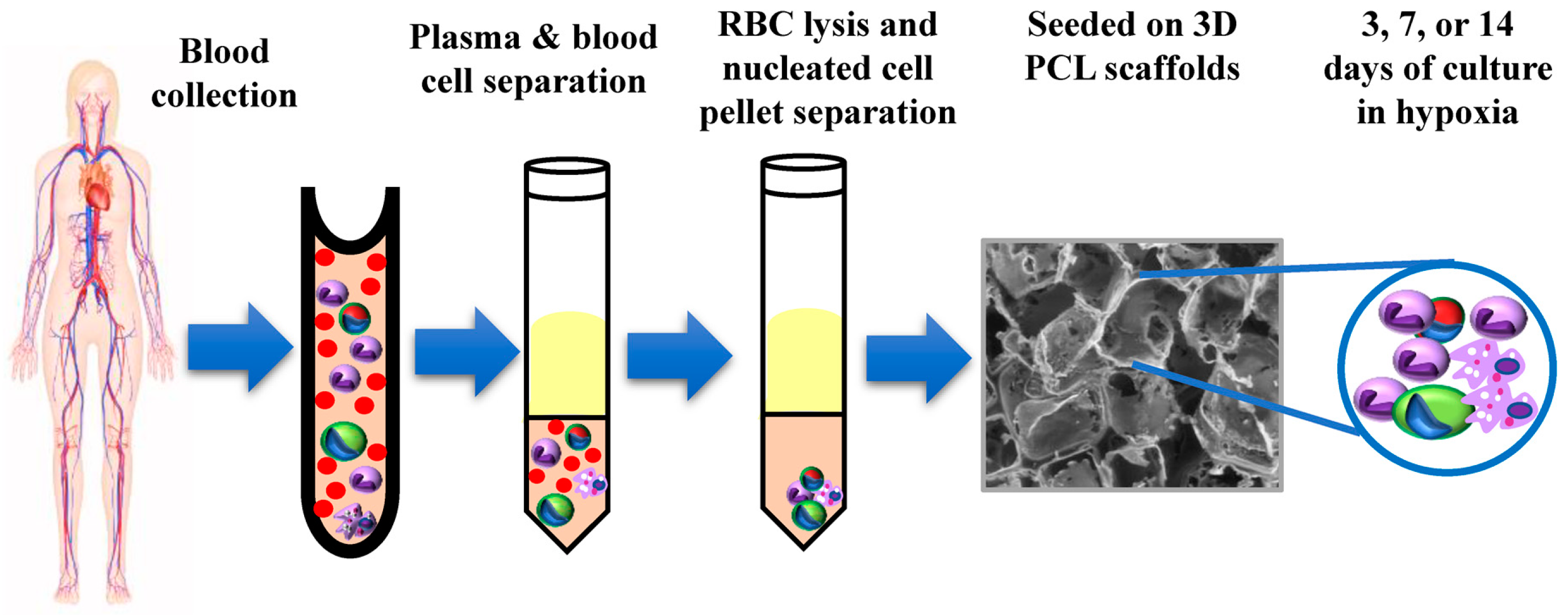
In this study, we have exploited the 3D porous PCL scaffold to enrich CTCs derived from breast cancer patient blood samples without any prior enrichment. We have previously shown that culture of patient blood cells under hypoxic conditions for 14 days in laser ablated microwells helps in the enrichment of CTC subpopulation from RBC lysed nucleated cell fraction [
14] Using various lineage-specific markers, we further showed gradual depletion of other blood cell lineages with time [
14] More recently, we used a similar strategy to expand CTCs in agar-microwells [
15]. However, being inert, agar does not allow deposition of extracellular matrix. Therefore, here, we established a 3D PCL scaffold-based method, which better mimics the native cellular in vivo environment and allows ECM deposition, for culturing CTCs from RBC-depleted nucleated cell pellets of advanced breast cancer patient samples under hypoxic conditions. We detected CK-positive and CD45-negative CTCs in 12/16 patient samples using this culture method. We detected the deposition of thread-like and sheet-like ECM on the scaffold providing a substratum for cells to adhere and proliferate. Our study shows intra-patient and inter-patient heterogeneity with respect to the epithelial (E) and mesenchymal (M) characteristics suggesting a dynamic EMT spectrum in CTCs. Thus, we established a unique model system by creating in vivo-like functionality to culture CTCs from whole blood samples without prior enrichment. Our model may provide a versatile system for studying CTC biology as well as in the use of a number of downstream exciting applications for the clinical utility of CTCs.
2. Materials and Methods
2.1. Patients Sample, Blood Collection and Processing
Blood samples were collected from 16 chemo-naïve metastatic breast cancer patients. This study was approved by the institutional review board at Indian Institute of Science (IISc) and at the Kidwai Memorial Institute of Oncology (KMIO/MEC/017/23.March.2017, KMIO/MEC/018/23.March.2017, KMIO/MEC/011.November.2016). All patients gave their informed consent for the completion of the study. Among 16 patients (median age of breast cancer patients = 50.5), 31 patients were ER/PR positive (81.25%), 2 patients were triple negative (12.5%), and 8 patients were HER-2 positive (50%). Out of all these 16 patients with breast cancer, 1 patient had only brain metastasis (6.25%), 14 patients had liver, lung and bone metastasis (87.5%), and 3 patient had contralateral axillary and lymph node metastasis along with lung/liver/bone metastasis (
Supplementary Table S1). Clinicopathological information was recorded for each patient. Blood sample (~10 mL) was collected at a single draw in chemo-naïve conditions. All the samples were collected in sterile EDTA-coated vacutainer tubes (BD) and maintained at 4 °C until processing.
Blood samples were processed within 3–6 h of withdrawal to avoid blood clotting and to maintain cell viability. In order to isolate nucleated cells, plasma and blood cells were separated from whole blood by centrifuging at 1200 rpm for 10 min. Blood cells were then treated with chilled red blood cell (RBC) lysis buffer (154 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA) at a ratio of 1:5 with a gentle-mixing followed by incubation at room temperature (25 °C) for 15 min. Next, the whole content was centrifuged at 1200 rpm for 10 min at room temperature to remove lysed RBC fragments. The leftover, largely nucleated cells following RBS lysis were resuspended in fresh Dulbecco’s modified Eagle’s medium (DMEM; Sigma-Aldrich, Saint Louis, MO, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, Invitrogen, Carlsbad, CA, USA) containing antibiotics streptomycin sulphate and benzylpenicillin at final concentrations of 100 μg/mL and 100 U/mL, respectively. Each processed sample was split into multiple wells of a 96-well plate (~10 mL RBC-lysed blood was distributed into 5 or 6 wells).
2.2. Preparation of 3D Scaffold System
Fabrication of 3D PCL porous scaffolds is mentioned in detail elsewhere [
13]. Briefly, sodium chloride (Fisher scientific, Hampton, Hampshire, USA) crystals of a defined size range of 250–425 μm were used as the porogen. PCL (average molecular weight M
n = 80,000 g/mol; Sigma) was dissolved in chloroform and added on to the salt bed in 96 polypropylene plate. Finally, it was vacuum dried, and the salt was leached in dH
2O for three consecutive days with daily change of water to completely leach the salt. Subsequently, the morphology, pore size and pore interconnectivity were confirmed by scanning electron microscopy (SEM). Finally, scaffolds were made sterile using 70% ethanol wash followed by UV exposure for 30 min before using for tissue culture.
2.3. Culture of CTCs in 3D PCL Scaffolds
The RBC-lysed nucleated cells were seeded in scaffolds and maintained at 37 °C in 5% (v/v) CO2 and 1.1% O2 under humidified conditions. Each processed sample was split into multiple wells of a 96-well plate (~10 mL RBC-lysed blood was distributed into 5 or 6 wells). The culture medium (DMEM + 10% FBS) was changed after 72 h the first time after seeding, followed by every alternative day up to 14 days.
2.4. Immunophenotyping of Cells
Cells and scaffolds were fixed with 3.7% formaldehyde for 15 min and permeabilized with 0.2% Triton X-100 for maximum 10 min followed by three washes with PBST (0.05% Tween 20 in 1× PBS) each for 5 min. Blocking was done with blocking buffer (0.2% fish skin gelatin: FSG, 0.01% Tween 20, 0.2% sodium azide: NaN3) for 45 min. Primary antibodies were used at 1:200 dilutions for overnight at 4 °C. Excess antibodies were removed by giving three washes with PBST each for 5 min. Secondary antibodies were used at 1:200 dilutions and incubated at dark for 2 h at room temperature. In some studies, Phalloidin conjugated with Alexa Fluor™ 488 and Alexa Fluor™ 546 were used for 2 h in dark. Counterstaining for nucleus with Hoechst 33342 was done by incubating for 5 min in dark. All the samples were imaged using epifluorescence (Olympus IX71) or confocal laser (Olympus FV10i/Olympus FV3000/Leica SP8) microscope. Image processing was performed by ImageJ software.
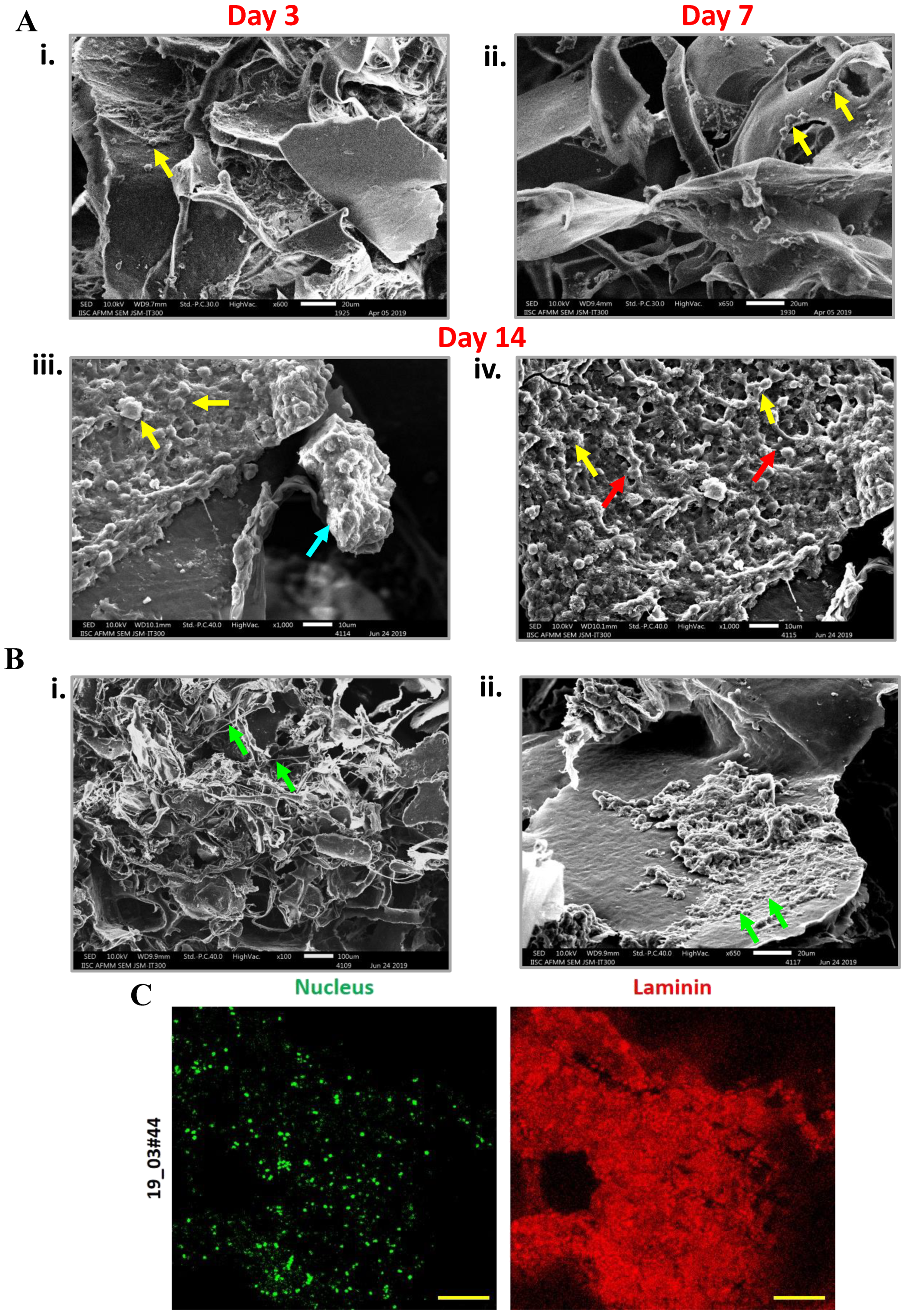
2.5. Study of Cell Morphology by Scanning Electron Microscopy (SEM)
RBC-depleted nucleated cells from patient blood sample cultured in scaffolds were fixed with 2.5% glutaraldehyde for 12 h at 4 °C. The cell-laden scaffolds were then dehydrated in a gradient of ethanol, 30, 50, 70, 90, and 100%, each for a period of 10 min. The samples were completely air-dried and gold-coated by means of sputtering apparatus before observation to avoid charging under the electron beam. The samples were analyzed using SEM microscopy (JEOL SEM, Peabody, MA, USA).
2.6. Bromodeoxyuridine (BrdU) Assay for Cell Proliferation in 3D PCL Scaffolds
2.6.1. Immunofluorescence-Based BrdU Assay
For immunostaining, RBC-depleted nucleated cells cultured in 3D PCL scaffolds (at day 7, day 14) were permeabilized with 0.2% Triton X-100 on ice for 90 sec followed by fixation with 3.7% formaldehyde at RT. Cells were denatured with denaturing solution (2N HCl, 0.5% Triton X-100) for 30 min at RT, followed by blocking (0.5% BSA, 0.5% Triton X-100) for 30 min at RT. Primary anti-BrdU was used at 1:1000 dilution for 2 h at RT. Excess antibodies were removed by washing thrice with PBST. Secondary antibody was used at 1:200 dilution for 1 h at RT in dark, followed by three washes with PBST. Finally, counterstaining for nucleus was done with Sytox green (Invitrogen, Carlsbad, CA, USA) for 5 min at RT in dark. Imaging was done in Leica SP8 confocal laser microscope. Images were processed with ImageJ software.
2.6.2. Colorimetry-Based BrdU Assay
RBC-depleted nucleated cells from blood samples of breast cancer patients were cultured in 3D PCL scaffolds for different day points (day 3, day 7, day 14) under hypoxic condition. MDA-MB-231 cells were cultured under a similar condition which was taken as control. Further, similar cells were cultured in 2D culture system for 24 h under normoxia which was taken as the 2D control. On the particular day point, BrdU label (1:2000 dilution in tissue culture media) was added and left for 48 h for incubation inside the incubator. Scaffolds containing cells were then fixed and denatured using fixative or denaturing solution (Calbiochem QIA58) by incubating for 30 min at room temperature (RT). Primary anti-BrdU antibody was used at 1:100 dilutions for 1 h at RT. Excess antibody was removed by using 1× wash buffer, provided with the kit. HRP conjugated secondary antibody was used at 1:1000 dilution for 30 min at RT followed by three washes with wash buffer. Wells were flooded with ddH2O. Next, the substrate (TMB-tetramethylbenzidine) was added in the well and was incubated in dark for 15 min at 37 °C which resulted in a change of color of the solution to blue. Finally, stop solution was added which turns the solution yellow colored. Absorbance was measured at dual wavelengths, 450 nm and 595 nm, within 30 min of adding stop solution.
4. Discussion
CTCs are considered as surrogate markers for monitoring and evaluating patient treatment responses. Current advances in technology have enabled us to isolate these rare CTC populations. However, the real challenge lies in the successful expansion of these cells in culture conditions. Our group has previously established laser-ablated microwells-based method for the rapid expansion of CTCs [
14]. Recently, we improvised the method using agar rather than tapered microwells to culture these cells [
15]. However, 2D culture methods are incompetent to mimic natural structural organization. They also exhibit compromised morphology and differentiation. Also, deposition of the extracellular matrix is critical for proper attachment and proliferation. Several 3D scaffolds either use a material like agar, which fails to allow ECM deposition, or matrigel, which has an abundant ECM that might not mimic the environment faced by CTCs at the secondary site. To overcome these concerns, we developed a 3D culture method using 3D porous PCL scaffolds which allowed deposition of ECM from the cells derived from patient samples and enabled the culture of CTCs.
When RBC-depleted nucleated cell pellets from whole blood from breast cancer patient samples were cultured in the 3D porous PCL scaffolds, we observed cell clusters which were positive for F actin. Our SEM micrographs revealed the presence of ECM deposition on the PCL scaffolds. This observation was confirmed by immunostaining for Laminin protein, a component of the extracellular matrix. In this study, SEM micrographs have revealed the formation of inter-cellular connections and tumor like masses or tumoroids derived from nucleated patient-blood samples. CTCs are typically identified based on the expression of epithelial markers such as keratins, EpCAM and the absence of common leukocyte marker CD45 [
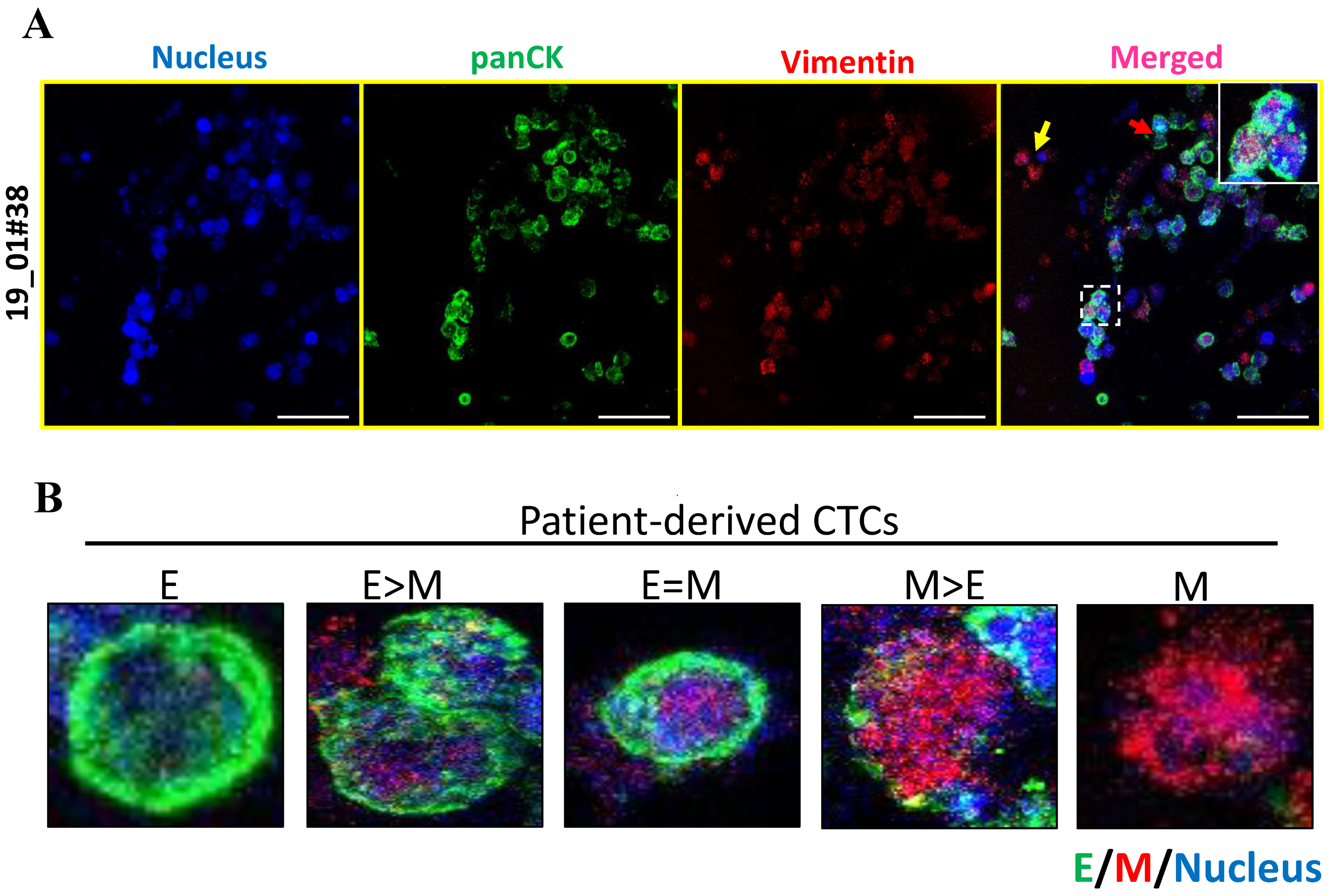
23]. In accordance with this, we noted the presence of pan-cytokeratin positive and CD45 negative cells in our 3D cultures. We cannot rule out the possibility that these are epithelial tumor cells engulfed by fibroblasts and macrophages; further characterization needs to be done to rule this out. However, recent reports have suggested the existence of E-M heterogeneity based on the expression of both epithelial and mesenchymal markers [
24]. We further characterized the epithelial and mesenchymal properties of CTCs in 3D PCL scaffolds. Our results indicated dual expression of both epithelial (pan CK) and mesenchymal markers (vimentin), revealing the presence of intermediate/hybrid EMT phenotype. Moreover, we also observed differential expression holds true for several other markers including E-cadherin, ZO-1, N cadherin and vimentin as well. Further investigation is required to validate the same with various histological subtypes of breast cancers (based on ER/PR/HER2 status).
The breast cancer patient-derived CTCs effectively reflect inter- and intra-patient heterogeneity in terms of E or M markers expression. Additionally, cell proliferation assays confirmed the expansion of viable nucleated cells in 3D culture system. Although our data showed proliferating cell population, and we have previously demonstrated the gradual depletion of other lineage cells [
14], it still remains to be confirmed if CTCs are indeed proliferating in these scaffolds by undertaking a co-staining staining for Pan-CK, CD45, and Ki67. The proliferating cells as observed by Ki67 and BrdU positivity in our study could include both CTCs as well as other cell types, such as cancer associated fibroblasts and macrophages, which may provide a suitable microenvironment for the enrichment of CTCs. Nevertheless, our study revealed visualization of viable cytokeratin positive/CD45 negative cells, and the detection of ZO-1 alone and ZO-1/N-cad double positive cells in these scaffolds by the end of 14 days in culture. Thus, our study shows that culture of RBC-lysed, nucleated cells from patient blood in 3D-PCL scaffold can serve as a novel system for culturing CTCs. Taken together, our study has demonstrated an easy but effective 3D culture method for in vitro culture and possible expansion of CTCs. Hence, this could serve as an excellent platform for a deeper study of CTC biology and metastasis. Taken together, we conclude that our novel ex vivo 3D culture system would provide a more reliable system for future personalized drug-screening for cancer patients.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





