Mucins as a New Frontier in Pulmonary Fibrosis

Abstract
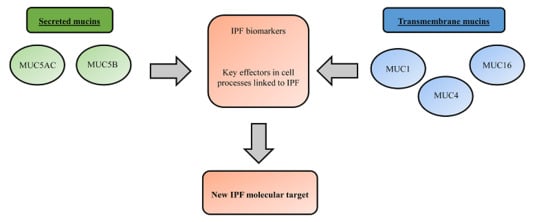
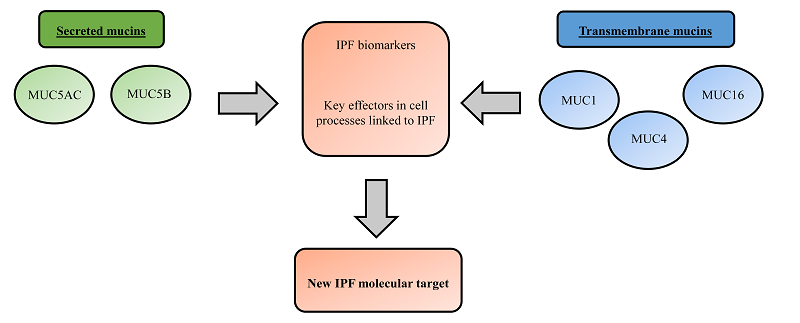
1. Introduction
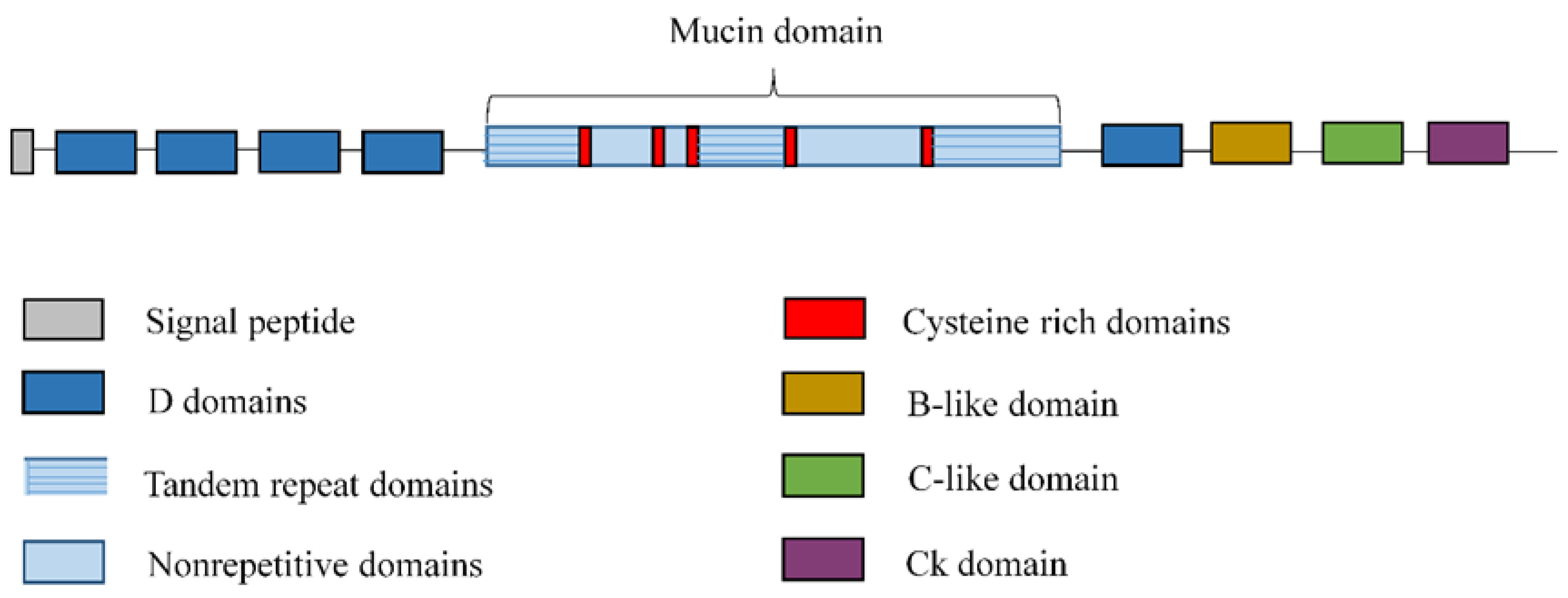
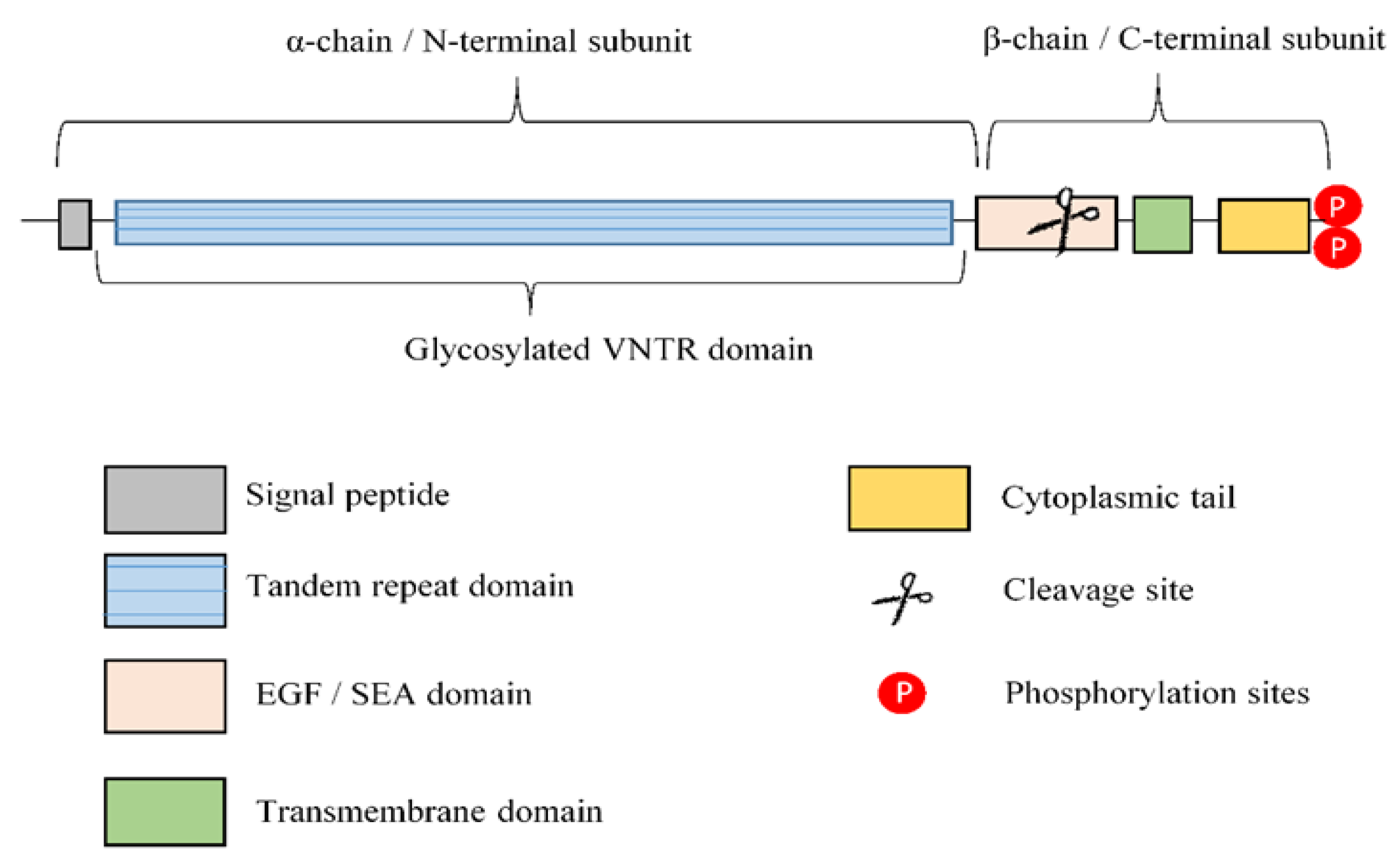
2. Overview of Lung Mucins
2.1. Secreted Mucins
2.2. Transmembrane Mucins
3. Mucins as Potential Idiopathic Pulmonary Fibrosis (IPF) Drug Targets
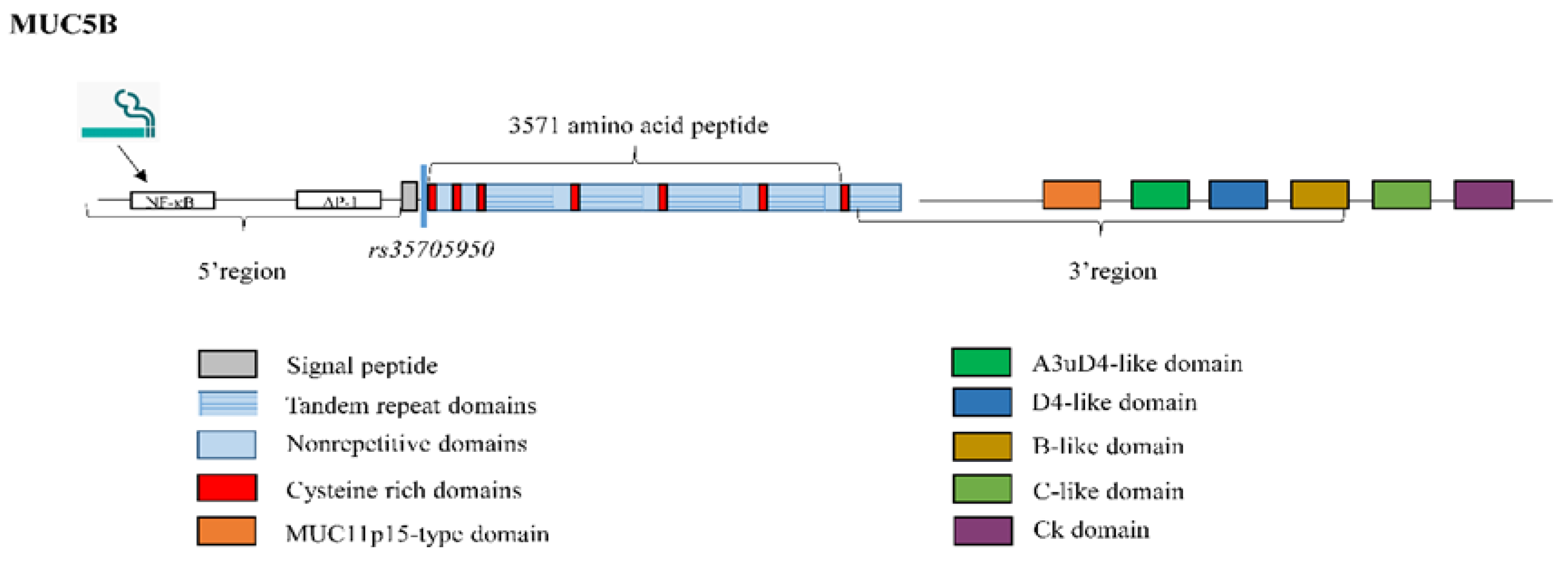
3.1. Mucin 5B
3.2. Mucin 5AC
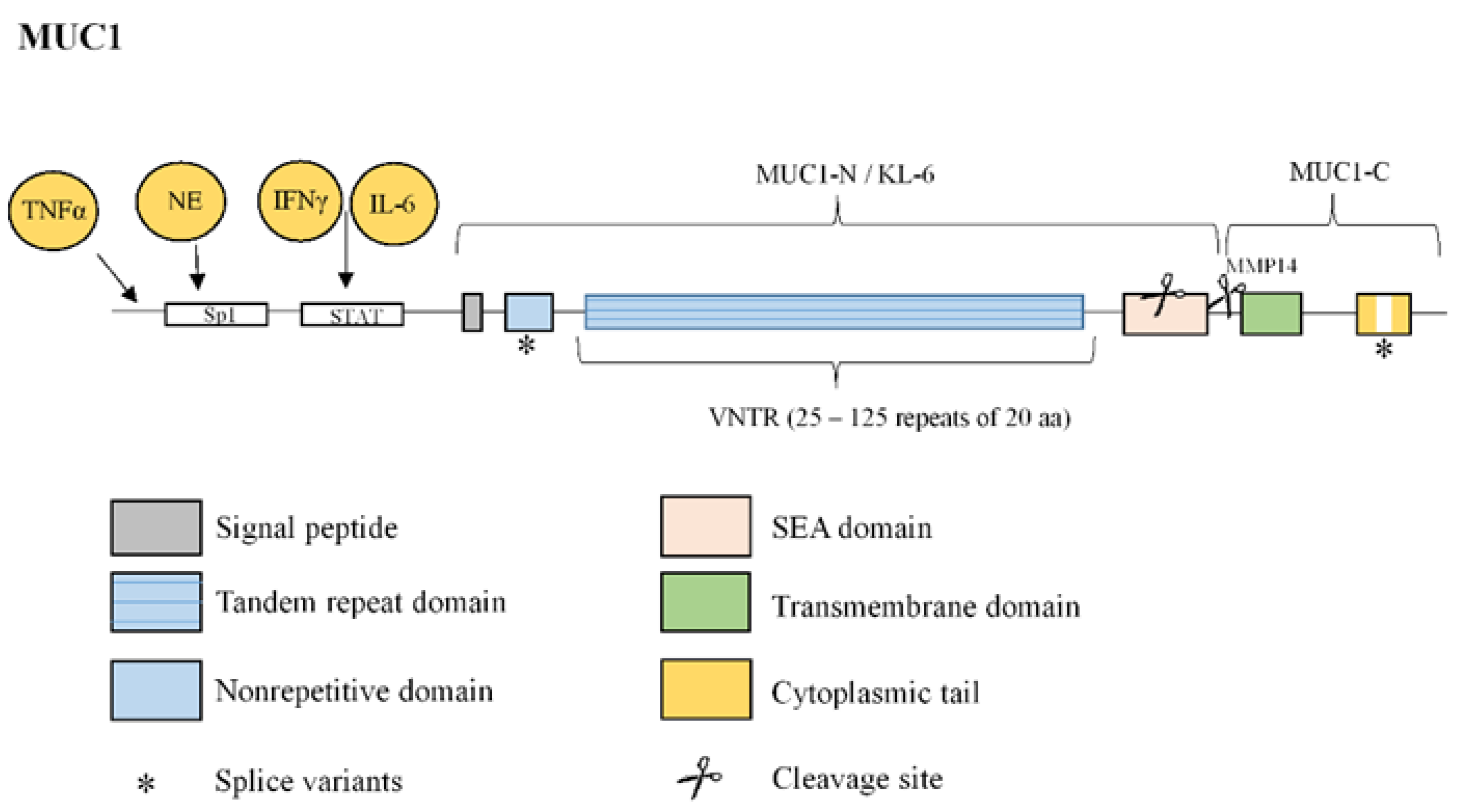
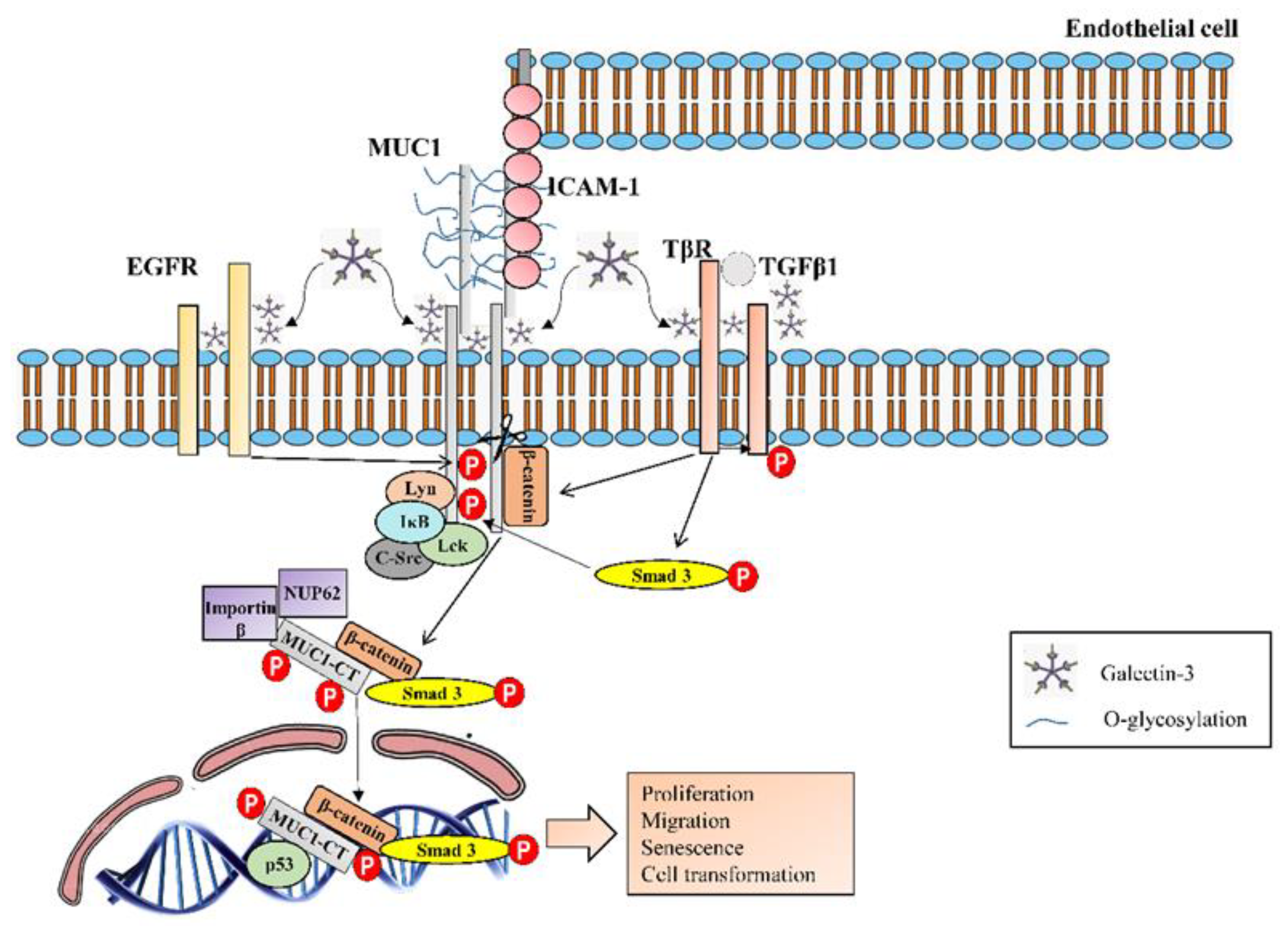
3.3. Mucin 1
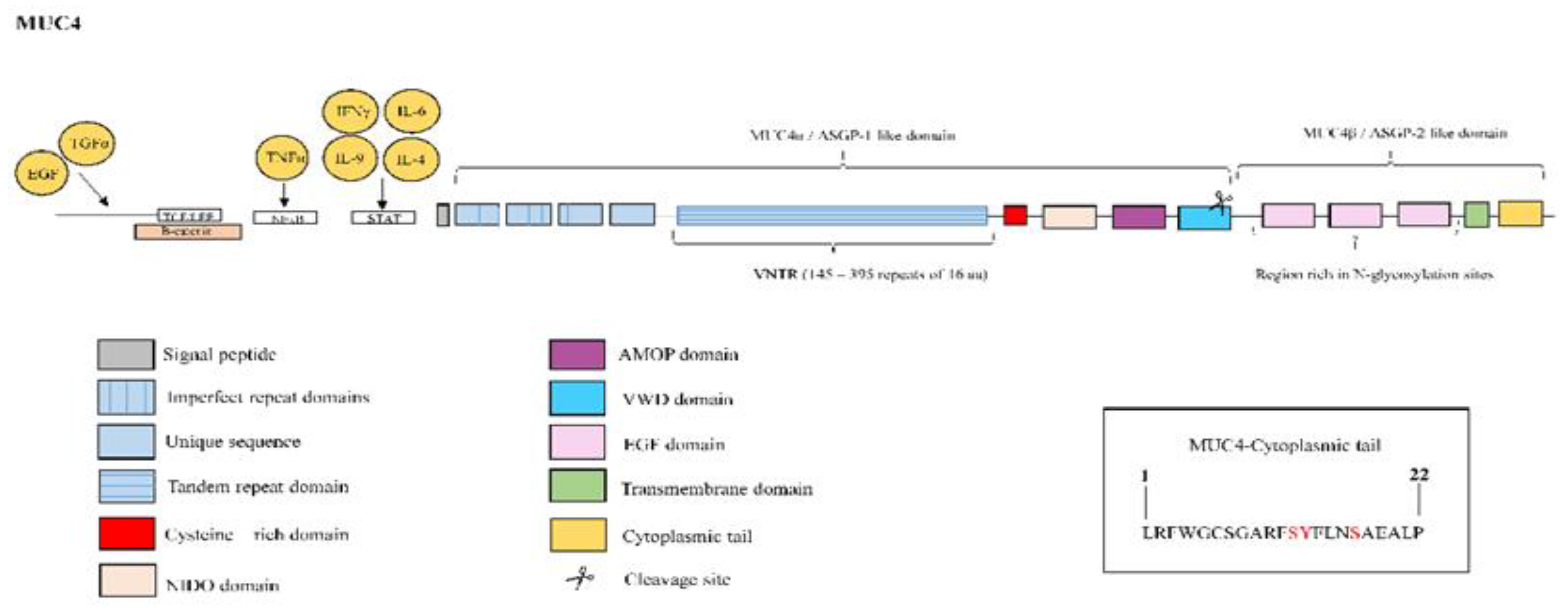
3.4. Mucin 4
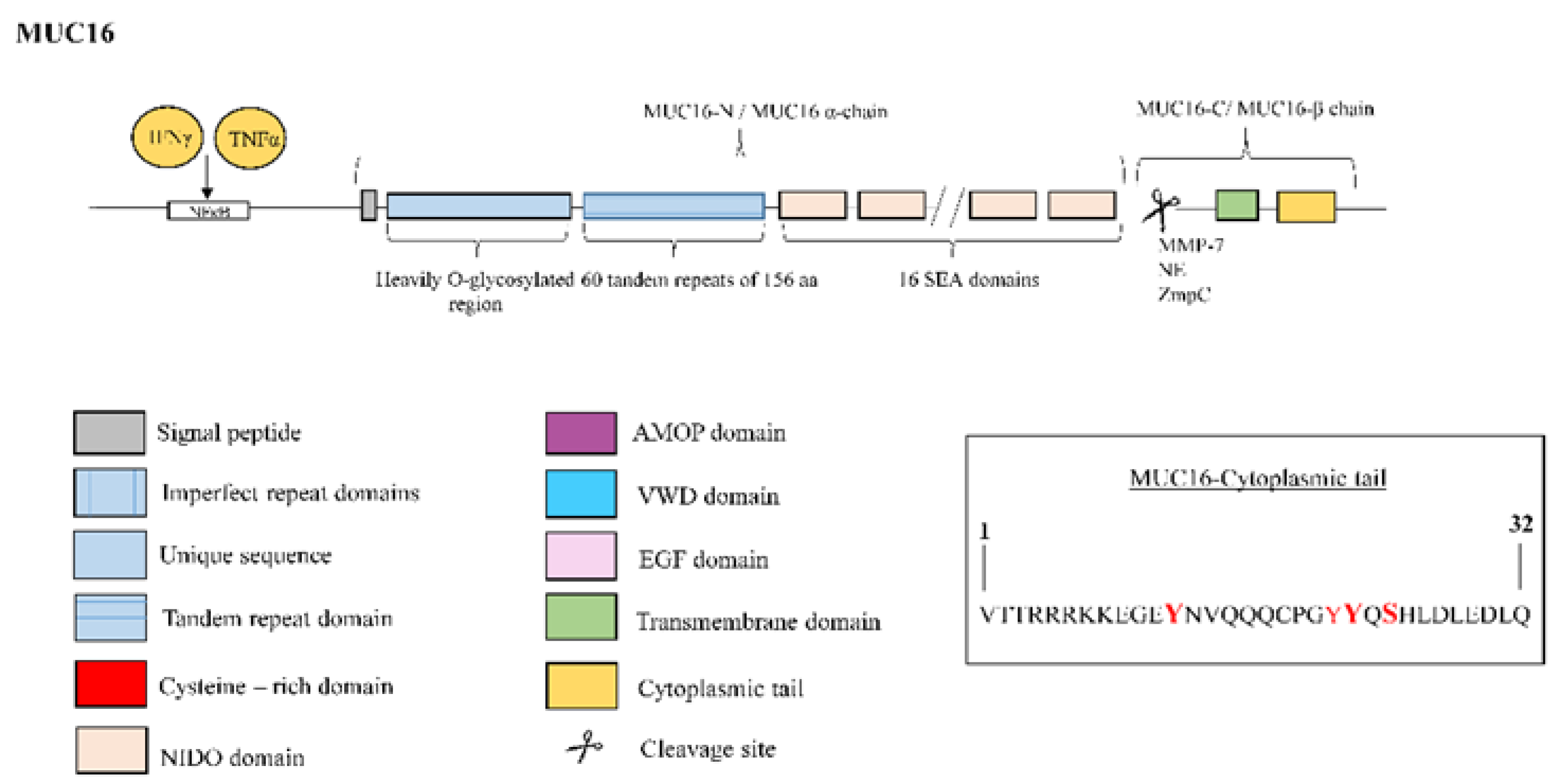
3.5. Mucin 16
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef] [PubMed]
- Fernandez Fabrellas, E.; Peris Sanchez, R.; Sabater Abad, C.; Juan Samper, G. Prognosis and Follow-Up of Idiopathic Pulmonary Fibrosis. Med. Sci. 2018, 6, 51. [Google Scholar] [CrossRef] [PubMed]
- Drakopanagiotakis, F.; Wujak, L.; Wygrecka, M.; Markart, P. Biomarkers in idiopathic pulmonary fibrosis. Matrix Biol. 2018, 68–69, 404–421. [Google Scholar] [CrossRef] [PubMed]
- Inchingolo, R.; Varone, F.; Sgalla, G.; Richeldi, L. Existing and emerging biomarkers for disease progression in idiopathic pulmonary fibrosis. Expert Rev. Respir. Med. 2019, 13, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Guiot, J.; Moermans, C.; Henket, M.; Corhay, J.L.; Louis, R. Blood Biomarkers in Idiopathic Pulmonary Fibrosis. Lung 2017, 195, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Jee, A.S.; Sahhar, J.; Youssef, P.; Bleasel, J.; Adelstein, S.; Nguyen, M.; Corte, T.J. Review: Serum biomarkers in idiopathic pulmonary fibrosis and systemic sclerosis associated interstitial lung disease—Frontiers and horizons. Pharmacol. Ther. 2019. [Google Scholar] [CrossRef] [PubMed]
- Maher, T.M.; Oballa, E.; Simpson, J.K.; Porte, J.; Habgood, A.; Fahy, W.A.; Flynn, A.; Molyneaux, P.L.; Braybrooke, R.; Divyateja, H.; et al. An epithelial biomarker signature for idiopathic pulmonary fibrosis: An analysis from the multicentre PROFILE cohort study. Lancet Respir. Med. 2017, 5, 946–955. [Google Scholar] [CrossRef]
- Ricci, A.; Mariotta, S.; Bronzetti, E.; Bruno, P.; Vismara, L.; De Dominicis, C.; Lagana, B.; Paone, G.; Mura, M.; Rogliani, P.; et al. Serum CA 15-3 is increased in pulmonary fibrosis. Sarcoidosis Vasc. Diffus. Lung Dis. 2009, 26, 54–63. [Google Scholar]
- Ishikawa, N.; Hattori, N.; Yokoyama, A.; Kohno, N. Utility of KL-6/MUC1 in the clinical management of interstitial lung diseases. Respir. Investig. 2012, 50, 3–13. [Google Scholar] [CrossRef]
- Wakamatsu, K.; Nagata, N.; Kumazoe, H.; Oda, K.; Ishimoto, H.; Yoshimi, M.; Takata, S.; Hamada, M.; Koreeda, Y.; Takakura, K.; et al. Prognostic value of serial serum KL-6 measurements in patients with idiopathic pulmonary fibrosis. Respir. Investig. 2017, 55, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Bergantini, L.; Bargagli, E.; Cameli, P.; Cekorja, B.; Lanzarone, N.; Pianigiani, L.; Vietri, L.; Bennett, D.; Sestini, P.; Rottoli, P. Serial KL-6 analysis in patients with idiopathic pulmonary fibrosis treated with nintedanib. Respir. Investig. 2019, 57, 290–291. [Google Scholar] [CrossRef] [PubMed]
- Ohshimo, S.; Yokoyama, A.; Hattori, N.; Ishikawa, N.; Hirasawa, Y.; Kohno, N. KL-6, a human MUC1 mucin, promotes proliferation and survival of lung fibroblasts. Biochem. Biophys. Res. Commun. 2005, 338, 1845–1852. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Yan, D.R.; Zhu, S.L.; Gu, J.; Bian, W.; Rong, Z.H.; Shen, C. KL-6 regulated the expression of HGF, collagen and myofibroblast differentiation. Eur. Rev. Med. Pharmacol. Sci. 2013, 17, 3073–3077. [Google Scholar]
- Greene, K.E.; King, T.E., Jr.; Kuroki, Y.; Bucher-Bartelson, B.; Hunninghake, G.W.; Newman, L.S.; Nagae, H.; Mason, R.J. Serum surfactant proteins-A and -D as biomarkers in idiopathic pulmonary fibrosis. Eur. Respir. J. 2002, 19, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Kinder, B.W.; Brown, K.K.; McCormack, F.X.; Ix, J.H.; Kervitsky, A.; Schwarz, M.I.; King, T.E., Jr. Serum surfactant protein-A is a strong predictor of early mortality in idiopathic pulmonary fibrosis. Chest 2009, 135, 1557–1563. [Google Scholar] [CrossRef] [PubMed]
- Barlo, N.P.; van Moorsel, C.H.; Ruven, H.J.; Zanen, P.; van den Bosch, J.M.; Grutters, J.C. Surfactant protein-D predicts survival in patients with idiopathic pulmonary fibrosis. Sarcoidosis Vasc. Diffus. Lung Dis. 2009, 26, 155–161. [Google Scholar]
- Mora, A.L.; Rojas, M.; Pardo, A.; Selman, M. Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease. Nat. Rev. Drug Discov. 2017, 16, 810. [Google Scholar] [CrossRef]
- Thornton, D.J.; Rousseau, K.; McGuckin, M.A. Structure and function of the polymeric mucins in airways mucus. Annu. Rev. Physiol. 2008, 70, 459–486. [Google Scholar] [CrossRef]
- Seibold, M.A.; Wise, A.L.; Speer, M.C.; Steele, M.P.; Brown, K.K.; Loyd, J.E.; Fingerlin, T.E.; Zhang, W.; Gudmundsson, G.; Groshong, S.D.; et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N. Engl. J. Med. 2011, 364, 1503–1512. [Google Scholar] [CrossRef]
- Rosas, I.O.; Richards, T.J.; Konishi, K.; Zhang, Y.; Gibson, K.; Lokshin, A.E.; Lindell, K.O.; Cisneros, J.; Macdonald, S.D.; Pardo, A.; et al. MMP1 and MMP7 as potential peripheral blood biomarkers in idiopathic pulmonary fibrosis. PLoS Med. 2008, 5, e93. [Google Scholar] [CrossRef] [PubMed]
- Chien, J.W.; Richards, T.J.; Gibson, K.F.; Zhang, Y.; Lindell, K.O.; Shao, L.; Lyman, S.K.; Adamkewicz, J.I.; Smith, V.; Kaminski, N.; et al. Serum lysyl oxidase-like 2 levels and idiopathic pulmonary fibrosis disease progression. Eur. Respir. J. 2014, 43, 1430–1438. [Google Scholar] [CrossRef] [PubMed]
- Naik, P.K.; Bozyk, P.D.; Bentley, J.K.; Popova, A.P.; Birch, C.M.; Wilke, C.A.; Fry, C.D.; White, E.S.; Sisson, T.H.; Tayob, N.; et al. Periostin promotes fibrosis and predicts progression in patients with idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 303, L1046–L1056. [Google Scholar] [CrossRef] [PubMed]
- Hieshima, K.; Imai, T.; Baba, M.; Shoudai, K.; Ishizuka, K.; Nakagawa, T.; Tsuruta, J.; Takeya, M.; Sakaki, Y.; Takatsuki, K.; et al. A novel human CC chemokine PARC that is most homologous to macrophage-inflammatory protein-1 alpha/LD78 alpha and chemotactic for T lymphocytes, but not for monocytes. J. Immunol. 1997, 159, 1140–1149. [Google Scholar] [PubMed]
- Prasse, A.; Pechkovsky, D.V.; Toews, G.B.; Jungraithmayr, W.; Kollert, F.; Goldmann, T.; Vollmer, E.; Muller-Quernheim, J.; Zissel, G. A vicious circle of alveolar macrophages and fibroblasts perpetuates pulmonary fibrosis via CCL18. Am. J. Respir. Crit. Care Med. 2006, 173, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Prasse, A.; Probst, C.; Bargagli, E.; Zissel, G.; Toews, G.B.; Flaherty, K.R.; Olschewski, M.; Rottoli, P.; Muller-Quernheim, J. Serum CC-chemokine ligand 18 concentration predicts outcome in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2009, 179, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Ziegenhagen, M.W.; Zabel, P.; Zissel, G.; Schlaak, M.; Muller-Quernheim, J. Serum level of interleukin 8 is elevated in idiopathic pulmonary fibrosis and indicates disease activity. Am. J. Respir. Crit. Care Med. 1998, 157, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Richards, T.J.; Kaminski, N.; Baribaud, F.; Flavin, S.; Brodmerkel, C.; Horowitz, D.; Li, K.; Choi, J.; Vuga, L.J.; Lindell, K.O.; et al. Peripheral blood proteins predict mortality in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2012, 185, 67–76. [Google Scholar] [CrossRef]
- Korthagen, N.M.; van Moorsel, C.H.; Barlo, N.P.; Ruven, H.J.; Kruit, A.; Heron, M.; van den Bosch, J.M.; Grutters, J.C. Serum and BALF YKL-40 levels are predictors of survival in idiopathic pulmonary fibrosis. Respir. Med. 2011, 105, 106–113. [Google Scholar] [CrossRef]
- O’Dwyer, D.N.; Armstrong, M.E.; Kooblall, M.; Donnelly, S.C. Targeting defective Toll-like receptor-3 function and idiopathic pulmonary fibrosis. Expert Opin. Ther. Targets 2015, 19, 507–514. [Google Scholar] [CrossRef]
- O’Dwyer, D.N.; Armstrong, M.E.; Trujillo, G.; Cooke, G.; Keane, M.P.; Fallon, P.G.; Simpson, A.J.; Millar, A.B.; McGrath, E.E.; Whyte, M.K.; et al. The Toll-like receptor 3 L412F polymorphism and disease progression in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2013, 188, 1442–1450. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, G.; Meneghin, A.; Flaherty, K.R.; Sholl, L.M.; Myers, J.L.; Kazerooni, E.A.; Gross, B.H.; Oak, S.R.; Coelho, A.L.; Evanoff, H.; et al. TLR9 differentiates rapidly from slowly progressing forms of idiopathic pulmonary fibrosis. Sci. Transl. Med. 2010, 2, 57ra82. [Google Scholar] [CrossRef] [PubMed]
- Oldham, J.M.; Ma, S.F.; Martinez, F.J.; Anstrom, K.J.; Raghu, G.; Schwartz, D.A.; Valenzi, E.; Witt, L.; Lee, C.; Vij, R.; et al. TOLLIP, MUC5B, and the Response to N-Acetylcysteine among Individuals with Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2015, 192, 1475–1482. [Google Scholar] [CrossRef] [PubMed]
- Noth, I.; Zhang, Y.; Ma, S.F.; Flores, C.; Barber, M.; Huang, Y.; Broderick, S.M.; Wade, M.S.; Hysi, P.; Scuirba, J.; et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: A genome-wide association study. Lancet Respir. Med. 2013, 1, 309–317. [Google Scholar] [CrossRef]
- Van Putten, J.P.M.; Strijbis, K. Transmembrane Mucins: Signaling Receptors at the Intersection of Inflammation and Cancer. J. Innate Immun. 2017, 9, 281–299. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Anton, A.; Debolos, C.; Garrido, M.; Roca-Ferrer, J.; Barranco, C.; Alobid, I.; Xaubet, A.; Picado, C.; Mullol, J. Mucin genes have different expression patterns in healthy and diseased upper airway mucosa. Clin. Exp. Allergy 2006, 36, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Lillehoj, E.P.; Kato, K.; Lu, W.; Kim, K.C. Cellular and molecular biology of airway mucins. Int. Rev. Cell Mol. Biol. 2013, 303, 139–202. [Google Scholar] [PubMed]
- Rose, M.C.; Voynow, J.A. Respiratory tract mucin genes and mucin glycoproteins in health and disease. Physiol. Rev. 2006, 86, 245–278. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Rubin, B.K.; Voynow, J.A. Mucins, Mucus, and Goblet Cells. Chest 2018, 154, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Corfield, A.P. Mucins: A biologically relevant glycan barrier in mucosal protection. Biochim. Biophys. Acta 2015, 1850, 236–252. [Google Scholar] [CrossRef] [PubMed]
- Dhanisha, S.S.; Guruvayoorappan, C.; Drishya, S.; Abeesh, P. Mucins: Structural diversity, biosynthesis, its role in pathogenesis and as possible therapeutic targets. Crit. Rev. Oncol. Hematol. 2018, 122, 98–122. [Google Scholar] [CrossRef] [PubMed]
- Hattrup, C.L.; Gendler, S.J. Structure and function of the cell surface (tethered) mucins. Annu. Rev. Physiol. 2008, 70, 431–457. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.S.; Pearson, J.P. Upper airway mucin gene expression: A review. Laryngoscope 2007, 117, 932–938. [Google Scholar] [CrossRef] [PubMed]
- Kirkham, S.; Sheehan, J.K.; Knight, D.; Richardson, P.S.; Thornton, D.J. Heterogeneity of airways mucus: Variations in the amounts and glycoforms of the major oligomeric mucins MUC5AC and MUC5B. Biochem. J. 2002, 361, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Rogers, D.F. Mucus pathophysiology in COPD: Differences to asthma, and pharmacotherapy. Monaldi Arch. Chest Dis. 2000, 55, 324–332. [Google Scholar] [PubMed]
- Kolahian, S.; Fernandez, I.E.; Eickelberg, O.; Hartl, D. Immune Mechanisms in Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2016, 55, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Desai, T.J.; Brownfield, D.G.; Krasnow, M.A. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature 2014, 507, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Mathai, S.K.; Schwartz, D.A. Genetics in Idiopathic Pulmonary Fibrosis Pathogenesis, Prognosis, and Treatment. Front. Med. 2017, 4, 154. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.M.; Ngor, W.M.; Gordish-Dressman, H.; Freishtat, R.J.; Rose, M.C. MUC7 polymorphisms are associated with a decreased risk of a diagnosis of asthma in an African American population. J. Investig. Med. 2009, 57, 882–886. [Google Scholar] [CrossRef]
- Kirkbride, H.J.; Bolscher, J.G.; Nazmi, K.; Vinall, L.E.; Nash, M.W.; Moss, F.M.; Mitchell, D.M.; Swallow, D.M. Genetic polymorphism of MUC7: Allele frequencies and association with asthma. Eur. J. Hum. Genet. 2001, 9, 347–354. [Google Scholar] [CrossRef]
- Wu, A.M.; Csako, G.; Herp, A. Structure, biosynthesis, and function of salivary mucins. Mol. Cell Biochem. 1994, 137, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Bafna, S.; Kaur, S.; Batra, S.K. Membrane-bound mucins: The mechanistic basis for alterations in the growth and survival of cancer cells. Oncogene 2010, 29, 2893–2904. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, M.A.; Swanson, B.J. Mucins in cancer: Protection and control of the cell surface. Nat. Rev. Cancer 2004, 4, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Blalock, T.D.; Spurr-Michaud, S.J.; Tisdale, A.S.; Gipson, I.K. Release of membrane-associated mucins from ocular surface epithelia. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1864–1871. [Google Scholar] [CrossRef]
- Ponnusamy, M.P.; Seshacharyulu, P.; Lakshmanan, I.; Vaz, A.P.; Chugh, S.; Batra, S.K. Emerging role of mucins in epithelial to mesenchymal transition. Curr. Cancer Drug Targets 2013, 13, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Peljto, A.L.; Zhang, Y.; Fingerlin, T.E.; Ma, S.F.; Garcia, J.G.; Richards, T.J.; Silveira, L.J.; Lindell, K.O.; Steele, M.P.; Loyd, J.E.; et al. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA 2013, 309, 2232–2239. [Google Scholar] [CrossRef]
- Seibold, M.A.; Smith, R.W.; Urbanek, C.; Groshong, S.D.; Cosgrove, G.P.; Brown, K.K.; Schwarz, M.I.; Schwartz, D.A.; Reynolds, S.D. The idiopathic pulmonary fibrosis honeycomb cyst contains a mucocilary pseudostratified epithelium. PLoS ONE 2013, 8, e58658. [Google Scholar] [CrossRef]
- Plantier, L.; Crestani, B.; Wert, S.E.; Dehoux, M.; Zweytick, B.; Guenther, A.; Whitsett, J.A. Ectopic respiratory epithelial cell differentiation in bronchiolised distal airspaces in idiopathic pulmonary fibrosis. Thorax 2011, 66, 651–657. [Google Scholar] [CrossRef]
- Ballester, B.; Milara, J.; Sanz, C.; González, S.; Guijarro, R.; Martínez, C.; Cortijo, J. Role of MUC1 in idiopathic pulmonary fibrosis. Eur. Respir. J. 2016. [Google Scholar] [CrossRef]
- Kam, J.L.; Regimbald, L.H.; Hilgers, J.H.; Hoffman, P.; Krantz, M.J.; Longenecker, B.M.; Hugh, J.C. MUC1 synthetic peptide inhibition of intercellular adhesion molecule-1 and MUC1 binding requires six tandem repeats. Cancer Res. 1998, 58, 5577–5581. [Google Scholar]
- Tsoutsou, P.G.; Gourgoulianis, K.I.; Petinaki, E.; Mpaka, M.; Efremidou, S.; Maniatis, A.; Molyvdas, P.A. ICAM-1, ICAM-2 and ICAM-3 in the sera of patients with idiopathic pulmonary fibrosis. Inflammation 2004, 28, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, S.; Duraisamy, S.; Barbashov, S.; Kawano, T.; Kharbanda, S.; Kufe, D. The MUC1 and galectin-3 oncoproteins function in a microRNA-dependent regulatory loop. Mol. Cell 2007, 27, 992–1004. [Google Scholar] [CrossRef] [PubMed]
- Kufe, D.W. Mucins in cancer: Function, prognosis and therapy. Nat. Rev. Cancer 2009, 9, 874–885. [Google Scholar] [CrossRef] [PubMed]
- Ballester, B.; Roger, I.; Contreras, S.; Montero, P.; Milara, J. Role of MUC1 in idiopathic pulmonary fibrosis: Mechanistic insights. Eur. Respir. J. 2017. [Google Scholar] [CrossRef]
- Senapati, S.; Das, S.; Batra, S.K. Mucin-interacting proteins: From function to therapeutics. Trends Biochem. Sci. 2010, 35, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Ballester, B.; Milara, J.; Guijarro, R.; Morcillo, E.; Cortijo, J. Role of MUC4 in idiopathic pulmonary fibrosis. Eur. Respir. J. 2018. [Google Scholar] [CrossRef]
- Zhi, X.; Tao, J.; Xie, K.; Zhu, Y.; Li, Z.; Tang, J.; Wang, W.; Xu, H.; Zhang, J.; Xu, Z. MUC4-induced nuclear translocation of beta-catenin: A novel mechanism for growth, metastasis and angiogenesis in pancreatic cancer. Cancer Lett. 2014, 346, 104–113. [Google Scholar] [CrossRef]
- Chaturvedi, P.; Singh, A.P.; Chakraborty, S.; Chauhan, S.C.; Bafna, S.; Meza, J.L.; Singh, P.K.; Hollingsworth, M.A.; Mehta, P.P.; Batra, S.K. MUC4 mucin interacts with and stabilizes the HER2 oncoprotein in human pancreatic cancer cells. Cancer Res. 2008, 68, 2065–2070. [Google Scholar] [CrossRef]
- Lakshmanan, I.; Ponnusamy, M.P.; Das, S.; Chakraborty, S.; Haridas, D.; Mukhopadhyay, P.; Lele, S.M.; Batra, S.K. MUC16 induced rapid G2/M transition via interactions with JAK2 for increased proliferation and anti-apoptosis in breast cancer cells. Oncogene 2012, 31, 805–817. [Google Scholar] [CrossRef]
- Matte, I.; Lane, D.; Boivin, M.; Rancourt, C.; Piche, A. MUC16 mucin (CA125) attenuates TRAIL-induced apoptosis by decreasing TRAIL receptor R2 expression and increasing c-FLIP expression. BMC Cancer 2014, 14, 234. [Google Scholar] [CrossRef]
- Akita, K.; Tanaka, M.; Tanida, S.; Mori, Y.; Toda, M.; Nakada, H. CA125/MUC16 interacts with Src family kinases, and over-expression of its C-terminal fragment in human epithelial cancer cells reduces cell-cell adhesion. Eur. J. Cell Biol. 2013, 92, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Seelenmeyer, C.; Wegehingel, S.; Lechner, J.; Nickel, W. The cancer antigen CA125 represents a novel counter receptor for galectin-1. J. Cell Sci. 2003, 116, 1305–1318. [Google Scholar] [CrossRef] [PubMed]
- Argueso, P.; Guzman-Aranguez, A.; Mantelli, F.; Cao, Z.; Ricciuto, J.; Panjwani, N. Association of cell surface mucins with galectin-3 contributes to the ocular surface epithelial barrier. J. Biol. Chem. 2009, 284, 23037–23045. [Google Scholar] [CrossRef] [PubMed]
- Desseyn, J.L.; Guyonnet-Duperat, V.; Porchet, N.; Aubert, J.P.; Laine, A. Human mucin gene MUC5B, the 10.7-kb large central exon encodes various alternate subdomains resulting in a super-repeat. Structural evidence for a 11p15.5 gene family. J. Biol. Chem. 1997, 272, 3168–3178. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhao, Y.H.; Di, Y.P.; Wu, R. Characterization of human mucin 5B gene expression in airway epithelium and the genomic clone of the amino-terminal and 5’-flanking region. Am. J. Respir. Cell Mol. Biol. 2001, 25, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Vinall, L.E.; Hill, A.S.; Pigny, P.; Pratt, W.S.; Toribara, N.; Gum, J.R.; Kim, Y.S.; Porchet, N.; Aubert, J.P.; Swallow, D.M. Variable number tandem repeat polymorphism of the mucin genes located in the complex on 11p15.5. Hum. Genet. 1998, 102, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Thornton, D.J.; Howard, M.; Khan, N.; Sheehan, J.K. Identification of two glycoforms of the MUC5B mucin in human respiratory mucus. Evidence for a cysteine-rich sequence repeated within the molecule. J. Biol. Chem. 1997, 272, 9561–9566. [Google Scholar] [CrossRef] [PubMed]
- Desseyn, J.L.; Buisine, M.P.; Porchet, N.; Aubert, J.P.; Laine, A. Genomic organization of the human mucin gene MUC5B. cDNA and genomic sequences upstream of the large central exon. J. Biol. Chem. 1998, 273, 30157–30164. [Google Scholar] [CrossRef] [PubMed]
- Desseyn, J.L.; Aubert, J.P.; Van Seuningen, I.; Porchet, N.; Laine, A. Genomic organization of the 3’ region of the human mucin gene MUC5B. J. Biol. Chem. 1997, 272, 16873–16883. [Google Scholar] [CrossRef]
- Preciado, D.; Kuo, E.; Ashktorab, S.; Manes, P.; Rose, M. Cigarette smoke activates NFkappaB-mediated Tnf-alpha release from mouse middle ear cells. Laryngoscope 2010, 120, 2508–2515. [Google Scholar] [CrossRef]
- Fujimoto, H.; D’Alessandro-Gabazza, C.N.; Palanki, M.S.; Erdman, P.E.; Takagi, T.; Gabazza, E.C.; Bruno, N.E.; Yano, Y.; Hayashi, T.; Tamaki, S.; et al. Inhibition of nuclear factor-kappaB in T cells suppresses lung fibrosis. Am. J. Respir. Crit. Care Med. 2007, 176, 1251–1260. [Google Scholar] [CrossRef]
- Preciado, D.; Lin, J.; Wuertz, B.; Rose, M. Cigarette smoke activates NF kappa B and induces Muc5b expression in mouse middle ear cells. Laryngoscope 2008, 118, 464–471. [Google Scholar] [CrossRef]
- Arakawa, H.; Honma, K. Honeycomb lung: History and current concepts. AJR Am. J. Roentgenol. 2011, 196, 773–782. [Google Scholar] [CrossRef]
- Boucher, R.C. Idiopathic pulmonary fibrosis--a sticky business. N. Engl. J. Med. 2011, 364, 1560–1561. [Google Scholar] [CrossRef]
- Button, B.; Cai, L.H.; Ehre, C.; Kesimer, M.; Hill, D.B.; Sheehan, J.K.; Boucher, R.C.; Rubinstein, M. A periciliary brush promotes the lung health by separating the mucus layer from airway epithelia. Science 2012, 337, 937–941. [Google Scholar] [CrossRef]
- Roy, M.G.; Livraghi-Butrico, A.; Fletcher, A.A.; McElwee, M.M.; Evans, S.E.; Boerner, R.M.; Alexander, S.N.; Bellinghausen, L.K.; Song, A.S.; Petrova, Y.M.; et al. Muc5b is required for airway defence. Nature 2014, 505, 412–416. [Google Scholar] [CrossRef]
- Lahdaoui, F.; Messager, M.; Vincent, A.; Hec, F.; Gandon, A.; Warlaumont, M.; Renaud, F.; Leteurtre, E.; Piessen, G.; Jonckheere, N.; et al. Depletion of MUC5B mucin in gastrointestinal cancer cells alters their tumorigenic properties: Implication of the Wnt/beta-catenin pathway. Biochem. J. 2017, 474, 3733–3746. [Google Scholar] [CrossRef]
- Guo, Y.; Xiao, L.; Sun, L.; Liu, F. Wnt/beta-catenin signaling: A promising new target for fibrosis diseases. Physiol. Res. 2012, 61, 337–346. [Google Scholar]
- Helling, B.A.; Gerber, A.N.; Kadiyala, V.; Sasse, S.K.; Pedersen, B.S.; Sparks, L.; Nakano, Y.; Okamoto, T.; Evans, C.M.; Yang, I.V.; et al. Regulation of MUC5B Expression in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2017, 57, 91–99. [Google Scholar] [CrossRef]
- Chen, G.; Ribeiro, C.M.P.; Sun, L.; Okuda, K.; Kato, T.; Gilmore, R.C.; Martino, M.B.; Dang, H.; Abzhanova, A.; Lin, J.M.; et al. XBP1S Regulates MUC5B in a Promoter Variant-Dependent Pathway in Idiopathic Pulmonary Fibrosis Airway Epithelia. Am. J. Respir. Crit. Care Med. 2019, 200, 220–234. [Google Scholar] [CrossRef]
- Nakano, Y.; Yang, I.V.; Walts, A.D.; Watson, A.M.; Helling, B.A.; Fletcher, A.A.; Lara, A.R.; Schwarz, M.I.; Evans, C.M.; Schwartz, D.A. MUC5B Promoter Variant rs35705950 Affects MUC5B Expression in the Distal Airways in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2016, 193, 464–466. [Google Scholar] [CrossRef]
- Molyneaux, P.L.; Cox, M.J.; Willis-Owen, S.A.; Mallia, P.; Russell, K.E.; Russell, A.M.; Murphy, E.; Johnston, S.L.; Schwartz, D.A.; Wells, A.U.; et al. The role of bacteria in the pathogenesis and progression of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 906–913. [Google Scholar] [CrossRef]
- Evans, C.M.; Fingerlin, T.E.; Schwarz, M.I.; Lynch, D.; Kurche, J.; Warg, L.; Yang, I.V.; Schwartz, D.A. Idiopathic Pulmonary Fibrosis: A Genetic Disease That Involves Mucociliary Dysfunction of the Peripheral Airways. Physiol. Rev. 2016, 96, 1567–1591. [Google Scholar] [CrossRef]
- Buisine, M.P.; Devisme, L.; Copin, M.C.; Durand-Reville, M.; Gosselin, B.; Aubert, J.P.; Porchet, N. Developmental mucin gene expression in the human respiratory tract. Am. J. Respir. Cell Mol. Biol. 1999, 20, 209–218. [Google Scholar] [CrossRef]
- Buisine, M.P.; Desreumaux, P.; Leteurtre, E.; Copin, M.C.; Colombel, J.F.; Porchet, N.; Aubert, J.P. Mucin gene expression in intestinal epithelial cells in Crohn’s disease. Gut 2001, 49, 544–551. [Google Scholar] [CrossRef]
- Young, H.W.; Williams, O.W.; Chandra, D.; Bellinghausen, L.K.; Perez, G.; Suarez, A.; Tuvim, M.J.; Roy, M.G.; Alexander, S.N.; Moghaddam, S.J.; et al. Central role of Muc5ac expression in mucous metaplasia and its regulation by conserved 5’ elements. Am. J. Respir. Cell Mol. Biol. 2007, 37, 273–290. [Google Scholar] [CrossRef]
- Tao, J.; Zhang, M.; Wen, Z.; Wang, B.; Zhang, L.; Ou, Y.; Tang, X.; Yu, X.; Jiang, Q. Inhibition of EP300 and DDR1 synergistically alleviates pulmonary fibrosis in vitro and in vivo. Biomed. Pharmacother. 2018, 106, 1727–1733. [Google Scholar] [CrossRef]
- Xu, S.; Hui, Y.; Shu, J.; Qian, J.; Li, L. Characterization of the human mucin 5AC promoter and its regulation by the histone acetyltransferase P300. Int. J. Mol. Med. 2019, 43, 1263–1270. [Google Scholar] [CrossRef]
- Hu, B.; Phan, S.H. Notch in fibrosis and as a target of anti-fibrotic therapy. Pharmacol. Res. 2016, 108, 57–64. [Google Scholar] [CrossRef]
- Ou-Yang, H.F.; Wu, C.G.; Qu, S.Y.; Li, Z.K. Notch signaling downregulates MUC5AC expression in airway epithelial cells through Hes1-dependent mechanisms. Respiration 2013, 86, 341–346. [Google Scholar] [CrossRef]
- Araya, J.; Kojima, J.; Takasaka, N.; Ito, S.; Fujii, S.; Hara, H.; Yanagisawa, H.; Kobayashi, K.; Tsurushige, C.; Kawaishi, M.; et al. Insufficient autophagy in idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 304, L56–L69. [Google Scholar] [CrossRef]
- Patel, A.S.; Lin, L.; Geyer, A.; Haspel, J.A.; An, C.H.; Cao, J.; Rosas, I.O.; Morse, D. Autophagy in idiopathic pulmonary fibrosis. PLoS ONE 2012, 7, e41394. [Google Scholar] [CrossRef]
- Sosulski, M.L.; Gongora, R.; Danchuk, S.; Dong, C.; Luo, F.; Sanchez, C.G. Deregulation of selective autophagy during aging and pulmonary fibrosis: The role of TGFbeta1. Aging Cell 2015, 14, 774–783. [Google Scholar] [CrossRef]
- Ye, Y.; Zhao, J.; Ye, J.; Jiang, X.; Liu, H.; Xie, Y.; Zhang, J.; Luo, Q. The role of autophagy in the overexpression of MUC5AC in patients with chronic rhinosinusitis. Int. Immunopharmacol. 2019, 71, 169–180. [Google Scholar] [CrossRef]
- Fujisawa, T.; Velichko, S.; Thai, P.; Hung, L.Y.; Huang, F.; Wu, R. Regulation of airway MUC5AC expression by IL-1beta and IL-17A; the NF-kappaB paradigm. J. Immunol. 2009, 183, 6236–6243. [Google Scholar] [CrossRef]
- Wilson, M.S.; Madala, S.K.; Ramalingam, T.R.; Gochuico, B.R.; Rosas, I.O.; Cheever, A.W.; Wynn, T.A. Bleomycin and IL-1beta-mediated pulmonary fibrosis is IL-17A dependent. J. Exp. Med. 2010, 207, 535–552. [Google Scholar] [CrossRef]
- Sugimoto, N.; Suzukawa, M.; Nagase, H.; Koizumi, Y.; Ro, S.; Kobayashi, K.; Yoshihara, H.; Kojima, Y.; Kamiyama-Hara, A.; Hebisawa, A.; et al. IL-9 Blockade Suppresses Silica-induced Lung Inflammation and Fibrosis in Mice. Am. J. Respir. Cell Mol. Biol. 2019, 60, 232–243. [Google Scholar] [CrossRef]
- Longphre, M.; Li, D.; Gallup, M.; Drori, E.; Ordonez, C.L.; Redman, T.; Wenzel, S.; Bice, D.E.; Fahy, J.V.; Basbaum, C. Allergen-induced IL-9 directly stimulates mucin transcription in respiratory epithelial cells. J. Clin. Investig. 1999, 104, 1375–1382. [Google Scholar] [CrossRef]
- Gregory, A.D.; Kliment, C.R.; Metz, H.E.; Kim, K.H.; Kargl, J.; Agostini, B.A.; Crum, L.T.; Oczypok, E.A.; Oury, T.A.; Houghton, A.M. Neutrophil elastase promotes myofibroblast differentiation in lung fibrosis. J. Leukoc. Biol. 2015, 98, 143–152. [Google Scholar] [CrossRef]
- Voynow, J.A.; Young, L.R.; Wang, Y.; Horger, T.; Rose, M.C.; Fischer, B.M. Neutrophil elastase increases MUC5AC mRNA and protein expression in respiratory epithelial cells. Am. J. Physiol. 1999, 276, L835–L843. [Google Scholar]
- Lorenzo-Salazar, J.M.; Ma, S.F.; Jou, J.; Hou, P.C.; Guillen-Guio, B.; Allen, R.J.; Jenkins, R.G.; Wain, L.V.; Oldham, J.M.; Noth, I.; et al. Novel idiopathic pulmonary fibrosis susceptibility variants revealed by deep sequencing. ERJ Open Res. 2019, 5. [Google Scholar] [CrossRef]
- Patton, S.; Gendler, S.J.; Spicer, A.P. The epithelial mucin, MUC1, of milk, mammary gland and other tissues. Biochim. Biophys. Acta 1995, 1241, 407–423. [Google Scholar] [CrossRef]
- Taylor-Papadimitriou, J.; Burchell, J.; Miles, D.W.; Dalziel, M. MUC1 and cancer. Biochim. Biophys. Acta 1999, 1455, 301–313. [Google Scholar] [CrossRef]
- Duffy, M.J.; Shering, S.; Sherry, F.; McDermott, E.; O’Higgins, N. CA 15-3: A prognostic marker in breast cancer. Int. J. Biol. Markers 2000, 15, 330–333. [Google Scholar] [CrossRef]
- Gion, M.; Mione, R.; Leon, A.E.; Dittadi, R. Comparison of the diagnostic accuracy of CA27.29 and CA15.3 in primary breast cancer. Clin. Chem. 1999, 45, 630–637. [Google Scholar]
- JafariNezhad, A.; YektaKooshali, M.H. Lung cancer in idiopathic pulmonary fibrosis: A systematic review and meta-analysis. PLoS ONE 2018, 13, e0202360. [Google Scholar] [CrossRef]
- Kufe, D. Oncogenic function of the MUC1 receptor subunit in gene regulation. Oncogene 2010, 29, 5663–5666. [Google Scholar] [CrossRef]
- Nath, S.; Mukherjee, P. MUC1: A multifaceted oncoprotein with a key role in cancer progression. Trends Mol. Med. 2014, 20, 332–342. [Google Scholar] [CrossRef]
- Palmai-Pallag, T.; Khodabukus, N.; Kinarsky, L.; Leir, S.H.; Sherman, S.; Hollingsworth, M.A.; Harris, A. The role of the SEA (sea urchin sperm protein, enterokinase and agrin) module in cleavage of membrane-tethered mucins. FEBS J. 2005, 272, 2901–2911. [Google Scholar] [CrossRef]
- Thathiah, A.; Blobel, C.P.; Carson, D.D. Tumor necrosis factor-alpha converting enzyme/ADAM 17 mediates MUC1 shedding. J. Biol. Chem. 2003, 278, 3386–3394. [Google Scholar] [CrossRef]
- Thathiah, A.; Carson, D.D. MT1-MMP mediates MUC1 shedding independent of TACE/ADAM17. Biochem. J. 2004, 382, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Pardo, A.; Cabrera, S.; Maldonado, M.; Selman, M. Role of matrix metalloproteinases in the pathogenesis of idiopathic pulmonary fibrosis. Respir. Res. 2016, 17, 23. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Yang, D.; Zhu, S.; Gu, J.; Ding, F.; Bian, W.; Rong, Z.; Shen, C. Bleomycin-induced pulmonary fibrosis is attenuated by an antibody against KL-6. Exp. Lung Res. 2013, 39, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.; McGuckin, M.A.; Hurst, T.; Ward, B.G. Effect of interferon-gamma and TNF-alpha on MUC1 mucin expression in ovarian carcinoma cell lines. Dis. Markers 1994, 12, 43–50. [Google Scholar] [CrossRef] [PubMed]
- King, T.E., Jr.; Albera, C.; Bradford, W.Z.; Costabel, U.; Hormel, P.; Lancaster, L.; Noble, P.W.; Sahn, S.A.; Szwarcberg, J.; Thomeer, M.; et al. Effect of interferon gamma-1b on survival in patients with idiopathic pulmonary fibrosis (INSPIRE): A multicentre, randomised, placebo-controlled trial. Lancet 2009, 374, 222–228. [Google Scholar] [CrossRef]
- Gaemers, I.C.; Vos, H.L.; Volders, H.H.; van der Valk, S.W.; Hilkens, J. A stat-responsive element in the promoter of the episialin/MUC1 gene is involved in its overexpression in carcinoma cells. J. Biol. Chem. 2001, 276, 6191–6199. [Google Scholar] [CrossRef]
- Shirasaki, H.; Kanaizumi, E.; Watanabe, K.; Konno, N.; Sato, J.; Narita, S.; Himi, T. Tumor necrosis factor increases MUC1 mRNA in cultured human nasal epithelial cells. Acta Otolaryngol. 2003, 123, 524–531. [Google Scholar] [CrossRef]
- Kuwahara, I.; Lillehoj, E.P.; Hisatsune, A.; Lu, W.; Isohama, Y.; Miyata, T.; Kim, K.C. Neutrophil elastase stimulates MUC1 gene expression through increased Sp1 binding to the MUC1 promoter. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 289, L355–L362. [Google Scholar] [CrossRef]
- Rahn, J.J.; Shen, Q.; Mah, B.K.; Hugh, J.C. MUC1 initiates a calcium signal after ligation by intercellular adhesion molecule-1. J. Biol. Chem. 2004, 279, 29386–29390. [Google Scholar] [CrossRef]
- Mackinnon, A.C.; Gibbons, M.A.; Farnworth, S.L.; Leffler, H.; Nilsson, U.J.; Delaine, T.; Simpson, A.J.; Forbes, S.J.; Hirani, N.; Gauldie, J.; et al. Regulation of transforming growth factor-beta1-driven lung fibrosis by galectin-3. Am. J. Respir. Crit. Care Med. 2012, 185, 537–546. [Google Scholar] [CrossRef]
- Singh, P.K.; Wen, Y.; Swanson, B.J.; Shanmugam, K.; Kazlauskas, A.; Cerny, R.L.; Gendler, S.J.; Hollingsworth, M.A. Platelet-derived growth factor receptor beta-mediated phosphorylation of MUC1 enhances invasiveness in pancreatic adenocarcinoma cells. Cancer Res. 2007, 67, 5201–5210. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Raina, D.; Chen, W.; Li, G.; Huang, L.; Kufe, D. MUC1 oncoprotein functions in activation of fibroblast growth factor receptor signaling. Mol. Cancer Res. 2006, 4, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, J.A.; Masri, A.A.; Adriance, M.C.; Tessier, J.C.; Kotlarczyk, K.L.; Thompson, M.C.; Gendler, S.J. MUC1 overexpression results in mammary gland tumorigenesis and prolonged alveolar differentiation. Oncogene 2004, 23, 5739–5747. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Chen, D.; Liu, D.; Yin, L.; Kharbanda, S.; Kufe, D. MUC1 oncoprotein blocks glycogen synthase kinase 3beta-mediated phosphorylation and degradation of beta-catenin. Cancer Res. 2005, 65, 10413–10422. [Google Scholar] [CrossRef] [PubMed]
- Behrens, M.E.; Grandgenett, P.M.; Bailey, J.M.; Singh, P.K.; Yi, C.H.; Yu, F.; Hollingsworth, M.A. The reactive tumor microenvironment: MUC1 signaling directly reprograms transcription of CTGF. Oncogene 2010, 29, 5667–5677. [Google Scholar] [CrossRef] [PubMed]
- Konigshoff, M.; Kramer, M.; Balsara, N.; Wilhelm, J.; Amarie, O.V.; Jahn, A.; Rose, F.; Fink, L.; Seeger, W.; Schaefer, L.; et al. WNT1-inducible signaling protein-1 mediates pulmonary fibrosis in mice and is upregulated in humans with idiopathic pulmonary fibrosis. J. Clin. Investig. 2009, 119, 772–787. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Primers 2017, 3, 17074. [Google Scholar] [CrossRef]
- Roy, L.D.; Sahraei, M.; Subramani, D.B.; Besmer, D.; Nath, S.; Tinder, T.L.; Bajaj, E.; Shanmugam, K.; Lee, Y.Y.; Hwang, S.I.; et al. MUC1 enhances invasiveness of pancreatic cancer cells by inducing epithelial to mesenchymal transition. Oncogene 2011, 30, 1449–1459. [Google Scholar] [CrossRef]
- Singh, P.K.; Hollingsworth, M.A. Cell surface-associated mucins in signal transduction. Trends Cell Biol. 2006, 16, 467–476. [Google Scholar] [CrossRef]
- Carson, D.D. The cytoplasmic tail of MUC1: A very busy place. Sci. Signal 2008, 1, pe35. [Google Scholar] [CrossRef]
- Arriola, E.; Ottensmeier, C. TG4010: A vaccine with a therapeutic role in cancer. Immunotherapy 2016, 8, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Sangha, R.; Butts, C. L-BLP25: A peptide vaccine strategy in non small cell lung cancer. Clin. Cancer Res. 2007, 13, s4652–s4654. [Google Scholar] [CrossRef] [PubMed]
- Heery, C.R.; Ibrahim, N.K.; Arlen, P.M.; Mohebtash, M.; Murray, J.L.; Koenig, K.; Madan, R.A.; McMahon, S.; Marte, J.L.; Steinberg, S.M.; et al. Docetaxel Alone or in Combination With a Therapeutic Cancer Vaccine (PANVAC) in Patients With Metastatic Breast Cancer: A Randomized Clinical Trial. JAMA Oncol. 2015, 1, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Swartz, M.J.; Batra, S.K.; Varshney, G.C.; Hollingsworth, M.A.; Yeo, C.J.; Cameron, J.L.; Wilentz, R.E.; Hruban, R.H.; Argani, P. MUC4 expression increases progressively in pancreatic intraepithelial neoplasia. Am. J. Clin. Pathol. 2002, 117, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.P.; Moniaux, N.; Chauhan, S.C.; Meza, J.L.; Batra, S.K. Inhibition of MUC4 expression suppresses pancreatic tumor cell growth and metastasis. Cancer Res. 2004, 64, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Llinares, K.; Escande, F.; Aubert, S.; Buisine, M.P.; de Bolos, C.; Batra, S.K.; Gosselin, B.; Aubert, J.P.; Porchet, N.; Copin, M.C. Diagnostic value of MUC4 immunostaining in distinguishing epithelial mesothelioma and lung adenocarcinoma. Mod. Pathol. 2004, 17, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Shibahara, H.; Tamada, S.; Higashi, M.; Goto, M.; Batra, S.K.; Hollingsworth, M.A.; Imai, K.; Yonezawa, S. MUC4 is a novel prognostic factor of intrahepatic cholangiocarcinoma-mass forming type. Hepatology 2004, 39, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Fowler, J.; Vinall, L.; Swallow, D. Polymorphism of the human muc genes. Front. Biosci. 2001, 6, D1207–D1215. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, A.; Moniaux, N.; Winpenny, J.P.; Hollingsworth, M.A.; Aubert, J.P.; Batra, S.K. Human MUC4 mucin cDNA and its variants in pancreatic carcinoma. J. Biochem. 2000, 128, 233–243. [Google Scholar] [CrossRef]
- Escande, F.; Lemaitre, L.; Moniaux, N.; Batra, S.K.; Aubert, J.P.; Buisine, M.P. Genomic organization of MUC4 mucin gene. Towards the characterization of splice variants. Eur. J. Biochem. 2002, 269, 3637–3644. [Google Scholar] [CrossRef]
- Carraway, K.L.; Price-Schiavi, S.A.; Komatsu, M.; Jepson, S.; Perez, A.; Carraway, C.A. Muc4/sialomucin complex in the mammary gland and breast cancer. J. Mammary Gland Biol. Neoplasia 2001, 6, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, J.; Wilhelm, J.; Marsh, L.M.; Ghanim, B.; Klepetko, W.; Kovacs, G.; Olschewski, H.; Olschewski, A.; Kwapiszewska, G. Distinct differences in gene expression patterns in pulmonary arteries of patients with chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis with pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2014, 190, 98–111. [Google Scholar] [CrossRef] [PubMed]
- King, T.E., Jr.; Brown, K.K.; Raghu, G.; du Bois, R.M.; Lynch, D.A.; Martinez, F.; Valeyre, D.; Leconte, I.; Morganti, A.; Roux, S.; et al. BUILD-3: A randomized, controlled trial of bosentan in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2011, 184, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Behr, J.; Brown, K.K.; Egan, J.J.; Kawut, S.M.; Flaherty, K.R.; Martinez, F.J.; Nathan, S.D.; Wells, A.U.; Collard, H.R.; et al. Treatment of idiopathic pulmonary fibrosis with ambrisentan: A parallel, randomized trial. Ann. Intern. Med. 2013, 158, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Million-Rousseau, R.; Morganti, A.; Perchenet, L.; Behr, J.; Group, M.S. Macitentan for the treatment of idiopathic pulmonary fibrosis: The randomised controlled MUSIC trial. Eur. Respir. J. 2013, 42, 1622–1632. [Google Scholar] [CrossRef]
- Perrais, M.; Pigny, P.; Ducourouble, M.P.; Petitprez, D.; Porchet, N.; Aubert, J.P.; Van Seuningen, I. Characterization of human mucin gene MUC4 promoter: Importance of growth factors and proinflammatory cytokines for its regulation in pancreatic cancer cells. J. Biol. Chem. 2001, 276, 30923–30933. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.P.; Chauhan, S.C.; Andrianifahanana, M.; Moniaux, N.; Meza, J.L.; Copin, M.C.; van Seuningen, I.; Hollingsworth, M.A.; Aubert, J.P.; Batra, S.K. MUC4 expression is regulated by cystic fibrosis transmembrane conductance regulator in pancreatic adenocarcinoma cells via transcriptional and post-translational mechanisms. Oncogene 2007, 26, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Damera, G.; Xia, B.; Ancha, H.R.; Sachdev, G.P. IL-9 modulated MUC4 gene and glycoprotein expression in airway epithelial cells. Biosci. Rep. 2006, 26, 55–67. [Google Scholar] [CrossRef]
- Damera, G.; Xia, B.; Sachdev, G.P. IL-4 induced MUC4 enhancement in respiratory epithelial cells in vitro is mediated through JAK-3 selective signaling. Respir. Res. 2006, 7, 39. [Google Scholar] [CrossRef]
- Mejias-Luque, R.; Peiro, S.; Vincent, A.; Van Seuningen, I.; de Bolos, C. IL-6 induces MUC4 expression through gp130/STAT3 pathway in gastric cancer cell lines. Biochim. Biophys. Acta 2008, 1783, 1728–1736. [Google Scholar] [CrossRef]
- Huaux, F.; Liu, T.; McGarry, B.; Ullenbruch, M.; Phan, S.H. Dual roles of IL-4 in lung injury and fibrosis. J. Immunol. 2003, 170, 2083–2092. [Google Scholar] [CrossRef] [PubMed]
- Papiris, S.A.; Tomos, I.P.; Karakatsani, A.; Spathis, A.; Korbila, I.; Analitis, A.; Kolilekas, L.; Kagouridis, K.; Loukides, S.; Karakitsos, P.; et al. High levels of IL-6 and IL-8 characterize early-on idiopathic pulmonary fibrosis acute exacerbations. Cytokine 2018, 102, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Baughman, R.P.; Lower, E.E.; Miller, M.A.; Bejarano, P.A.; Heffelfinger, S.C. Overexpression of transforming growth factor-alpha and epidermal growth factor-receptor in idiopathic pulmonary fibrosis. Sarcoidosis Vasc. Diffus. Lung Dis. 1999, 16, 57–61. [Google Scholar]
- Pai, P.; Rachagani, S.; Lakshmanan, I.; Macha, M.A.; Sheinin, Y.; Smith, L.M.; Ponnusamy, M.P.; Batra, S.K. The canonical Wnt pathway regulates the metastasis-promoting mucin MUC4 in pancreatic ductal adenocarcinoma. Mol. Oncol. 2016, 10, 224–239. [Google Scholar] [CrossRef] [PubMed]
- Fischer, B.M.; Cuellar, J.G.; Diehl, M.L.; deFreytas, A.M.; Zhang, J.; Carraway, K.L.; Voynow, J.A. Neutrophil elastase increases MUC4 expression in normal human bronchial epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2003, 284, L671–L679. [Google Scholar] [CrossRef]
- Ponnusamy, M.P.; Singh, A.P.; Jain, M.; Chakraborty, S.; Moniaux, N.; Batra, S.K. MUC4 activates HER2 signalling and enhances the motility of human ovarian cancer cells. Br. J. Cancer 2008, 99, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Rachagani, S.; Macha, M.A.; Ponnusamy, M.P.; Haridas, D.; Kaur, S.; Jain, M.; Batra, S.K. MUC4 potentiates invasion and metastasis of pancreatic cancer cells through stabilization of fibroblast growth factor receptor 1. Carcinogenesis 2012, 33, 1953–1964. [Google Scholar] [CrossRef] [PubMed]
- Morgado, M.; Sutton, M.N.; Simmons, M.; Warren, C.R.; Lu, Z.; Constantinou, P.E.; Liu, J.; Francis, L.L.; Conlan, R.S.; Bast, R.C., Jr.; et al. Tumor necrosis factor-alpha and interferon-gamma stimulate MUC16 (CA125) expression in breast, endometrial and ovarian cancers through NFkappaB. Oncotarget 2016, 7, 14871–14884. [Google Scholar] [CrossRef]
- Yilmaz, M.B.; Zorlu, A.; Dogan, O.T.; Karahan, O.; Tandogan, I.; Akkurt, I. Role of CA-125 in identification of right ventricular failure in chronic obstructive pulmonary disease. Clin. Cardiol. 2011, 34, 244–248. [Google Scholar] [CrossRef]
- Yin, B.W.; Lloyd, K.O. Molecular cloning of the CA125 ovarian cancer antigen: Identification as a new mucin, MUC16. J. Biol. Chem. 2001, 276, 27371–27375. [Google Scholar] [CrossRef]
- O’Brien, T.J.; Beard, J.B.; Underwood, L.J.; Dennis, R.A.; Santin, A.D.; York, L. The CA 125 gene: An extracellular superstructure dominated by repeat sequences. Tumour Biol. 2001, 22, 348–366. [Google Scholar] [PubMed]
- Duraisamy, S.; Ramasamy, S.; Kharbanda, S.; Kufe, D. Distinct evolution of the human carcinoma-associated transmembrane mucins, MUC1, MUC4 AND MUC16. Gene 2006, 373, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Wreschner, D.H.; McGuckin, M.A.; Williams, S.J.; Baruch, A.; Yoeli, M.; Ziv, R.; Okun, L.; Zaretsky, J.; Smorodinsky, N.; Keydar, I.; et al. Generation of ligand-receptor alliances by “SEA” module-mediated cleavage of membrane-associated mucin proteins. Protein Sci. 2002, 11, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Majhi, P.D.; Al-Mugotir, M.H.; Rachagani, S.; Sorgen, P.; Batra, S.K. Membrane proximal ectodomain cleavage of MUC16 occurs in the acidifying Golgi/post-Golgi compartments. Sci. Rep. 2015, 5, 9759. [Google Scholar] [CrossRef] [PubMed]
- Govindarajan, B.; Menon, B.B.; Spurr-Michaud, S.; Rastogi, K.; Gilmore, M.S.; Argueso, P.; Gipson, I.K. A metalloproteinase secreted by Streptococcus pneumoniae removes membrane mucin MUC16 from the epithelial glycocalyx barrier. PLoS ONE 2012, 7, e32418. [Google Scholar] [CrossRef] [PubMed]
- Milara, J.; Ballester, B.; Morell, A.; Ortiz, J.L.; Escriva, J.; Fernandez, E.; Perez-Vizcaino, F.; Cogolludo, A.; Pastor, E.; Artigues, E.; et al. JAK2 mediates lung fibrosis, pulmonary vascular remodelling and hypertension in idiopathic pulmonary fibrosis: An experimental study. Thorax 2018, 73, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Rachagani, S.; Torres-Gonzalez, M.P.; Lakshmanan, I.; Majhi, P.D.; Smith, L.M.; Wagner, K.U.; Batra, S.K. Carboxyl-terminal domain of MUC16 imparts tumorigenic and metastatic functions through nuclear translocation of JAK2 to pancreatic cancer cells. Oncotarget 2015, 6, 5772–5787. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Horowitz, J.C. Evolving concepts of apoptosis in idiopathic pulmonary fibrosis. Proc. Am. Thorac. Soc. 2006, 3, 350–356. [Google Scholar] [CrossRef]
- Akram, K.M.; Lomas, N.J.; Forsyth, N.R.; Spiteri, M.A. Alveolar epithelial cells in idiopathic pulmonary fibrosis display upregulation of TRAIL, DR4 and DR5 expression with simultaneous preferential over-expression of pro-apoptotic marker p53. Int. J. Clin. Exp. Pathol. 2014, 7, 552–564. [Google Scholar]
- Comamala, M.; Pinard, M.; Theriault, C.; Matte, I.; Albert, A.; Boivin, M.; Beaudin, J.; Piche, A.; Rancourt, C. Downregulation of cell surface CA125/MUC16 induces epithelial-to-mesenchymal transition and restores EGFR signalling in NIH:OVCAR3 ovarian carcinoma cells. Br. J. Cancer 2011, 104, 989–999. [Google Scholar] [CrossRef]
- Giannakouros, P.; Comamala, M.; Matte, I.; Rancourt, C.; Piche, A. MUC16 mucin (CA125) regulates the formation of multicellular aggregates by altering beta-catenin signaling. Am. J. Cancer Res. 2015, 5, 219–230. [Google Scholar] [PubMed]
- Fujita, Y.; Krause, G.; Scheffner, M.; Zechner, D.; Leddy, H.E.; Behrens, J.; Sommer, T.; Birchmeier, W. Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat. Cell Biol. 2002, 4, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Roura, S.; Miravet, S.; Piedra, J.; Garcia de Herreros, A.; Dunach, M. Regulation of E-cadherin/Catenin association by tyrosine phosphorylation. J. Biol. Chem. 1999, 274, 36734–36740. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Hung, W.C.; Wang, P.; Paul, C.; Konstantopoulos, K. Mesothelin binding to CA125/MUC16 promotes pancreatic cancer cell motility and invasion via MMP-7 activation. Sci. Rep. 2013, 3, 1870. [Google Scholar] [CrossRef] [PubMed]
- Zolak, J.S.; Jagirdar, R.; Surolia, R.; Karki, S.; Oliva, O.; Hock, T.; Guroji, P.; Ding, Q.; Liu, R.M.; Bolisetty, S.; et al. Pleural mesothelial cell differentiation and invasion in fibrogenic lung injury. Am. J. Pathol. 2013, 182, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Nasreen, N.; Mohammed, K.A.; Mubarak, K.K.; Baz, M.A.; Akindipe, O.A.; Fernandez-Bussy, S.; Antony, V.B. Pleural mesothelial cell transformation into myofibroblasts and haptotactic migration in response to TGF-beta1 in vitro. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L115–L124. [Google Scholar] [CrossRef] [PubMed]
- Mubarak, K.K.; Montes-Worboys, A.; Regev, D.; Nasreen, N.; Mohammed, K.A.; Faruqi, I.; Hensel, E.; Baz, M.A.; Akindipe, O.A.; Fernandez-Bussy, S.; et al. Parenchymal trafficking of pleural mesothelial cells in idiopathic pulmonary fibrosis. Eur. Respir. J. 2012, 39, 133–140. [Google Scholar] [CrossRef]
- Kathiriya, J.J.; Nakra, N.; Nixon, J.; Patel, P.S.; Vaghasiya, V.; Alhassani, A.; Tian, Z.; Allen-Gipson, D.; Dave, V. Galectin-1 inhibition attenuates profibrotic signaling in hypoxia-induced pulmonary fibrosis. Cell Death Discov. 2017, 3, 17010. [Google Scholar] [CrossRef] [PubMed]
- Berek, J.S.; Taylor, P.T.; Gordon, A.; Cunningham, M.J.; Finkler, N.; Orr, J., Jr.; Rivkin, S.; Schultes, B.C.; Whiteside, T.L.; Nicodemus, C.F. Randomized, placebo-controlled study of oregovomab for consolidation of clinical remission in patients with advanced ovarian cancer. J. Clin. Oncol. 2004, 22, 3507–3516. [Google Scholar] [CrossRef] [PubMed]
- Sabbatini, P.; Harter, P.; Scambia, G.; Sehouli, J.; Meier, W.; Wimberger, P.; Baumann, K.H.; Kurzeder, C.; Schmalfeldt, B.; Cibula, D.; et al. Abagovomab as maintenance therapy in patients with epithelial ovarian cancer: A phase III trial of the AGO OVAR, COGI, GINECO, and GEICO—The MIMOSA study. J. Clin. Oncol. 2013, 31, 1554–1561. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Batra, S.K. Understanding the Unique Attributes of MUC16 (CA125): Potential Implications in Targeted Therapy. Cancer Res. 2015, 75, 4669–4674. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biomarker | Definition | Significance |
|---|---|---|
| Alveolar Epithelial Cell Dysfunction | ||
| KL-6 1/MUC1 | Glycoprotein mainly expressed at the extracellular membrane surface of type II pneumocytes [10]. | Increased serum levels during IPF acute exacerbation [11]. Serum levels correlate with IPF severity and prognosis [11]. Biomarker indicative of the response to nintedanib treatment [12]. Promotion of lung fibroblast migration and proliferation, FMT 2 and EMT 3 [13,14]. |
| CA15-3 | Central protein core of MUC1 | Serum levels significantly higher in patients with IPF [9]. Elevated serum levels correlate with decreased total lung capacity, decreased diffusing capacity of carbon monoxide and high resolution computed tomography findings [9]. |
| CA125 | Peptide epitope of MUC16 | Rising concentrations over 3 months are associated with increased risk of IPF mortality [8]. |
| Surfactant proteins (SP-A, SP-D and SP-C) | Lipoprotein complexes synthesized and secreted by type II pneumocytes. | Elevated serum levels in IPF patients [15]. Serum SP-A and SP-D levels are predictors of IPF prognosis [16,17]. Mutations on the genes encoding for SP-C and SP-A2 have been described within families of patients with pulmonary fibrosis [18]. |
| MUC5B | Secreted mucin produced mainly in mucous cells of the submucosal glands [19]. | A common gain-of-function promoter variant (rs35705950) has been reported in 30–35% of IPF patients [20]. |
| Extracellular Matrix Remodeling and Fibroproliferation | ||
| Matrix metalloproteinases (MMP-1 and MMP-7) | Zinc-dependent peptidases that are mainly responsible for ECM 4 degradation. | Elevated levels in the plasma, BALF 5 and tissue of IPF patients [21]. Elevated MMP-7 serum levels correlate with disease severity [21] |
| LOXL2 6 | Enzymes that facilitate the cross-linking of type 1 collagen molecules and stabilizes ECM. | Serum levels are correlated to IPF progression [22]. |
| Periostin | Protein secreted by bronchial epithelial cells and that promotes ECM deposition and mesenchymal cell proliferation. | Elevated serum levels in IPF patients. Serum levels correlate with IPF physiological progression [23] |
| Immune Disfunction | ||
| CCL18 7 | Small protein mainly secreted by monocytes, macrophages and dendritic cells that acts as a chemoattractant [24] and has an important role stimulating fibroblasts to synthesise collagen in fibrotic lung diseases [25]. | Serum level is a predictor of IPF outcome and mortality [26]. |
| IL-8 8 | Cytokine highly chemo-attractant for neutrophils | Negative correlation between IL-8, pulmonary function tests [27] and survival [28]. |
| YKL-40 | Chitinase-like protein produced from alveolar macrophages and type II pneumocytes which regulate proliferation of different cell types. | Serum and BALF YKL-40 levels are predictors of IPF survival [29] |
| TLR3 9 | Receptors that mediate the innate immune response to infection and tissue injury [30]. | TLR3 L412F polymorphism is associated with a significantly greater risk of mortality and an accelerated decline in FVC 10 [31]. |
| TLR9 11 | Receptors that mediate the innate immune response to infection and tissue injury [30]. | Higher concentrations of TLR9 in surgical lung biopsies from IPF rapidly progressive patients than in tissue from IPF slowly progressing patients [32]. |
| TOLLIP 12 | Inhibitory adaptor protein within TLRs involved in the regulation of the innate immune system. | Significant correlation between response to N-acetylcysteine therapy and the rs3750920 polymorphism [33]. The rs5743890 minor allele is protective and associated with reduced susceptibility to IPF [34]. |
| Mucin | Chromosome | Main Tissue Expression |
|---|---|---|
| Secreted Mucins-Gel-Forming | ||
| MUC2 | 11p15.5 | Jejunum, ileum, colon, endometrium, respiratory tact |
| MUC5AC | 11p15.5 | Respiratory tract, stomach, conjunctiva, endocervix, endometrium |
| MUC5B | 11p15.5 | Respiratory tract, submandibular glands, endocervix |
| MUC6 | 11p15.5 | Stomach, ileum, gall bladder, endocervix, endometrium |
| MUC19 | 12q12 | Sublingual gland, submandibular gland, respiratory tract, eye, middle ear epithelium |
| Secreted Mucins-Non-Gel-Forming | ||
| MUC7 | 4q13-q21 | Respiratory tract, sublingual and submandibular glands |
| MUC8 | 12q24.3 | Respiratory tract, uterus, endocervix, endometrium |
| MUC9 | 1p13 | Fallopian tubes |
| Transmembrane Mucins | ||
| MUC1 | 1q21 | Breast, pancreas, duodenum, ileum, colon, trachea, bronchi, cornea, conjunctiva, fallopian tubes, uterus, endometrium, endocervix, ectocervix, vagina |
| MUC3A/B | 7q22 | Small intestine, colon, gall bladder |
| MUC4 | 3q29 | Breast, respiratory tract, small intestine, colon, conjunctiva, cornea, endocervix, ectocervix, vagina, endometrium, lungs |
| MUC12 | 7q22 | Colon, small intestine, stomach, pancreas, lung, kidney, prostate, uterus |
| MUC13 | 3q21.2 | Colon, trachea, kidney, small intestine |
| MUC14 | 4q24 | Heart, kidney, lungs |
| MUC15 | 11p14.3 | Colon, respiratory tract, small intestine, prostate |
| MUC16 | 19p13.2 | Ovary, cornea, conjunctiva, respiratory tract, endometrium |
| MUC17 | 7q22 | Stomach, duodenum, colon |
| MUC20 | 3q29 | Placenta, colon, respiratory tract, prostate, liver |
| MUC21 | 6p21 | Respiratory tract, thymus, colon |
| MUC22 | 6p21.22 | Lungs, placenta, testis |
| Mucin | Participation in IPF |
|---|---|
| Secreted Mucins-Gel-Forming | |
| MUC5B | Expression 14.1 higher in IPF patients than in control subjects [20]. rs35705950: risk allele for IPF development (30%–35% of IPF patients) [20]. rs35705950: survival advantage allele [56]. |
| MUC5AC | Reduced expression in globet cells from IPF lesions in comparison with controls [57,58]. rs34474233 and rs34815853: risk alleles for IPF development. |
| Transmembrane Mucins | |
| MUC1 | Overexpression in lung tissue from IPF patients [59]. Secreted KL-6 1/MUC1 is proposed as a useful biomarker to evaluate disease activity and predict the clinical outcomes in IPF [10]. Secreted MUC1/KL-6 promotes lung fibroblast migration, proliferation, EMT 2 and FMT 3 [13,14]. MUC1 is activated by the extracellular endothelial ICAM-1 4 [60], elevated in serum of IPF patients [61]. MUC1-C terminal subunit interacts with the fibrotic galectin-3, serving as a bridge to associate MUC1-C with cell surface growth receptors involved in IPF [62]. Cell surface growth factor receptors involved in IPF (such as EGFR 5, FGFR3 6, PDGFR 7 and TGβR 8) phosphorylate and activate MUC1-CT 9 [63,64]. MUC1-CT is phosphorylated and activated by intracellular kinases such as c-Src, Lyn and Lck, which are activated by growth factors elevated in IPF [65] MUC1-CT interacts with the fibrotic transcription factor β-catenin and they both together translocate into the nucleus [64] MUC1-CT nuclear translocation promotes fibrotic processes [63]. |
| MUC4 | MUC4 is overexpressed in lung tissue from IPF patients [66]. MUC4 collaborates with the fibrotic TGFβ1 to induce EMT and FMT cellular transformations [66]. MUC4 induces nuclear translocation of the fibrotic transcription factor β-catenin [67]. MUC4 serves as an intramembrane ligand for ErbB2, inducing activation of ERK ½ 10 and regulating cell proliferation, growth, survival and differentiation [68]. |
| MUC16 | MUC16/CA125 antigen has been identified as a serum biomarker to predict disease progression and death in IPF patients [8]. MUC16-C terminal subunit associates with JAK2 11 and promotes cell proliferation [69]. MUC16-C terminal subunit decreases TRAIL 12-induced apoptosis [70]. MUC16 interacts with c-Src and the E-cadherin/β-catenin complex, leading to deregulation of E-Cadherin and enhancing cell migration [71]. MUC16 binds to mesothelin, enhancing epithelial mesothelial to mesenchymal transition, cell motility and invasion [42]. MUC16 binds to galectin-1 [72] and galectin-3 [73]. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ballester, B.; Milara, J.; Cortijo, J. Mucins as a New Frontier in Pulmonary Fibrosis. J. Clin. Med. 2019, 8, 1447. https://doi.org/10.3390/jcm8091447
Ballester B, Milara J, Cortijo J. Mucins as a New Frontier in Pulmonary Fibrosis. Journal of Clinical Medicine. 2019; 8(9):1447. https://doi.org/10.3390/jcm8091447
Chicago/Turabian StyleBallester, Beatriz, Javier Milara, and Julio Cortijo. 2019. "Mucins as a New Frontier in Pulmonary Fibrosis" Journal of Clinical Medicine 8, no. 9: 1447. https://doi.org/10.3390/jcm8091447
APA StyleBallester, B., Milara, J., & Cortijo, J. (2019). Mucins as a New Frontier in Pulmonary Fibrosis. Journal of Clinical Medicine, 8(9), 1447. https://doi.org/10.3390/jcm8091447