Systematic Evaluation of Light-Activatable Biohybrids for Anti-Glioma Photodynamic Therapy

 ,
,
Abstract
1. Introduction
2. Methods
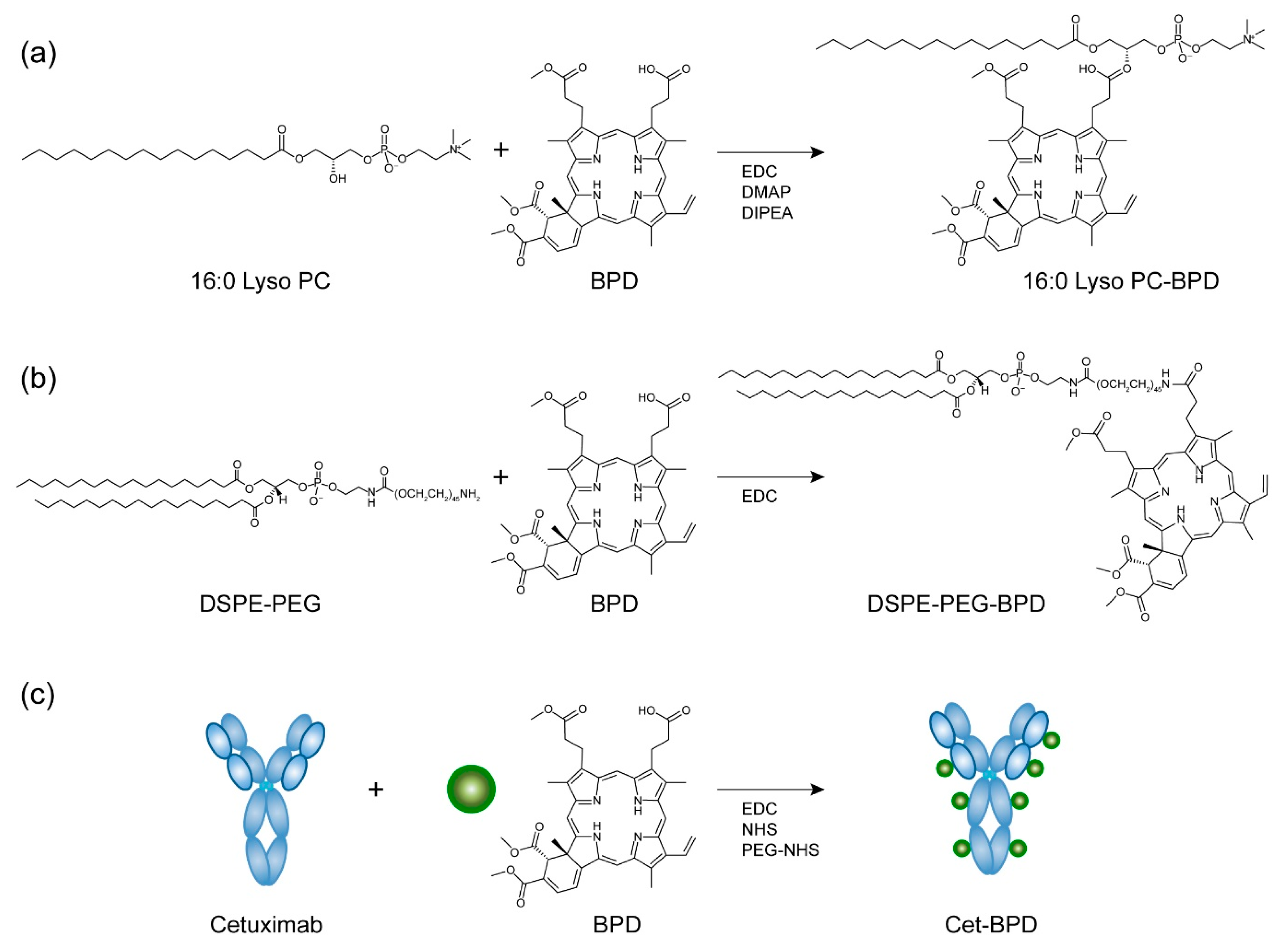
2.1. PSBM Preparation and Purification
2.2. Photophysical and Photochemical Characterizations
2.3. Cell Culture
2.4. Photodynamic Therapy (PDT) of Glioblastoma Cells Using PSBMs
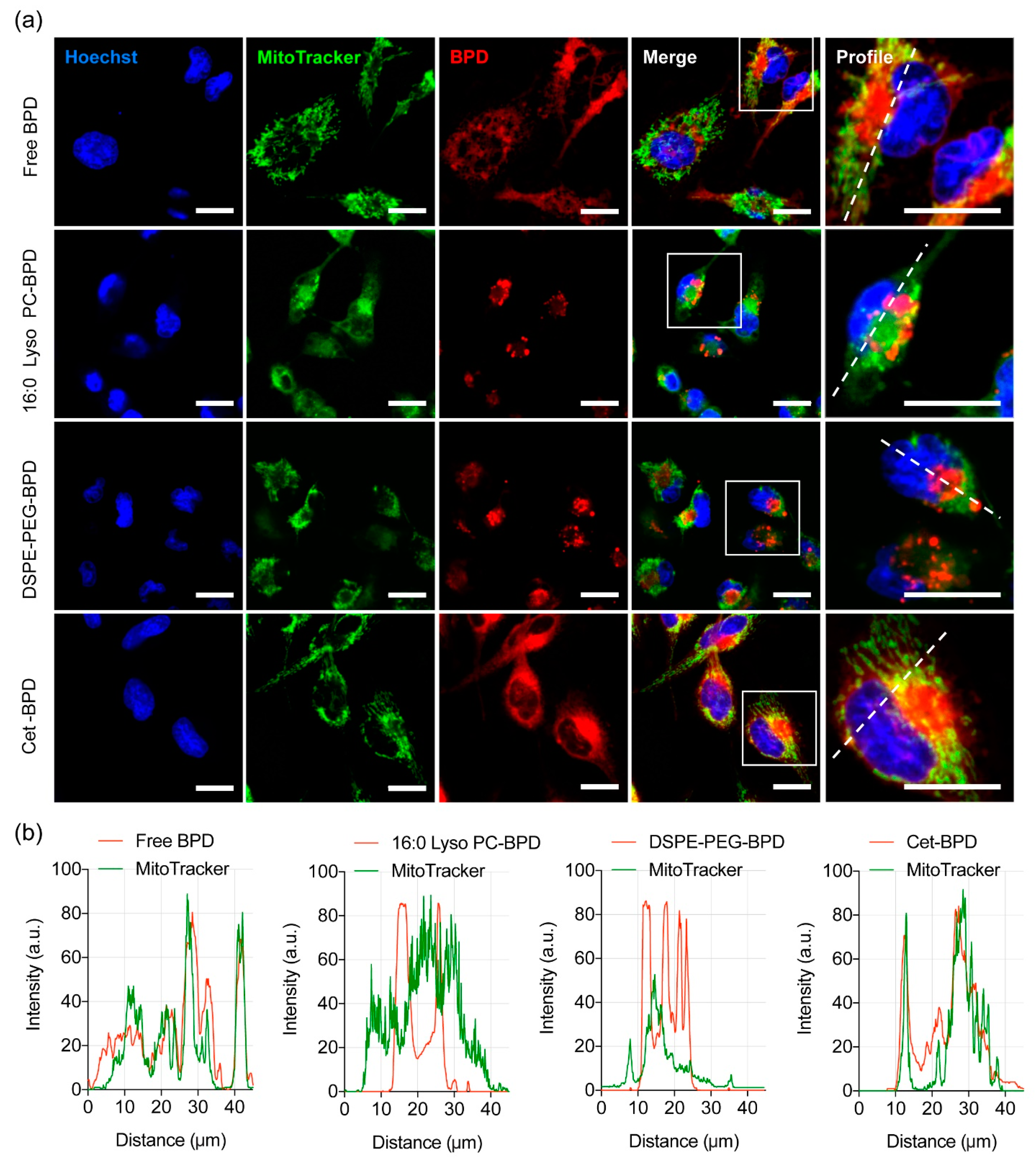
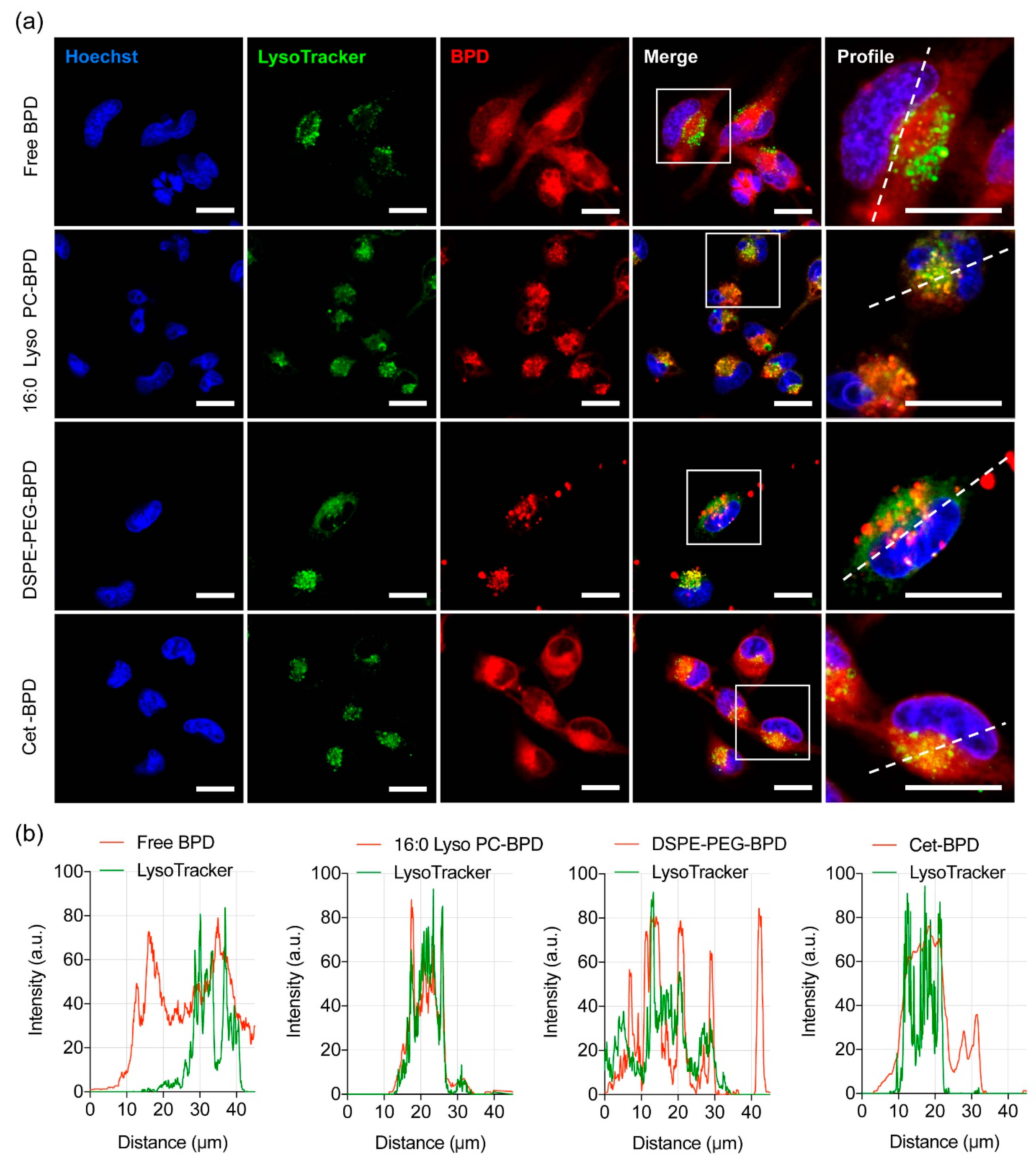
2.5. PSBM Uptake and Localization Studies in Glioblastoma Cells
2.6. Immunoblotting
2.7. Statistical Analyses
2.8. Ethical Approval and Informed Consent
3. Results and Discussion
3.1. Synthesis and Characterization of PSBMs
3.2. Photoactivity of PSBMs
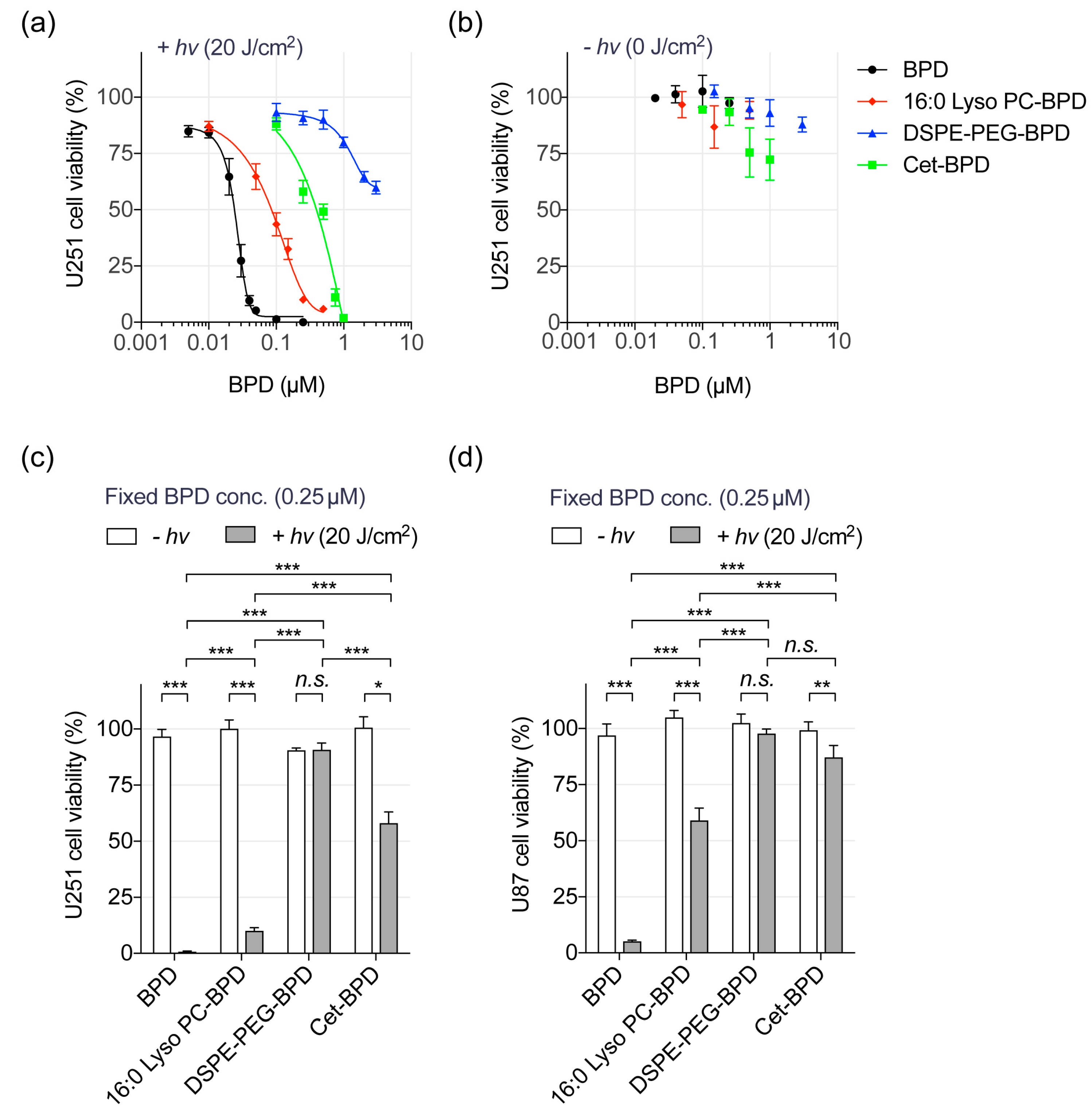
3.3. Near-Infrared (NIR) Light-Mediated Photodestruction of Glioblastoma Cells Using PSBMs
3.4. Extraction Method and Fluorescence Imaging to Quantify PSBM Uptake
3.5. Altered Subcellular Localization PSBM Leads to a New Combination PDT Approach for Glioblastoma.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide | (EDC) |
| 1-palmitoyl-2-hydroxy-sn-glycero-3-phosphocholine | (16:0 Lyso PC) |
| 4-(dimethylamino) pyridine | (DMAP) |
| aminolevulinic acid hydrochloride | (ALA HCL) |
| benzoporphyrin derivative | (BPD) |
| carbonilcyanide p-triflouromethoxyphenylhydrazone | (FCCP) |
| cetuximab | (Cet) |
| dimethyl sulfoxide | (DMSO) |
| distearoyl-phosphoethanolamine-polyethylene-glycol | (DSPE-PEG) |
| fluorescence-activated cell sorting | (FACS) |
| fluorescence intensity | (FI) |
| matrix-assisted laser desorption ionization-time of flight mass spectrometry | (MALDI-TOF MS) |
| mean fluorescence intensity | (MFI) |
| meso-tetrahydroxyphenyl chlorin | (mTHPC) |
| mitochondrial membrane potential | (ΔΨm) |
| N,N-diisopropylethylamine | (DIPEA) |
| N-hydroxysuccinimide | (NHS) |
| near-infrared | (NIR) |
| patient-derived xenograft | (PDX) |
| epidermal growth factor receptor | (EGFR) |
| phosphate buffered saline | (PBS) |
| photosensitizing biomolecules | (PSBM) |
| polyethylene glycol | (PEG) |
| protoporphyrin IX | (PpIX) |
| reactive molecular species | (RMS) |
| singlet oxygen | (1O2) |
| singlet oxygen sensor green | (SOSG) |
| standard error of the mean | (SEM) |
| sodium dodecyl sulfate-polyacrylamide gel electrophoresis | (SDS-PAGE) |
| The United States Food and Drug Administration | (FDA) |
References
- Almeida-Marrero, V.; van de Winckel, E.; Anaya-Plaza, E.; Torres, T.; de la Escosura, A. Porphyrinoid biohybrid materials as an emerging toolbox for biomedical light management. Chem. Soc. Rev. 2018, 47, 7369–7400. [Google Scholar] [CrossRef] [PubMed]
- Celli, J.P.; Spring, B.Q.; Rizvi, I.; Evans, C.L.; Samkoe, K.S.; Verma, S.; Pogue, B.W.; Hasan, T. Imaging and photodynamic therapy: mechanisms, monitoring, and optimization. Chem. Rev. 2010, 110, 2795–2838. [Google Scholar] [CrossRef] [PubMed]
- Dolmans, D.E.; Fukumura, D.; Jain, R.K. Photodynamic therapy for cancer. Nat. Rev. Cancer 2003, 3, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Castano, A.P.; Mroz, P.; Hamblin, M.R. Photodynamic therapy and anti-tumour immunity. Nat. Rev. Cancer 2006, 6, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.C.; Mallidi, S.; Liu, J.; Chiang, C.T.; Mai, Z.; Goldschmidt, R.; Ebrahim-Zadeh, N.; Rizvi, I.; Hasan, T. Photodynamic Therapy Synergizes with Irinotecan to Overcome Compensatory Mechanisms and Improve Treatment Outcomes in Pancreatic Cancer. Cancer Res. 2016, 76, 1066–1077. [Google Scholar] [PubMed]
- Spring, B.Q.; Bryan Sears, R.; Zheng, L.Z.; Mai, Z.; Watanabe, R.; Sherwood, M.E.; Schoenfeld, D.A.; Pogue, B.W.; Pereira, S.P.; Villa, E.; et al. A photoactivable multi-inhibitor nanoliposome for tumour control and simultaneous inhibition of treatment escape pathways. Nat. Nanotechnol. 2016, 11, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.C.; Rizvi, I.; Liu, J.; Anbil, S.; Kalra, A.; Lee, H.; Baglo, Y.; Paz, N.; Hayden, D.; Pereira, S.; et al. Photodynamic Priming Mitigates Chemotherapeutic Selection Pressures and Improves Drug Delivery. Cancer Res. 2018, 78, 558–571. [Google Scholar] [CrossRef] [PubMed]
- Spring, B.Q.; Rizvi, I.; Xu, N.; Hasan, T. The role of photodynamic therapy in overcoming cancer drug resistance. Photochem. Photobiol. Sci. 2015, 14, 1476–1491. [Google Scholar] [CrossRef]
- Pigula, M.; Huang, H.C.; Mallidi, S.; Anbil, S.; Liu, J.; Mai, Z.; Hasan, T. Size-dependent Tumor Response to Photodynamic Therapy and Irinotecan Monotherapies Revealed by Longitudinal Ultrasound Monitoring in an Orthotopic Pancreatic Cancer Model. Photochem. Photobiol. 2018, 95, 378–386. [Google Scholar] [CrossRef]
- Gallagher-Colombo, S.M.; Miller, J.; Cengel, K.A.; Putt, M.E.; Vinogradov, S.A.; Busch, T.M. Erlotinib Pretreatment Improves Photodynamic Therapy of Non-Small Cell Lung Carcinoma Xenografts via Multiple Mechanisms. Cancer Res. 2015, 75, 3118–3126. [Google Scholar] [CrossRef]
- Obaid, G.; Broekgaarden, M.; Bulin, A.-L.; Huang, H.-C.; Kuriakose, J.; Liu, J.; Hasan, T. Photonanomedicine: a convergence of photodynamic therapy and nanotechnology. Nanoscale 2016, 8, 12471–12503. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.C.; Hasan, T. The “Nano” World in Photodynamic Therapy. Austin J. Nanomed. Nanotechnol. 2014, 2, 1020. [Google Scholar]
- Treatment of Age-Related Macular Degeneration with Photodynamic Therapy (TAP) Study Group. Photodynamic therapy of subfoveal choroidal neovascularization in age-related macular degeneration with verteporfin: one-year results of 2 randomized clinical trials—TAP report. Treatment of age-related macular degeneration with photodynamic therapy (TAP) Study Group. Arch. Ophthalmol. 1999, 117, 1329–1345. [Google Scholar]
- Schmidt-Erfurth, U.; Hasan, T.; Gragoudas, E.; Michaud, N.; Flotte, T.J.; Birngruber, R. Vascular targeting in photodynamic occlusion of subretinal vessels. Ophthalmology 1994, 101, 1953–1961. [Google Scholar] [CrossRef]
- Huggett, M.T.; Jermyn, M.; Gillams, A.; Illing, R.; Mosse, S.; Novelli, M.; Kent, E.; Bown, S.G.; Hasan, T.; Pogue, B.W.; et al. Phase I/II study of verteporfin photodynamic therapy in locally advanced pancreatic cancer. Br. J. Cancer 2014, 110, 1698–1704. [Google Scholar] [CrossRef] [PubMed]
- Bown, S.G.; Rogowska, A.Z.; Whitelaw, D.E.; Lees, W.R.; Lovat, L.B.; Ripley, P.; Jones, L.; Wyld, P.; Gillams, A.; Hatfield, A.W.R. Photodynamic therapy for cancer of the pancreas. Gut 2002, 50, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.M.; Azzouzi, A.R.; Barret, E.; Villers, A.; Muir, G.H.; Barber, N.J.; Bott, S.; Trachtenberg, J.; Arumainayagam, N.; Gaillac, B.; et al. Determination of optimal drug dose and light dose index to achieve minimally invasive focal ablation of localised prostate cancer using WST11-vascular-targeted photodynamic (VTP) therapy. BJU Int. 2015, 116, 888–896. [Google Scholar] [CrossRef]
- Schmidt, S.; Wagner, U.; Oehr, P.; Krebs, D. Clinical use of photodynamic therapy in gynecologic tumor patients—Antibody-targeted photodynamic laser therapy as a new oncologic treatment procedure. Zentralbl. Gynakol. 1992, 114, 307–311. [Google Scholar]
- Mitsunaga, M.; Ogawa, M.; Kosaka, N.; Rosenblum, L.T.; Choyke, P.L.; Kobayashi, H. Cancer cell-selective in vivo near infrared photoimmunotherapy targeting specific membrane molecules. Nat. Med. 2011, 17, 1685–1691. [Google Scholar] [CrossRef]
- Komatsu, T.; Moritake, M.; Nakagawa, A.; Tsuchida, E. Self-organized lipid-porphyrin bilayer membranes in vesicular form: nanostructure, photophysical properties, and dioxygen coordination. Chemistry 2002, 8, 5469–5480. [Google Scholar] [CrossRef]
- Lovell, J.F.; Jin, C.S.; Huynh, E.; Jin, H.; Kim, C.; Rubinstein, J.L.; Chan, W.C.; Cao, W.; Wang, L.V.; Zheng, G. Porphysome nanovesicles generated by porphyrin bilayers for use as multimodal biophotonic contrast agents. Nat. Mater. 2011, 10, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Geng, J.; Ah Yi, H.; Gogia, S.; Neelamegham, S.; Jacobs, A.; Lovell, J.F. Functionalization of cobalt porphyrin-phospholipid bilayers with his-tagged ligands and antigens. Nat. Chem. 2015, 7, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Baglo, Y.; Liang, B.J.; Robey, R.W.; Ambudkar, S.V.; Gottesman, M.M.; Huang, H.C. Porphyrin-lipid assemblies and nanovesicles overcome ABC transporter-mediated photodynamic therapy resistance in cancer cells. Cancer Lett. 2019, 457, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Hamblin, M.R.; Miller, J.L.; Rizvi, I.; Ortel, B.; Maytin, E.V.; Hasan, T. Pegylation of a chlorin(e6) polymer conjugate increases tumor targeting of photosensitizer. Cancer Res. 2001, 61, 7155–7162. [Google Scholar] [PubMed]
- Hamblin, M.R.; Miller, J.L.; Rizvi, I.; Loew, H.G.; Hasan, T. Pegylation of charged polymer-photosensitiser conjugates: effects on photodynamic efficacy. Br. J. Cancer 2003, 89, 937–943. [Google Scholar] [CrossRef] [PubMed]
- Savellano, M.D.; Hasan, T. Targeting cells that overexpress the epidermal growth factor receptor with polyethylene glycolated BPD verteporfin photosensitizer immunoconjugates. Photochem. Photobiol. 2003, 77, 431–439. [Google Scholar] [CrossRef]
- Spring, B.Q.; Abu-Yousif, A.O.; Palanisami, A.; Rizvi, I.; Zheng, X.; Mai, Z.; Anbil, S.; Sears, R.B.; Mensah, L.B.; Goldschmidt, R.; et al. Selective treatment and monitoring of disseminated cancer micrometastases in vivo using dual-function, activatable immunoconjugates. Proc. Natl. Acad. Sci. USA 2014, 111, E933–E942. [Google Scholar] [CrossRef] [PubMed]
- Savellano, M.D.; Hasan, T. Photochemical targeting of epidermal growth factor receptor: A mechanistic study. Clin. Cancer Res. 2005, 11, 1658–1668. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.C.; Pigula, M.; Fang, Y.; Hasan, T. Immobilization of Photo-Immunoconjugates on Nanoparticles Leads to Enhanced Light-Activated Biological Effects. Small 2018. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, I.; Nath, S.; Obaid, G.; Ruhi, M.K.; Moore, K.; Bano, S.; Kessel, D.; Hasan, T. A Combination of Visudyne and a Lipid-anchored Liposomal Formulation of Benzoporphyrin Derivative Enhances Photodynamic Therapy Efficacy in a 3D Model for Ovarian Cancer. Photochem. Photobiol. 2019, 95, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Obaid, G.; Jin, W.; Bano, S.; Kessel, D.; Hasan, T. Nanolipid Formulations of Benzoporphyrin Derivative: Exploring the Dependence of Nanoconstruct Photophysics and Photochemistry on Their Therapeutic Index in Ovarian Cancer Cells. Photochem. Photobiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Sarkaria, J.N.; Carlson, B.L.; Schroeder, M.A.; Grogan, P.; Brown, P.D.; Giannini, C.; Ballman, K.V.; Kitange, G.J.; Guha, A.; Pandita, A.; et al. Use of an Orthotopic Xenograft Model for Assessing the Effect of Epidermal Growth Factor Receptor Amplification on Glioblastoma Radiation Response. Clin. Cancer Res. 2006, 12, 2264–2271. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.S.; Cui, L.; Wang, F.; Chen, J.; Zheng, G. Targeting-triggered porphysome nanostructure disruption for activatable photodynamic therapy. Adv. Healthc. Mater. 2014, 3, 1240–1249. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Geng, J.; Li, N.; Carter, K.A.; Shao, S.; Atilla-Gokcumen, G.E.; Lovell, J.F. Vessel-Targeted Chemophototherapy with Cationic Porphyrin-Phospholipid Liposomes. Mol. Cancer Ther. 2017, 16, 2452–2461. [Google Scholar] [CrossRef] [PubMed]
- Abrahamse, H.; Hamblin, M.R. New photosensitizers for photodynamic therapy. Biochem. J. 2016, 473, 347–364. [Google Scholar] [CrossRef] [PubMed]
- Immordino, M.L.; Dosio, F.; Cattel, L. Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential. Int. J. Nanomed. 2006, 1, 297–315. [Google Scholar]
- Kaufman, M.B. Pharmaceutical Approval Update. Pharm. Ther. 2017, 42, 673–683. [Google Scholar]
- Stepp, H.; Beck, T.; Pongratz, T.; Meinel, T.; Kreth, F.W.; Tonn, J.; Stummer, W. ALA and malignant glioma: Fluorescence-guided resection and photodynamic treatment. J. Environ. Pathol. Toxicol. Oncol. 2007, 26, 157–164. [Google Scholar] [CrossRef]
- Han, J.; Burgess, K. Fluorescent indicators for intracellular pH. Chem. Rev. 2010, 110, 2709–2728. [Google Scholar] [CrossRef]
- Zhu, H.; Fan, J.; Du, J.; Peng, X. Fluorescent Probes for Sensing and Imaging within Specific Cellular Organelles. Accounts Chem. Res. 2016, 49, 2115–2126. [Google Scholar] [CrossRef] [PubMed]
- Kessel, D.; Luo, Y. Mitochondrial photodamage and PDT-induced apoptosis. J. Photochem. Photobiol. B 1998, 42, 89–95. [Google Scholar] [CrossRef]
- Kessel, D.; Castelli, M. Evidence that bcl-2 is the target of three photosensitizers that induce a rapid apoptotic response. Photochem. Photobiol. 2001, 74, 318–322. [Google Scholar] [CrossRef]
- Bacellar, I.O.; Tsubone, T.M.; Pavani, C.; Baptista, M.S. Photodynamic Efficiency: From Molecular Photochemistry to Cell Death. Int. J. Mol. Sci. 2015, 16, 20523–20559. [Google Scholar] [CrossRef] [PubMed]
- Kessel, D.; Reiners, J.J., Jr. Promotion of Proapoptotic Signals by Lysosomal Photodamage. Photochem. Photobiol. 2015, 91, 931–936. [Google Scholar] [CrossRef] [PubMed]
- Tsubone, T.M.; Martins, W.K.; Pavani, C.; Junqueira, H.C.; Itri, R.; Baptista, M.S. Enhanced efficiency of cell death by lysosome-specific photodamage. Sci. Rep. 2017, 7, 6734. [Google Scholar] [CrossRef]
- Dos Santos, A.F.; Terra, L.F.; Wailemann, R.A.; Oliveira, T.C.; Gomes, V.M.; Mineiro, M.F.; Meotti, F.C.; Bruni-Cardoso, A.; Baptista, M.S.; Labriola, L. Methylene blue photodynamic therapy induces selective and massive cell death in human breast cancer cells. BMC Cancer 2017, 17, 194. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef]
- To, M.S.; Aromataris, E.C.; Castro, J.; Roberts, M.L.; Barritt, G.J.; Rychkov, G.Y. Mitochondrial uncoupler FCCP activates proton conductance but does not block store-operated Ca2+ current in liver cells. Arch. Biochem. Biophys. 2010, 495, 152–158. [Google Scholar] [CrossRef]
- Martins, W.K.; Santos, N.F.; Rocha, C.S.; Bacellar, I.O.L.; Tsubone, T.M.; Viotto, A.C.; Matsukuma, A.Y.; Abrantes, A.B.P.; Siani, P.; Dias, L.G.; et al. Parallel damage in mitochondria and lysosomes is an efficient way to photoinduce cell death. Autophagy 2018. [Google Scholar] [CrossRef]
- Cincotta, L.; Szeto, D.; Lampros, E.; Hasan, T.; Cincotta, A.H. Benzophenothiazine and benzoporphyrin derivative combination phototherapy effectively eradicates large murine sarcomas. Photochem. Photobiol. 1996, 63, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Kessel, D. Photodynamic therapy: Promotion of efficacy by a sequential protocol. J. Porphyr. Phthalocyanines 2016, 20, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Martins, W.K.; Costa, É.T.; Cruz, M.C.; Stolf, B.S.; Miotto, R.; Cordeiro, R.M.; Baptista, M.S. Parallel damage in mitochondrial and lysosomal compartments promotes efficient cell death with autophagy: The case of the pentacyclic triterpenoids. Sci. Rep. 2015, 5, 12425. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, I.; Obaid, G.; Bano, S.; Hasan, T.; Kessel, D. Photodynamic therapy: Promoting in vitro efficacy of photodynamic therapy by liposomal formulations of a photosensitizing agent. Lasers Surg. Med. 2018, 50, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Moor, A.C. Signaling pathways in cell death and survival after photodynamic therapy. J. Photochem. Photobiol. B 2000, 57, 1–13. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Lines | U251 | U87 | |
|---|---|---|---|
| Groups * | PDT Efficiency ** | PDT Efficiency ** | |
| Free BPD | 1.12% ± 0.00% | 1.07% ± 0.00% | |
| 16:0 Lyso PC-BPD | 2.30% ± 0.03% | 2.14% ± 0.29% | |
| DSPE-PEG-BPD | 0.29% ± 0.09% | 0.09% ± 0.08% | |
| Cet-BPD | 0.94% ± 0.28% | 0.57% ± 0.24% | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inglut, C.T.; Baglo, Y.; Liang, B.J.; Cheema, Y.; Stabile, J.; Woodworth, G.F.; Huang, H.-C. Systematic Evaluation of Light-Activatable Biohybrids for Anti-Glioma Photodynamic Therapy. J. Clin. Med. 2019, 8, 1269. https://doi.org/10.3390/jcm8091269
Inglut CT, Baglo Y, Liang BJ, Cheema Y, Stabile J, Woodworth GF, Huang H-C. Systematic Evaluation of Light-Activatable Biohybrids for Anti-Glioma Photodynamic Therapy. Journal of Clinical Medicine. 2019; 8(9):1269. https://doi.org/10.3390/jcm8091269
Chicago/Turabian StyleInglut, Collin T., Yan Baglo, Barry J. Liang, Yahya Cheema, Jillian Stabile, Graeme F. Woodworth, and Huang-Chiao Huang. 2019. "Systematic Evaluation of Light-Activatable Biohybrids for Anti-Glioma Photodynamic Therapy" Journal of Clinical Medicine 8, no. 9: 1269. https://doi.org/10.3390/jcm8091269
APA StyleInglut, C. T., Baglo, Y., Liang, B. J., Cheema, Y., Stabile, J., Woodworth, G. F., & Huang, H.-C. (2019). Systematic Evaluation of Light-Activatable Biohybrids for Anti-Glioma Photodynamic Therapy. Journal of Clinical Medicine, 8(9), 1269. https://doi.org/10.3390/jcm8091269