Inflammation as a Therapeutic Target in Atherosclerosis

 , , and
, , and
Abstract
1. Introduction
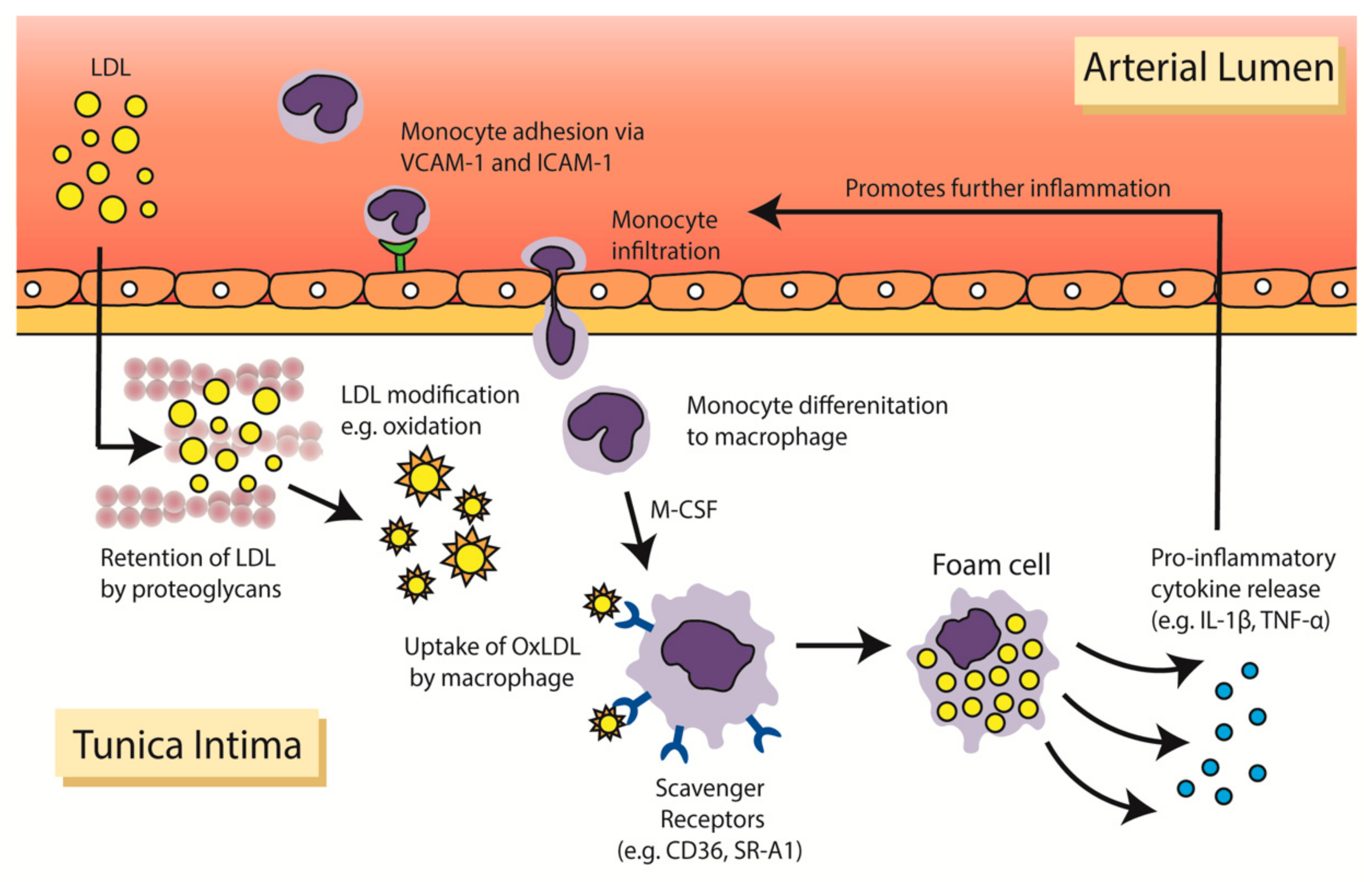
2. Inflammation in the Pathophysiology of Atherosclerosis
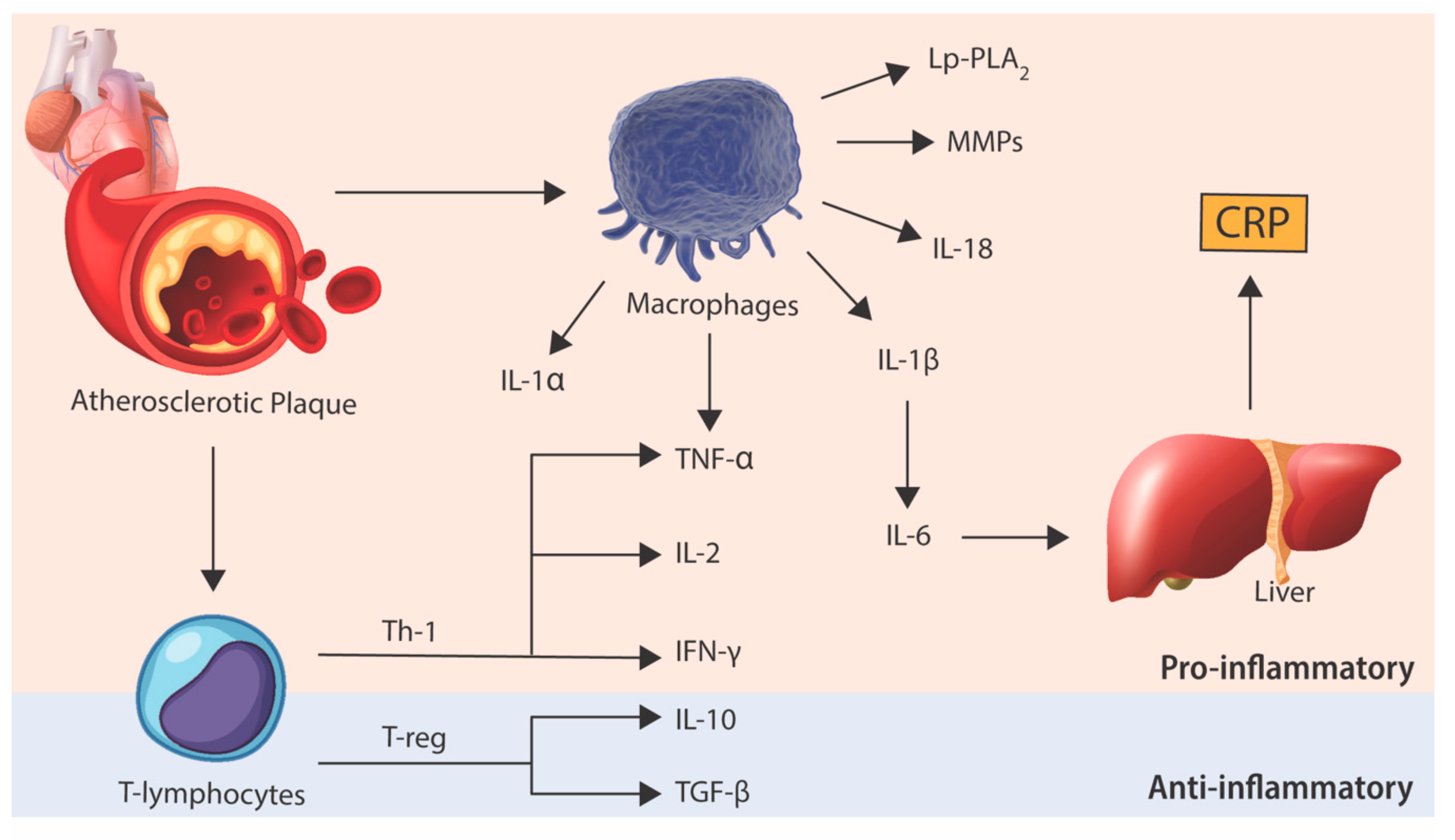
3. Burden of Cardiovascular Disease in Chronic Inflammatory Diseases
4. Beyond Statins: Where to Now for Anti-Atherosclerotic Therapies?
5. Anti-Cytokine Therapy in Atherosclerosis: The Basis for Human Trials
6. A New Era: Human Trials Targeting Cytokine Inhibition
6.1. Canakinumab: The CANTOS Trial
6.2. Methotrexate: The Cardiovascular Inflammation Reduction Trial (CIRT) Trial
6.3. Alternative Targets in the IL-1β/IL-6 Signaling Pathway
6.4. Colchicine: The LoDoCo Trial
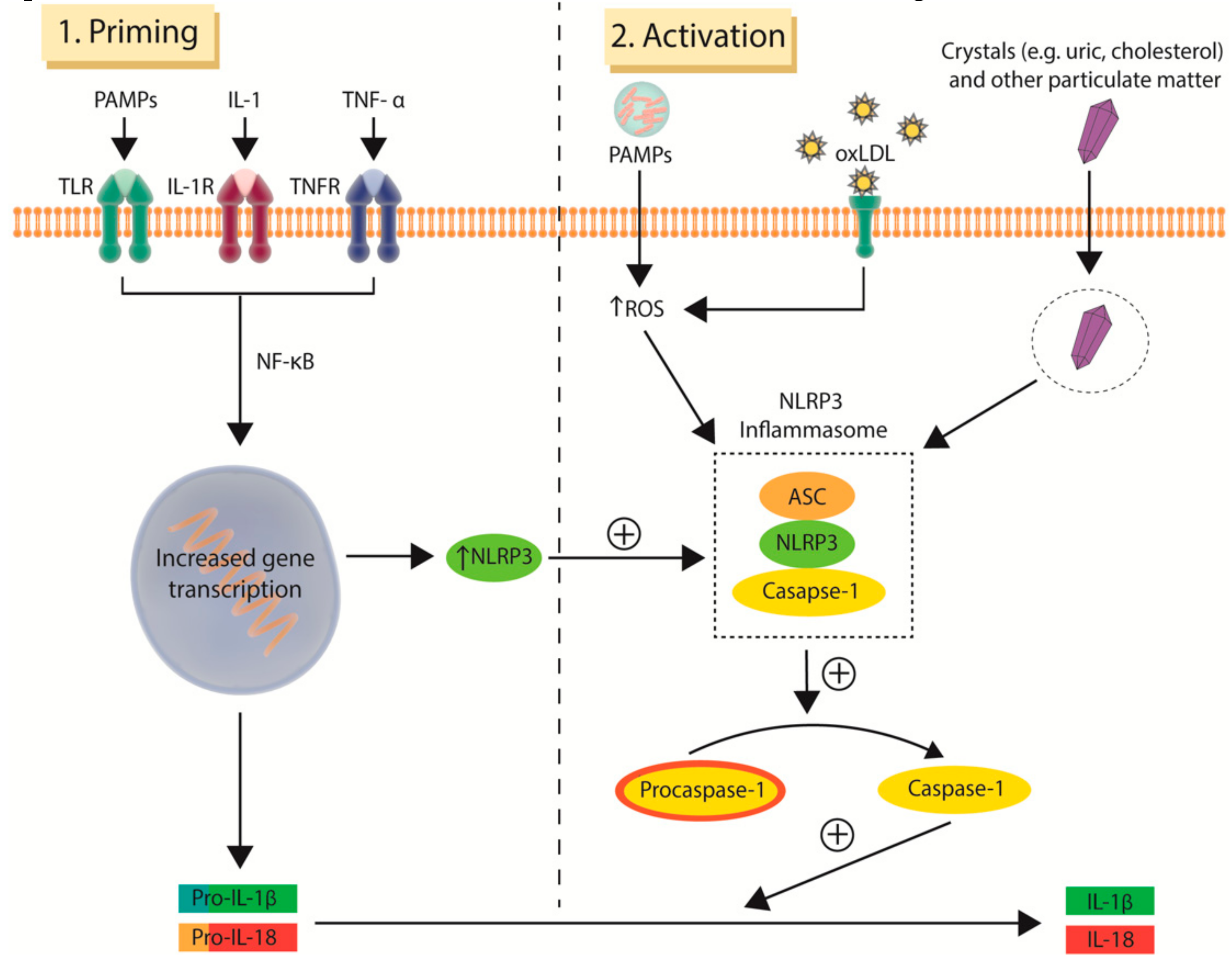
7. Clonal Haematopoiesis: A Novel Risk Factor for Atherosclerosis
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart disease and stroke statistics-2019 update: A report from the american heart association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef]
- Collins, R.; Reith, C.; Emberson, J.; Armitage, J.; Baigent, C.; Blackwell, L.; Blumenthal, R.; Danesh, J.; Smith, G.D.; DeMets, D.; et al. Interpretation of the evidence for the efficacy and safety of statin therapy. Lancet (Lond. Engl.) 2016, 388, 2532–2561. [Google Scholar] [CrossRef]
- Jernberg, T.; Hasvold, P.; Henriksson, M.; Hjelm, H.; Thuresson, M.; Janzon, M. Cardiovascular risk in post-myocardial infarction patients: Nationwide real world data demonstrate the importance of a long-term perspective. Eur. Heart J. 2015, 36, 1163–1170. [Google Scholar] [CrossRef]
- Libby, P.; Loscalzo, J.; Ridker, P.M.; Farkouh, M.E.; Hsue, P.Y.; Fuster, V.; Hasan, A.A.; Amar, S. Inflammation, immunity, and infection in atherothrombosis: Jacc review topic of the week. J. Am. Coll. Cardiol. 2018, 72, 2071–2081. [Google Scholar] [CrossRef]
- Hwang, S.J.; Ballantyne, C.M.; Sharrett, A.R.; Smith, L.C.; Davis, C.E.; Gotto, A.M., Jr.; Boerwinkle, E. Circulating adhesion molecules vcam-1, icam-1, and e-selectin in carotid atherosclerosis and incident coronary heart disease cases: The atherosclerosis risk in communities (aric) study. Circulation 1997, 96, 4219–4225. [Google Scholar] [CrossRef]
- Ridker, P.M.; Buring, J.E.; Rifai, N. Soluble p-selectin and the risk of future cardiovascular events. Circulation 2001, 103, 491–495. [Google Scholar] [CrossRef]
- Cybulsky, M.I.; Iiyama, K.; Li, H.; Zhu, S.; Chen, M.; Iiyama, M.; Davis, V.; Gutierrez-Ramos, J.C.; Connelly, P.W.; Milstone, D.S. A major role for vcam-1, but not icam-1, in early atherosclerosis. J. Clin. Investig. 2001, 107, 1255–1262. [Google Scholar] [CrossRef]
- Nageh, M.F.; Sandberg, E.T.; Marotti, K.R.; Lin, A.H.; Melchior, E.P.; Bullard, D.C.; Beaudet, A.L. Deficiency of inflammatory cell adhesion molecules protects against atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1517–1520. [Google Scholar] [CrossRef]
- Dong, Z.M.; Brown, A.A.; Wagner, D.D. Prominent role of p-selectin in the development of advanced atherosclerosis in apoe-deficient mice. Circulation 2000, 101, 2290–2295. [Google Scholar] [CrossRef]
- Collins, R.G.; Velji, R.; Guevara, N.V.; Hicks, M.J.; Chan, L.; Beaudet, A.L. P-selectin or intercellular adhesion molecule (icam)-1 deficiency substantially protects against atherosclerosis in apolipoprotein e-deficient mice. J. Exp. Med. 2000, 191, 189–194. [Google Scholar] [CrossRef]
- McGill, H.C., Jr.; McMahan, C.A.; Herderick, E.E.; Malcom, G.T.; Tracy, R.E.; Strong, J.P. Origin of atherosclerosis in childhood and adolescence. Am. J. Clin. Nutr. 2000, 72, 1307s–1315s. [Google Scholar]
- Strong, J.P.; Malcom, G.T.; Newman, W.P., 3rd; Oalmann, M.C. Early lesions of atherosclerosis in childhood and youth: Natural history and risk factors. J. Am. Coll. Nutr. 1992, 11 (Suppl. 1), 51s–54s. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Bobryshev, Y.V.; Orekhov, A.N. Macrophage-mediated cholesterol handling in atherosclerosis. J. Cell. Mol. Med. 2016, 20, 17–28. [Google Scholar] [CrossRef]
- Ohashi, R.; Mu, H.; Wang, X.; Yao, Q.; Chen, C. Reverse cholesterol transport and cholesterol efflux in atherosclerosis. QJM Mon. J. Assoc. Physicians 2005, 98, 845–856. [Google Scholar] [CrossRef]
- Tabas, I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat. Rev. Immunol. 2010, 10, 36–46. [Google Scholar] [CrossRef]
- Nadkarni, S.K.; Bouma, B.E.; de Boer, J.; Tearney, G.J. Evaluation of collagen in atherosclerotic plaques: The use of two coherent laser-based imaging methods. Lasers Med Sci. 2009, 24, 439–445. [Google Scholar] [CrossRef]
- Alexander, M.R.; Owens, G.K. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu. Rev. Physiol. 2012, 74, 13–40. [Google Scholar] [CrossRef]
- Hansson, G.K.; Jonasson, L. The discovery of cellular immunity in the atherosclerotic plaque. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1714–1717. [Google Scholar] [CrossRef]
- Wu, M.Y.; Li, C.J.; Hou, M.F.; Chu, P.Y. New insights into the role of inflammation in the pathogenesis of atherosclerosis. Int. J. Mol. Sci. 2017, 18, 2034. [Google Scholar] [CrossRef]
- Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef]
- Witztum, J.L.; Binder, C.J.; Chou, M.-Y.; Fogelstrand, L.; Hartvigsen, K.; Shaw, P.X.; Boullier, A. Natural antibodies in murine atherosclerosis. Curr. Drug Targets 2008, 9, 190–195. [Google Scholar] [CrossRef]
- Caligiuri, G.; Nicoletti, A.; Poirier, B.; Hansson, G.K. Protective immunity against atherosclerosis carried by b cells of hypercholesterolemic mice. J. Clin. Investig. 2002, 109, 745–753. [Google Scholar] [CrossRef]
- Shaw, P.X.; Horkko, S.; Chang, M.K.; Curtiss, L.K.; Palinski, W.; Silverman, G.J.; Witztum, J.L. Natural antibodies with the t15 idiotype may act in atherosclerosis, apoptotic clearance, and protective immunity. J. Clin. Investig. 2000, 105, 1731–1740. [Google Scholar] [CrossRef]
- Kyaw, T.; Tipping, P.; Bobik, A.; Toh, B.H. Opposing roles of b lymphocyte subsets in atherosclerosis. Autoimmunity 2017, 50, 52–56. [Google Scholar] [CrossRef]
- Tedgui, A.; Mallat, Z. Cytokines in atherosclerosis: Pathogenic and regulatory pathways. Physiol. Rev. 2006, 86, 515–581. [Google Scholar] [CrossRef]
- Turesson, C.; Jacobsson, L.T.H.; Matteson, E.L. Cardiovascular co-morbidity in rheumatic diseases. Vasc. Health Risk Manag. 2008, 4, 605–614. [Google Scholar] [CrossRef]
- Woolf, A.D.; Pfleger, B. Burden of major musculoskeletal conditions. Bull. World Health Organ. 2003, 81, 646–656. [Google Scholar]
- England, B.R.; Thiele, G.M.; Anderson, D.R.; Mikuls, T.R. Increased cardiovascular risk in rheumatoid arthritis: Mechanisms and implications. BMJ 2018, 361, k1036. [Google Scholar] [CrossRef]
- Widdifield, J.; Paterson, J.M.; Huang, A.; Bernatsky, S. Causes of death in rheumatoid arthritis: How do they compare to the general population? Arthritis Care Res. 2018, 70, 1748–1755. [Google Scholar] [CrossRef]
- Avina-Zubieta, J.A.; Thomas, J.; Sadatsafavi, M.; Lehman, A.J.; Lacaille, D. Risk of incident cardiovascular events in patients with rheumatoid arthritis: A meta-analysis of observational studies. Ann. Rheum. Dis. 2012, 71, 1524–1529. [Google Scholar] [CrossRef]
- Avina-Zubieta, J.A.; Choi, H.K.; Sadatsafavi, M.; Etminan, M.; Esdaile, J.M.; Lacaille, D. Risk of cardiovascular mortality in patients with rheumatoid arthritis: A meta-analysis of observational studies. Arthritis Rheum. 2008, 59, 1690–1697. [Google Scholar] [CrossRef]
- Peters, M.J.; van Halm, V.P.; Voskuyl, A.E.; Smulders, Y.M.; Boers, M.; Lems, W.F.; Visser, M.; Stehouwer, C.D.; Dekker, J.M.; Nijpels, G.; et al. Does rheumatoid arthritis equal diabetes mellitus as an independent risk factor for cardiovascular disease? A prospective study. Arthritis Rheum. 2009, 61, 1571–1579. [Google Scholar] [CrossRef]
- Pujades-Rodriguez, M.; Duyx, B.; Thomas, S.L.; Stogiannis, D.; Rahman, A.; Smeeth, L.; Hemingway, H. Rheumatoid arthritis and incidence of twelve initial presentations of cardiovascular disease: A population record-linkage cohort study in england. PLoS ONE 2016, 11, e0151245. [Google Scholar] [CrossRef]
- Del Rincon, I.D.; Williams, K.; Stern, M.P.; Freeman, G.L.; Escalante, A. High incidence of cardiovascular events in a rheumatoid arthritis cohort not explained by traditional cardiac risk factors. Arthritis Rheum. 2001, 44, 2737–2745. [Google Scholar] [CrossRef]
- Gonzalez-Gay, M.A.; Gonzalez-Juanatey, C.; Lopez-Diaz, M.J.; Pineiro, A.; Garcia-Porrua, C.; Miranda-Filloy, J.A.; Ollier, W.E.; Martin, J.; Llorca, J. Hla-drb1 and persistent chronic inflammation contribute to cardiovascular events and cardiovascular mortality in patients with rheumatoid arthritis. Arthritis Rheum. 2007, 57, 125–132. [Google Scholar] [CrossRef]
- Sattar, N.; McInnes, I.B. Vascular comorbidity in rheumatoid arthritis: Potential mechanisms and solutions. Curr. Opin. Rheumatol. 2005, 17, 286–292. [Google Scholar] [CrossRef]
- Dregan, A.; Charlton, J.; Chowienczyk, P.; Gulliford, M.C. Chronic inflammatory disorders and risk of type 2 diabetes mellitus, coronary heart disease, and stroke: A population-based cohort study. Circulation 2014, 130, 837–844. [Google Scholar] [CrossRef]
- Mason, J.C.; Libby, P. Cardiovascular disease in patients with chronic inflammation: Mechanisms underlying premature cardiovascular events in rheumatologic conditions. Eur. Heart J. 2015, 36, 482–489. [Google Scholar] [CrossRef]
- Teague, H.; Mehta, N.N. The link between inflammatory disorders and coronary heart disease: A look at recent studies and novel drugs in development. Curr. Atheroscler. Rep. 2016, 18, 3. [Google Scholar] [CrossRef]
- Armstrong, A.W.; Voyles, S.V.; Armstrong, E.J.; Fuller, E.N.; Rutledge, J.C. A tale of two plaques: Convergent mechanisms of t-cell-mediated inflammation in psoriasis and atherosclerosis. Exp. Dermatol. 2011, 20, 544–549. [Google Scholar] [CrossRef]
- Matsuura, E.; Atzeni, F.; Sarzi-Puttini, P.; Turiel, M.; Lopez, L.R.; Nurmohamed, M.T. Is atherosclerosis an autoimmune disease? BMC Med. 2014, 12, 47. [Google Scholar] [CrossRef]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.H.; Genest, J.; Gotto, A.M.; Kastelein, J.J.P.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; MacFadyen, J.G.; et al. Rosuvastatin to prevent vascular events in men and women with elevated c-reactive protein. N. Engl. J. Med. 2008, 359, 2195–2207. [Google Scholar] [CrossRef]
- Cannon, C.P.; Braunwald, E.; McCabe, C.H.; Rader, D.J.; Rouleau, J.L.; Belder, R.; Joyal, S.V.; Hill, K.A.; Pfeffer, M.A.; Skene, A.M. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N. Engl. J. Med. 2004, 350, 1495–1504. [Google Scholar] [CrossRef]
- Ridker, P.M.; Cannon, C.P.; Morrow, D.; Rifai, N.; Rose, L.M.; McCabe, C.H.; Pfeffer, M.A.; Braunwald, E. C-reactive protein levels and outcomes after statin therapy. N. Engl. J. Med. 2005, 352, 20–28. [Google Scholar] [CrossRef]
- Bohula, E.A.; Giugliano, R.P.; Cannon, C.P.; Zhou, J.; Murphy, S.A.; White, J.A.; Tershakovec, A.M.; Blazing, M.A.; Braunwald, E. Achievement of dual low-density lipoprotein cholesterol and high-sensitivity c-reactive protein targets more frequent with the addition of ezetimibe to simvastatin and associated with better outcomes in improve-it. Circulation 2015, 132, 1224–1233. [Google Scholar] [CrossRef]
- Cannon, C.P.; Blazing, M.A.; Giugliano, R.P.; McCagg, A.; White, J.A.; Theroux, P.; Darius, H.; Lewis, B.S.; Ophuis, T.O.; Jukema, J.W.; et al. Ezetimibe added to statin therapy after acute coronary syndromes. N. Engl. J. Med. 2015, 372, 2387–2397. [Google Scholar] [CrossRef]
- Tousoulis, D.; Oikonomou, E.; Economou, E.K.; Crea, F.; Kaski, J.C. Inflammatory cytokines in atherosclerosis: Current therapeutic approaches. Eur. Heart J. 2016, 37, 1723–1732. [Google Scholar] [CrossRef]
- Dinarello, C.A. Historical insights into cytokines. Eur. J. Immunol. 2007, 37 (Suppl. 1), S34–S45. [Google Scholar] [CrossRef]
- Dinarello, C.A. Anti-inflammatory agents: Present and future. Cell 2010, 140, 935–950. [Google Scholar] [CrossRef]
- Ridker, P.M.; Cushman, M.; Stampfer, M.J.; Tracy, R.P.; Hennekens, C.H. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N. Engl. J. Med. 1997, 336, 973–979. [Google Scholar] [CrossRef]
- Ridker, P.M.; Hennekens, C.H.; Buring, J.E.; Rifai, N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N. Engl. J. Med. 2000, 342, 836–843. [Google Scholar] [CrossRef]
- Ridker, P.M.; Hennekens, C.H.; Roitman-Johnson, B.; Stampfer, M.J.; Allen, J. Plasma concentration of soluble intercellular adhesion molecule 1 and risks of future myocardial infarction in apparently healthy men. Lancet (Lond. Engl.) 1998, 351, 88–92. [Google Scholar] [CrossRef]
- Ridker, P.M.; Rifai, N.; Stampfer, M.J.; Hennekens, C.H. Plasma concentration of interleukin-6 and the risk of future myocardial infarction among apparently healthy men. Circulation 2000, 101, 1767–1772. [Google Scholar] [CrossRef]
- Kaptoge, S.; Seshasai, S.R.; Gao, P.; Freitag, D.F.; Butterworth, A.S.; Borglykke, A.; Di Angelantonio, E.; Gudnason, V.; Rumley, A.; Lowe, G.D.; et al. Inflammatory cytokines and risk of coronary heart disease: New prospective study and updated meta-analysis. Eur. Heart J. 2014, 35, 578–589. [Google Scholar] [CrossRef]
- Swerdlow, D.I.; Holmes, M.V.; Kuchenbaecker, K.B.; Engmann, J.E.; Shah, T.; Sofat, R.; Guo, Y.; Chung, C.; Peasey, A.; Pfister, R.; et al. The interleukin-6 receptor as a target for prevention of coronary heart disease: A mendelian randomisation analysis. Lancet (Lond. Engl.) 2012, 379, 1214–1224. [Google Scholar]
- Danesh, J.; Kaptoge, S.; Mann, A.G.; Sarwar, N.; Wood, A.; Angleman, S.B.; Wensley, F.; Higgins, J.P.; Lennon, L.; Eiriksdottir, G.; et al. Long-term interleukin-6 levels and subsequent risk of coronary heart disease: Two new prospective studies and a systematic review. PLoS Med. 2008, 5, e78. [Google Scholar] [CrossRef]
- Sarwar, N.; Butterworth, A.S.; Freitag, D.F.; Gregson, J.; Willeit, P.; Gorman, D.N.; Gao, P.; Saleheen, D.; Rendon, A.; Nelson, C.P.; et al. Interleukin-6 receptor pathways in coronary heart disease: A collaborative meta-analysis of 82 studies. Lancet (Lond. Engl.) 2012, 379, 1205–1213. [Google Scholar]
- Tosato, G.; Jones, K.D. Interleukin-1 induces interleukin-6 production in peripheral blood monocytes. Blood 1990, 75, 1305–1310. [Google Scholar]
- Kirii, H.; Niwa, T.; Yamada, Y.; Wada, H.; Saito, K.; Iwakura, Y.; Asano, M.; Moriwaki, H.; Seishima, M. Lack of interleukin-1beta decreases the severity of atherosclerosis in apoe-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 656–660. [Google Scholar] [CrossRef]
- Bhaskar, V.; Yin, J.; Mirza, A.M.; Phan, D.; Vanegas, S.; Issafras, H.; Michelson, K.; Hunter, J.J.; Kantak, S.S. Monoclonal antibodies targeting il-1 beta reduce biomarkers of atherosclerosis in vitro and inhibit atherosclerotic plaque formation in apolipoprotein e-deficient mice. Atherosclerosis 2011, 216, 313–320. [Google Scholar] [CrossRef]
- Elhage, R.; Jawien, J.; Rudling, M.; Ljunggren, H.-G.; Takeda, K.; Akira, S.; Bayard, F.; Hansson, G.K. Reduced atherosclerosis in interleukin-18 deficient apolipoprotein e-knockout mice. Cardiovasc. Res. 2003, 59, 234–240. [Google Scholar] [CrossRef]
- Mallat, Z.; Corbaz, A.; Scoazec, A.; Graber, P.; Alouani, S.; Esposito, B.; Humbert, Y.; Chvatchko, Y.; Tedgui, A. Interleukin-18/interleukin-18 binding protein signaling modulates atherosclerotic lesion development and stability. Circ. Res. 2001, 89, e41–e45. [Google Scholar] [CrossRef]
- Ockene, I.S.; Matthews, C.E.; Rifai, N.; Ridker, P.M.; Reed, G.; Stanek, E. Variability and classification accuracy of serial high-sensitivity c-reactive protein measurements in healthy adults. Clin. Chem. 2001, 47, 444–450. [Google Scholar]
- Meier-Ewert, H.K.; Ridker, P.M.; Rifai, N.; Price, N.; Dinges, D.F.; Mullington, J.M. Absence of diurnal variation of c-reactive protein concentrations in healthy human subjects. Clin. Chem. 2001, 47, 426–430. [Google Scholar]
- Berk, B.C.; Weintraub, W.S.; Alexander, R.W. Elevation of c-reactive protein in “active” coronary artery disease. Am. J. Cardiol. 1990, 65, 168–172. [Google Scholar] [CrossRef]
- Ablij, H.; Meinders, A. C-reactive protein: History and revival. Eur. J. Intern. Med. 2002, 13, 412–422. [Google Scholar] [CrossRef]
- Albert, C.M.; Ma, J.; Rifai, N.; Stampfer, M.J.; Ridker, P.M. Prospective study of c-reactive protein, homocysteine, and plasma lipid levels as predictors of sudden cardiac death. Circulation 2002, 105, 2595–2599. [Google Scholar] [CrossRef]
- Rost, N.S.; Wolf, P.A.; Kase, C.S.; Kelly-Hayes, M.; Silbershatz, H.; Massaro, J.M.; D’Agostino, R.B.; Franzblau, C.; Wilson, P.W. Plasma concentration of c-reactive protein and risk of ischemic stroke and transient ischemic attack: The framingham study. Stroke 2001, 32, 2575–2579. [Google Scholar] [CrossRef]
- Ridker, P.M.; Rifai, N.; Rose, L.; Buring, J.E.; Cook, N.R. Comparison of c-reactive protein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular events. N. Engl. J. Med. 2002, 347, 1557–1565. [Google Scholar] [CrossRef]
- Kaptoge, S.; Di Angelantonio, E.; Lowe, G.; Pepys, M.B.; Thompson, S.G.; Collins, R.; Danesh, J. C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: An individual participant meta-analysis. Lancet (Lond. Engl.) 2010, 375, 132–140. [Google Scholar]
- Mann, D.L.; McMurray, J.J.; Packer, M.; Swedberg, K.; Borer, J.S.; Colucci, W.S.; Djian, J.; Drexler, H.; Feldman, A.; Kober, L.; et al. Targeted anticytokine therapy in patients with chronic heart failure: Results of the randomized etanercept worldwide evaluation (renewal). Circulation 2004, 109, 1594–1602. [Google Scholar] [CrossRef]
- Chung, E.S.; Packer, M.; Lo, K.H.; Fasanmade, A.A.; Willerson, J.T. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: Results of the anti-tnf therapy against congestive heart failure (attach) trial. Circulation 2003, 107, 3133–3140. [Google Scholar]
- Hassan, S.; Milman, U.; Feld, J.; Eder, L.; Lavi, I.; Cohen, S.; Zisman, D. Effects of anti-tnf-α treatment on lipid profile in rheumatic diseases: An analytical cohort study. Arthritis Res. Ther. 2016, 18, 261. [Google Scholar] [CrossRef]
- Curtis, J.R.; John, A.; Baser, O. Dyslipidemia and changes in lipid profiles associated with rheumatoid arthritis and initiation of anti-tumor necrosis factor therapy. Arthritis Care Res. 2012, 64, 1282–1291. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Pradhan, A.; MacFadyen, J.G.; Solomon, D.H.; Zaharris, E.; Mam, V.; Hasan, A.; Rosenberg, Y.; Iturriaga, E.; et al. Low-dose methotrexate for the prevention of atherosclerotic events. N. Engl. J. Med. 2019, 380, 752–762. [Google Scholar] [CrossRef]
- Nidorf, S.M.; Eikelboom, J.W.; Budgeon, C.A.; Thompson, P.L. Low-dose colchicine for secondary prevention of cardiovascular disease. J. Am. Coll. Cardiol. 2013, 61, 404–410. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; Braunwald, E.; White, H.D.; Steen, D.L.; Lukas, M.A.; Tarka, E.; Steg, P.G.; Hochman, J.S.; Bode, C.; Maggioni, A.P.; et al. Effect of darapladib on major coronary events after an acute coronary syndrome: The solid-timi 52 randomized clinical trialdarapladib and major coronary events after acsdarapladib and major coronary events after acs. JAMA 2014, 312, 1006–1015. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; Glaser, R.; Cavender, M.A.; Aylward, P.E.; Bonaca, M.P.; Budaj, A.; Davies, R.Y.; Dellborg, M.; Fox, K.A.A.; Gutierrez, J.A.T.; et al. Effect of losmapimod on cardiovascular outcomes in patients hospitalized with acute myocardial infarction: A randomized clinical trialeffect of losmapimod on cardiovascular outcomes in patients with acute mieffect of losmapimod on cardiovascular outcomes in patients with acute mi. JAMA 2016, 315, 1591–1599. [Google Scholar]
- Ridker, P.M.; Howard, C.P.; Walter, V.; Everett, B.; Libby, P.; Hensen, J.; Thuren, T. Effects of interleukin-1beta inhibition with canakinumab on hemoglobin a1c, lipids, c-reactive protein, interleukin-6, and fibrinogen: A phase iib randomized, placebo-controlled trial. Circulation 2012, 126, 2739–2748. [Google Scholar] [CrossRef]
- Ridker, P.M.; MacFadyen, J.G.; Everett, B.M.; Libby, P.; Thuren, T.; Glynn, R.J. Relationship of c-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: A secondary analysis from the cantos randomised controlled trial. Lancet (Lond. Engl.) 2018, 391, 319–328. [Google Scholar] [CrossRef]
- Ridker, P.M.; Libby, P.; MacFadyen, J.G.; Thuren, T.; Ballantyne, C.; Fonseca, F.; Koenig, W.; Shimokawa, H.; Everett, B.M.; Glynn, R.J. Modulation of the interleukin-6 signalling pathway and incidence rates of atherosclerotic events and all-cause mortality: Analyses from the canakinumab anti-inflammatory thrombosis outcomes study (cantos). Eur. Heart J. 2018, 39, 3499–3507. [Google Scholar] [CrossRef]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Everett, B.M.; Libby, P.; Glynn, R.J. Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: Exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet (Lond. Engl.) 2017, 390, 1833–1842. [Google Scholar] [CrossRef]
- Malaviya, A.N.; Sharma, A.; Agarwal, D.; Kapoor, S.; Garg, S.; Sawhney, S. Low-dose and high-dose methotrexate are two different drugs in practical terms. Int. J. Rheum. Dis. 2010, 13, 288–293. [Google Scholar] [CrossRef]
- Cutolo, M.; Sulli, A.; Pizzorni, C.; Seriolo, B.; Straub, R.H. Anti-inflammatory mechanisms of methotrexate in rheumatoid arthritis. Ann. Rheum. Dis. 2001, 60, 729–735. [Google Scholar] [CrossRef]
- Choi, H.K.; Hernan, M.A.; Seeger, J.D.; Robins, J.M.; Wolfe, F. Methotrexate and mortality in patients with rheumatoid arthritis: A prospective study. Lancet (Lond. Engl.) 2002, 359, 1173–1177. [Google Scholar] [CrossRef]
- Van Halm, V.P.; Nurmohamed, M.T.; Twisk, J.W.; Dijkmans, B.A.; Voskuyl, A.E. Disease-modifying antirheumatic drugs are associated with a reduced risk for cardiovascular disease in patients with rheumatoid arthritis: A case control study. Arthritis Res. Ther. 2006, 8, R151. [Google Scholar] [CrossRef]
- Naranjo, A.; Sokka, T.; Descalzo, M.A.; Calvo-Alen, J.; Horslev-Petersen, K.; Luukkainen, R.K.; Combe, B.; Burmester, G.R.; Devlin, J.; Ferraccioli, G.; et al. Cardiovascular disease in patients with rheumatoid arthritis: Results from the quest-ra study. Arthritis Res. Ther. 2008, 10, R30. [Google Scholar] [CrossRef]
- Micha, R.; Imamura, F.; Wyler von Ballmoos, M.; Solomon, D.H.; Hernan, M.A.; Ridker, P.M.; Mozaffarian, D. Systematic review and meta-analysis of methotrexate use and risk of cardiovascular disease. Am. J. Cardiol. 2011, 108, 1362–1370. [Google Scholar] [CrossRef]
- Mallat, Z.; Lambeau, G.; Tedgui, A. Lipoprotein-associated and secreted phospholipases a(2) in cardiovascular disease: Roles as biological effectors and biomarkers. Circulation 2010, 122, 2183–2200. [Google Scholar] [CrossRef]
- Tschopp, J.; Schroder, K. Nlrp3 inflammasome activation: The convergence of multiple signalling pathways on ros production? Nat. Rev. Immunol. 2010, 10, 210–215. [Google Scholar] [CrossRef]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nunez, G.; Schnurr, M.; et al. Nlrp3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef]
- Rajamaki, K.; Lappalainen, J.; Oorni, K.; Valimaki, E.; Matikainen, S.; Kovanen, P.T.; Eklund, K.K. Cholesterol crystals activate the nlrp3 inflammasome in human macrophages: A novel link between cholesterol metabolism and inflammation. PLoS ONE 2010, 5, e11765. [Google Scholar] [CrossRef]
- Martinon, F.; Petrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the nalp3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef]
- Zhou, W.; Chen, C.; Chen, Z.; Liu, L.; Jiang, J.; Wu, Z.; Zhao, M.; Chen, Y. Nlrp3: A novel mediator in cardiovascular disease. J. Immunol. Res. 2018, 2018, 5702103. [Google Scholar] [CrossRef]
- He, Y.; Hara, H.; Nunez, G. Mechanism and regulation of nlrp3 inflammasome activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef]
- Van Hout, G.P.; Bosch, L.; Ellenbroek, G.H.; de Haan, J.J.; van Solinge, W.W.; Cooper, M.A.; Arslan, F.; de Jager, S.C.; Robertson, A.A.; Pasterkamp, G.; et al. The selective nlrp3-inflammasome inhibitor mcc950 reduces infarct size and preserves cardiac function in a pig model of myocardial infarction. Eur. Heart J. 2017, 38, 828–836. [Google Scholar] [CrossRef]
- Van der Heijden, T.; Kritikou, E.; Venema, W.; van Duijn, J.; van Santbrink, P.J.; Slutter, B.; Foks, A.C.; Bot, I.; Kuiper, J. Nlrp3 inflammasome inhibition by mcc950 reduces atherosclerotic lesion development in apolipoprotein e-deficient mice-brief report. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1457–1461. [Google Scholar] [CrossRef]
- Huber, S.A.; Sakkinen, P.; Conze, D.; Hardin, N.; Tracy, R. Interleukin-6 exacerbates early atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2364–2367. [Google Scholar] [CrossRef]
- Akita, K.; Isoda, K.; Sato-Okabayashi, Y.; Kadoguchi, T.; Kitamura, K.; Ohtomo, F.; Shimada, K.; Daida, H. An interleukin-6 receptor antibody suppresses atherosclerosis in atherogenic mice. Front. Cardiovasc. Med. 2017, 4, 84. [Google Scholar] [CrossRef]
- Kleveland, O.; Kunszt, G.; Bratlie, M.; Ueland, T.; Broch, K.; Holte, E.; Michelsen, A.E.; Bendz, B.; Amundsen, B.H.; Espevik, T.; et al. Effect of a single dose of the interleukin-6 receptor antagonist tocilizumab on inflammation and troponin t release in patients with non-st-elevation myocardial infarction: A double-blind, randomized, placebo-controlled phase 2 trial. Eur. Heart J. 2016, 37, 2406–2413. [Google Scholar] [CrossRef]
- Hirabayashi, Y.; Ishii, T.; Harigae, H. Clinical efficacy of tocilizumab in patients with active rheumatoid arthritis in real clinical practice. Rheumatol. Int. 2010, 30, 1041–1048. [Google Scholar] [CrossRef][Green Version]
- Jones, S.A.; Scheller, J.; Rose-John, S. Therapeutic strategies for the clinical blockade of il-6/gp130 signaling. J. Clin. Investig. 2011, 121, 3375–3383. [Google Scholar] [CrossRef]
- Leung, Y.Y.; Yao Hui, L.L.; Kraus, V.B. Colchicine--update on mechanisms of action and therapeutic uses. Semin. Arthritis Rheum. 2015, 45, 341–350. [Google Scholar] [CrossRef]
- Demidowich, A.P.; Davis, A.I.; Dedhia, N.; Yanovski, J.A. Colchicine to decrease nlrp3-activated inflammation and improve obesity-related metabolic dysregulation. Med. Hypotheses 2016, 92, 67–73. [Google Scholar] [CrossRef]
- Verma, S.; Eikelboom, J.W.; Nidorf, S.M.; Al-Omran, M.; Gupta, N.; Teoh, H.; Friedrich, J.O. Colchicine in cardiac disease: A systematic review and meta-analysis of randomized controlled trials. BMC Cardiovasc. Disord. 2015, 15, 96. [Google Scholar] [CrossRef]
- Crittenden, D.B.; Lehmann, R.A.; Schneck, L.; Keenan, R.T.; Shah, B.; Greenberg, J.D.; Cronstein, B.N.; Sedlis, S.P.; Pillinger, M.H. Colchicine use is associated with decreased prevalence of myocardial infarction in patients with gout. J. Rheumatol. 2012, 39, 1458–1464. [Google Scholar] [CrossRef]
- Langevitz, P.; Livneh, A.; Neumann, L.; Buskila, D.; Shemer, J.; Amolsky, D.; Pras, M. Prevalence of ischemic heart disease in patients with familial mediterranean fever. Isr. Med Assoc. J. IMAJ 2001, 3, 9–12. [Google Scholar]
- Martínez, G.J.; Robertson, S.; Barraclough, J.; Xia, Q.; Mallat, Z.; Bursill, C.; Celermajer, D.S.; Patel, S. Colchicine acutely suppresses local cardiac production of inflammatory cytokines in patients with an acute coronary syndrome. J. Am. Heart Assoc. 2015, 4, e002128. [Google Scholar] [CrossRef]
- Vaidya, K.; Arnott, C.; Martínez, G.J.; Ng, B.; McCormack, S.; Sullivan, D.R.; Celermajer, D.S.; Patel, S. Colchicine therapy and plaque stabilization in patients with acute coronary syndrome. JACC Cardiovasc. Imaging 2018, 11, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Nidorf, M.; Thompson, P.L. Effect of colchicine (0.5 mg twice daily) on high-sensitivity c-reactive protein independent of aspirin and atorvastatin in patients with stable coronary artery disease. Am. J. Cardiol. 2007, 99, 805–807. [Google Scholar] [CrossRef] [PubMed]
- Hemkens, L.G.; Ewald, H.; Gloy, V.L.; Arpagaus, A.; Olu, K.K.; Nidorf, M.; Glinz, D.; Nordmann, A.J.; Briel, M. Colchicine for prevention of cardiovascular events. Cochrane Database Syst. Rev. 2016. [Google Scholar] [CrossRef] [PubMed]
- Pillinger, M.H.; Krasnokutsky, S.; Slobodnick, A.; Shah, B. Update on colchicine, 2017. Rheumatology 2017, 57, i4–i11. [Google Scholar]
- Heuser, M.; Thol, F.; Ganser, A. Clonal hematopoiesis of indeterminate potential. Dtsch. Arztebl. Int. 2016, 113, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef]
- Sano, S.; Oshima, K.; Wang, Y.; MacLauchlan, S.; Katanasaka, Y.; Sano, M.; Zuriaga, M.A.; Yoshiyama, M.; Goukassian, D.; Cooper, M.A.; et al. Tet2-mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the il-1beta/nlrp3 inflammasome. J. Am. Coll. Cardiol. 2018, 71, 875–886. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Trial Name | Study Design | Patient Number | Intervention | Primary Outcomes | Results | Benefit Observed |
|---|---|---|---|---|---|---|
| Broad-spectrum anti-inflammatory approach | ||||||
| Methotrexate | ||||||
| Cardiovascular Inflammation Reduction Trial (CIRT) [77] | Phase 3 multicentre, randomised, double-blind, placebo-controlled | 4786 | Oral low methotrexate (target dose of 15–20 mg weekly) vs. placebo | Non-fatal myocardial infarction, non-fatal stroke and cardiovascular death | HR 1.01; 95% CI 0.82–1.25; p = 0.91 |  |
| Colchicine | ||||||
| Low-dose colchicine for secondary prevention of cardiovascular disease (LoDoCo) [78] | Phase 3 multicentre, randomised, double-blind, placebo-controlled | 532 | Colchicine 0.5 mg/day vs. placebo | MI, fatal or non-fatal out-of-hospital cardiac arrest, or non-cardioembolic ischaemic stroke | HR 0.33; 95% CI 0.18–0.59; p < 0.001 |  |
| Narrow-spectrum anti-inflammatory approach | ||||||
| IL-1β | ||||||
| Anti-inflammatory Therapy with Cankinumab for Atherosclerosis (CANTOS) [76] | Phase 3 multicentre, randomised, double-blind, placebo-controlled | 10,061 | Subcutaneous injection of canakinumab (50 mg, 150 mg or 300 mg) every 3 months vs. placebo | Non-fatal MI, non-fatal stroke and cardiovascular death | HR 0.85; 95% CI 0.74–0.98; p = 0.021 in the 150 mg-treated group | |
| Lipoprotein-associated phospholipase A2 (Lp-PLA2) | ||||||
| SOLID-TIMI 52 [79] | Phase 3 multicentre, randomised, double-blind, placebo-controlled | 13,026 | Daily oral darapladib 160 mg vs. placebo | Coronary heart disease death, non-fatal MI and urgent revascularisation for myocardial ischaemia | HR 1.00; 95% CI 0.91–1.09; p = 0.93 | |
| P38 mitogen-activated protein kinase (MAPK) | ||||||
| LATITUDE-TIMI 60 [80] | Phase 3 multicentre, randomised, double-blind, placebo-controlled | 3503 | Oral losmapimod 7.5 mg twice daily vs. placebo | Non-fatal MI, severe recurrent ischaemia requiring urgent coronary artery revascularisation and cardiovascular death | HR 1.16; 95% CI 0.91–1.47; p = 0.24 | |
| Trial Name | Primary Site(s) | Study Design | Patient Number | Intervention | Primary Outcomes | Follow-up | Completion Date |
|---|---|---|---|---|---|---|---|
| Stable coronary artery disease | |||||||
| LoDoCo II: Low-dose Colchicine for Secondary Prevention of Cardiovascular Disease (ACTRN12614000093684) | Australia, Netherlands | Phase 3 multicentre, double blind, randomised placebo-controlled | 5500 | Colchicine 0.5 mg/day vs. placebo | ACS, cardiovascular death or stroke | 3 years | 20 Jan |
| Acute Coronary Syndrome (ACS) | |||||||
| COLCOT: Colchicine Cardiovascular Outcomes Trial (NCT02551094) | Canada | Phase 3 randomised placebo-controlled | 4745 | Colchicine 0.5 mg/day vs. placebo | MI, cardiovascular death, resuscitated cardiac arrest, stroke, or angina pectoris requiring revascularisation | 3–4 years | 19 Sep |
| COACS: Colchicine for Acute Coronary Syndromes (NCT01906749) | Italy | Phase 4 multicentre, double blind, randomised placebo- controlled | 500 | Colchicine 0.5 mg/day vs. placebo | ACS, ischaemic stroke, and overall mortality | 2 years | N/A |
| CLEAR-SYNERGY (OASIS-9): Colchicine and Spironolactone in Patients with STEMI/SYNERGY Stent Registry (NCT03048825) | Canada | Phase 3 multicentre, blinded, randomised placebo-controlled. 4 study arms, 2 × 2 factorial design | 4000 | Colchicine 1 mg/day and/or spironolactone 25 mg/day and/or placebo and/or SYNERGY stent | Cardiovascular death, recurrent MI, or stroke in the colchicine-treated group | 2 years | 21 Dec |
| Cerebrovascular disease | |||||||
| CONVINCE: Colchicine for Prevention of Vascular Inflammation in Non-cardio Embolic Stroke (NCT02898610) | Belgium, Ireland, Greece and Spain | Phase 3 multicentre, open-label, placebo controlled | 2623 | Colchicine 0.5 mg/day vs. placebo | Non-fatal major cardiac event and vascular death | 5 years | 21 Oct |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, M.T.; Fernando, S.; Schwarz, N.; Tan, J.T.; Bursill, C.A.; Psaltis, P.J. Inflammation as a Therapeutic Target in Atherosclerosis. J. Clin. Med. 2019, 8, 1109. https://doi.org/10.3390/jcm8081109
Nguyen MT, Fernando S, Schwarz N, Tan JT, Bursill CA, Psaltis PJ. Inflammation as a Therapeutic Target in Atherosclerosis. Journal of Clinical Medicine. 2019; 8(8):1109. https://doi.org/10.3390/jcm8081109
Chicago/Turabian StyleNguyen, Mau T, Sanuja Fernando, Nisha Schwarz, Joanne TM Tan, Christina A Bursill, and Peter J Psaltis. 2019. "Inflammation as a Therapeutic Target in Atherosclerosis" Journal of Clinical Medicine 8, no. 8: 1109. https://doi.org/10.3390/jcm8081109
APA StyleNguyen, M. T., Fernando, S., Schwarz, N., Tan, J. T., Bursill, C. A., & Psaltis, P. J. (2019). Inflammation as a Therapeutic Target in Atherosclerosis. Journal of Clinical Medicine, 8(8), 1109. https://doi.org/10.3390/jcm8081109