High-Risk Multiple Myeloma: Integrated Clinical and Omics Approach Dissects the Neoplastic Clone and the Tumor Microenvironment

 ,
,  ,
,  ,
,  , ,
, ,  , ,
, ,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Genetic Determinants of Multiple Myeloma and Clinical Prognostic Scores
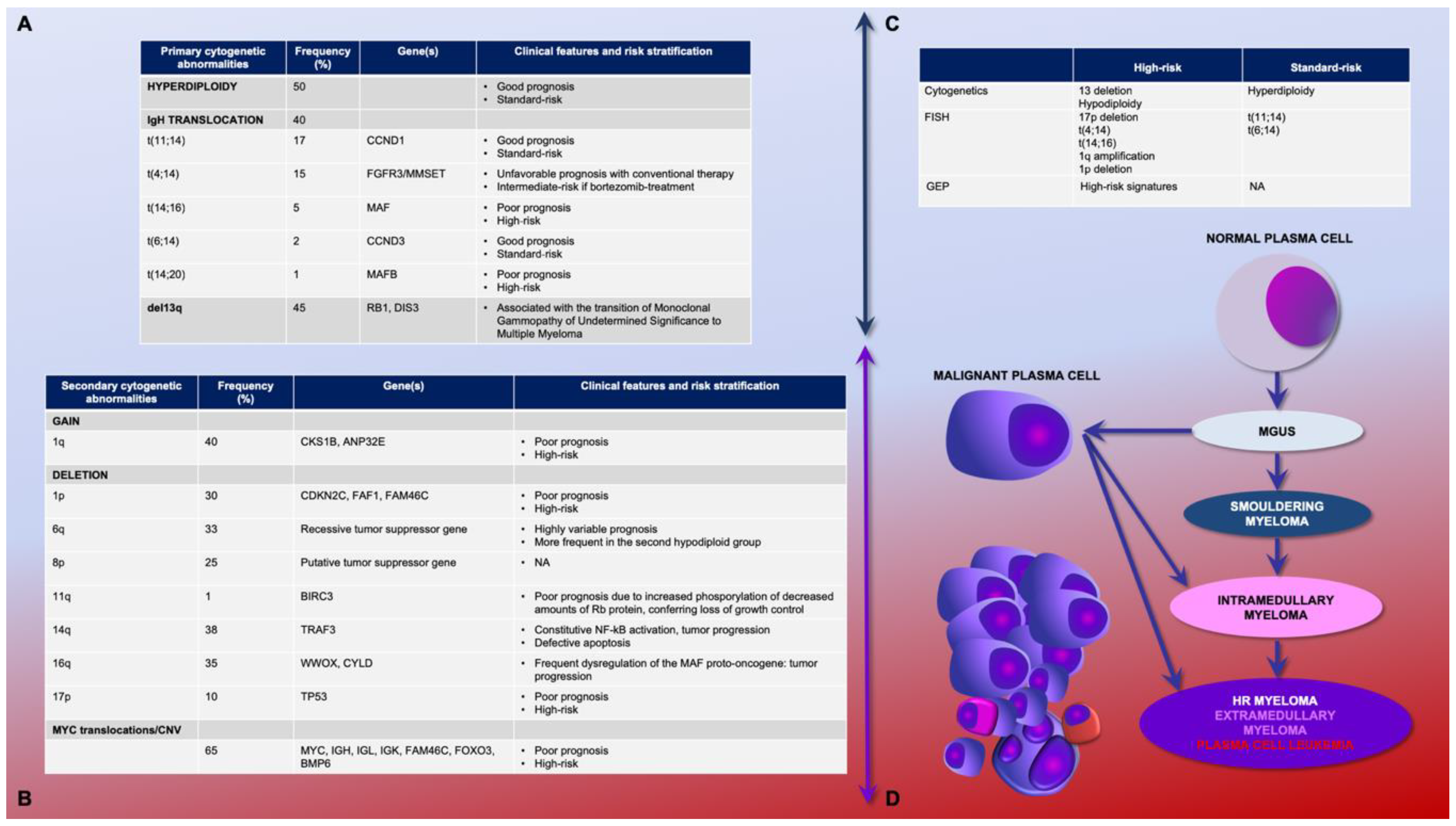
2.1. Primary and Secondary Genetic Events
2.2. Genetic Prognostic Relevance: Gene Expression Profiling and Cytogenetics
2.3. Combined Scores and Clinical Predictors of Prognosis
2.4. Minimal Residual Disease
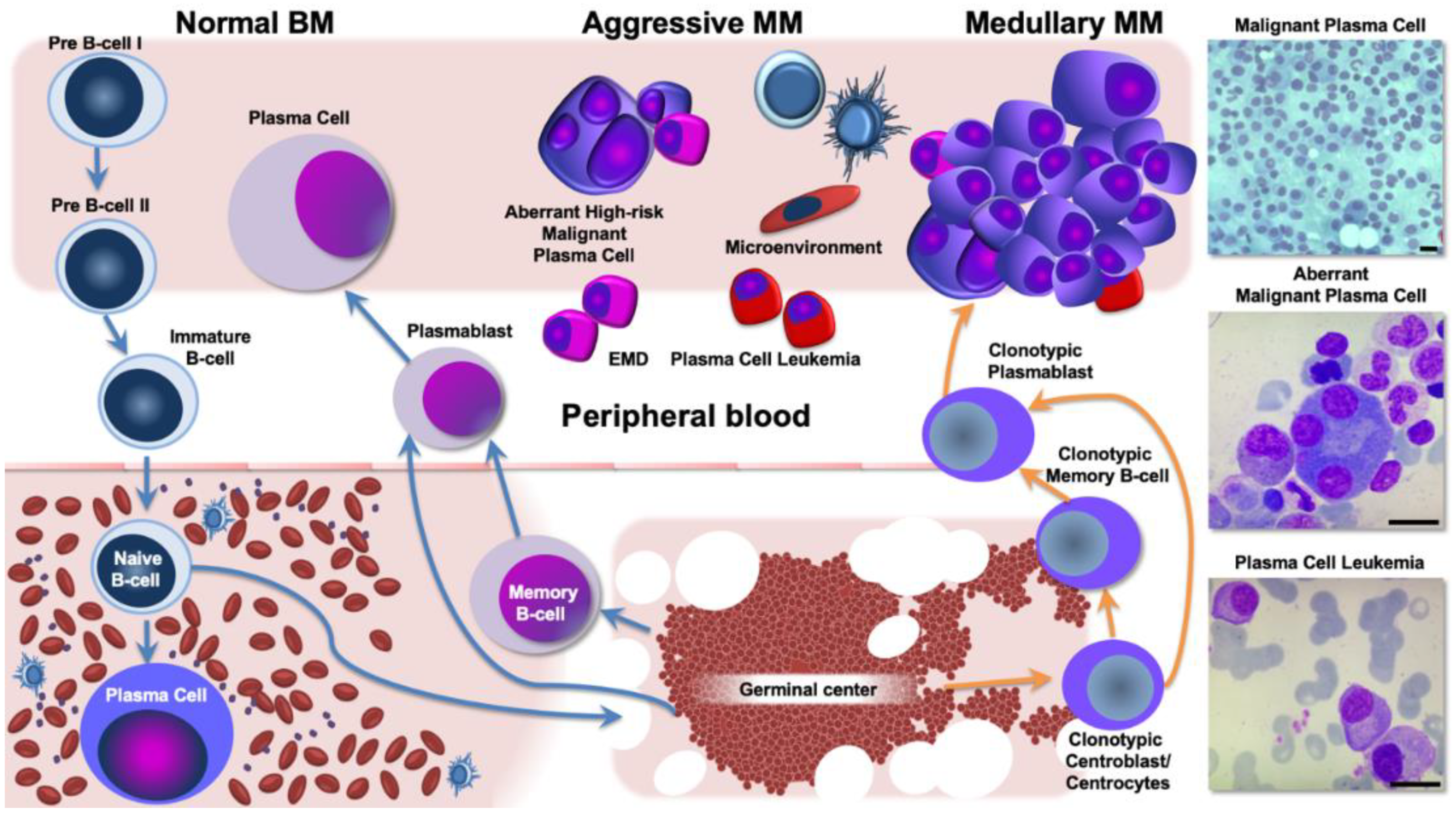
3. Aggressive and Refractory Multiple Myeloma Phenotypes: The Neoplastic Clone and the Interaction with the Tumor Microenvironment
3.1. The Angiogenic Trigger in Multiple Myeloma: Novel Perspectives from the Immune Microenvironment
3.2. Extramedullary Disease Characterization as a Paradigm for Corrupted Interaction between MM cells and Its Ecological Niche
3.3. Biological Background and Genomic Landscape of High-Risk Multiple Myeloma
4. Mechanisms of Drug Resistance in Aggressive Multiple Myeloma
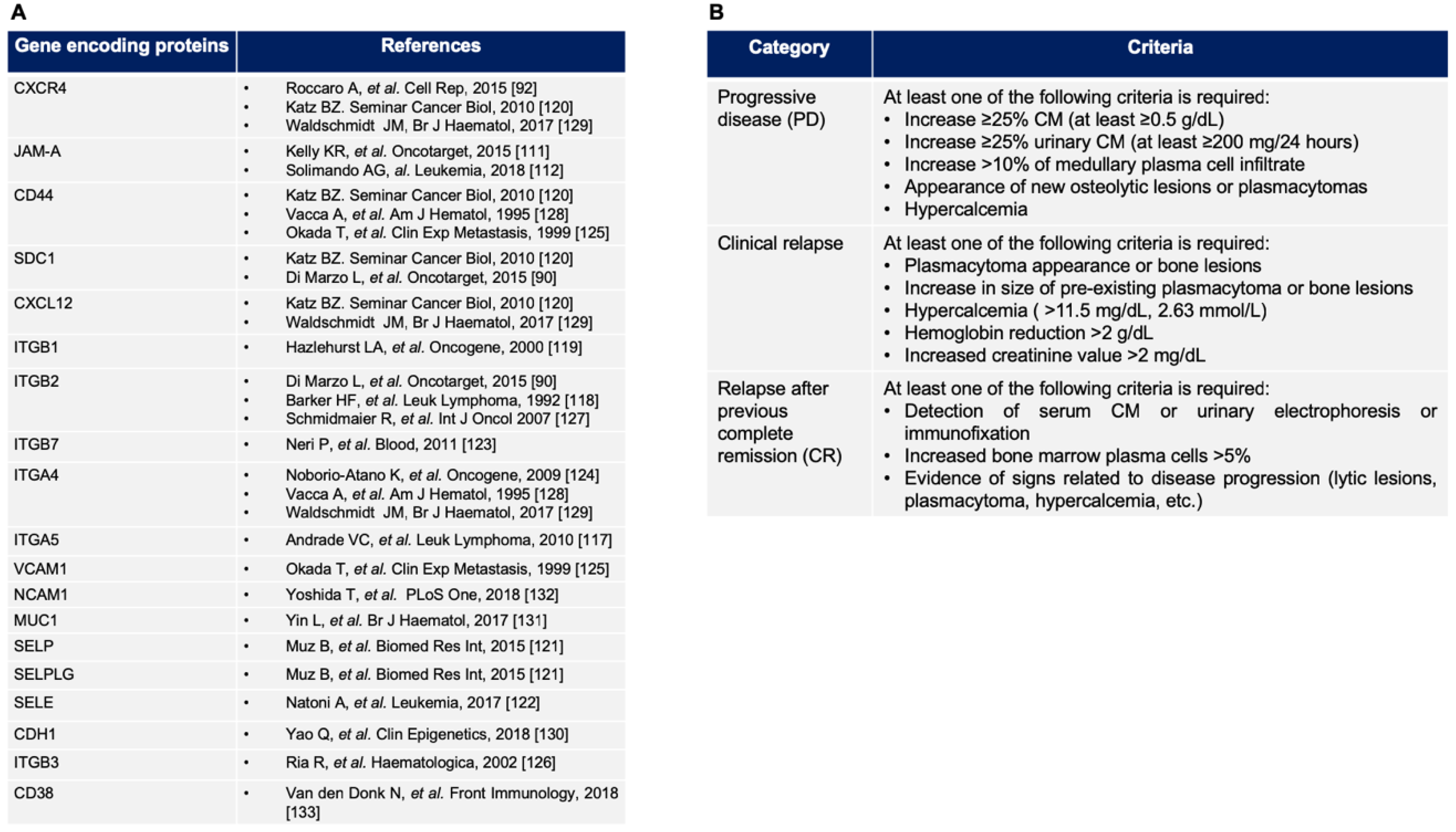
4.1. New Insights from the Bone Marrow Microenvironment Adhesion-Mediated Drug Resistance
4.2. Immuno-Modulatory Agents
4.3. Proteasome Inhibitors
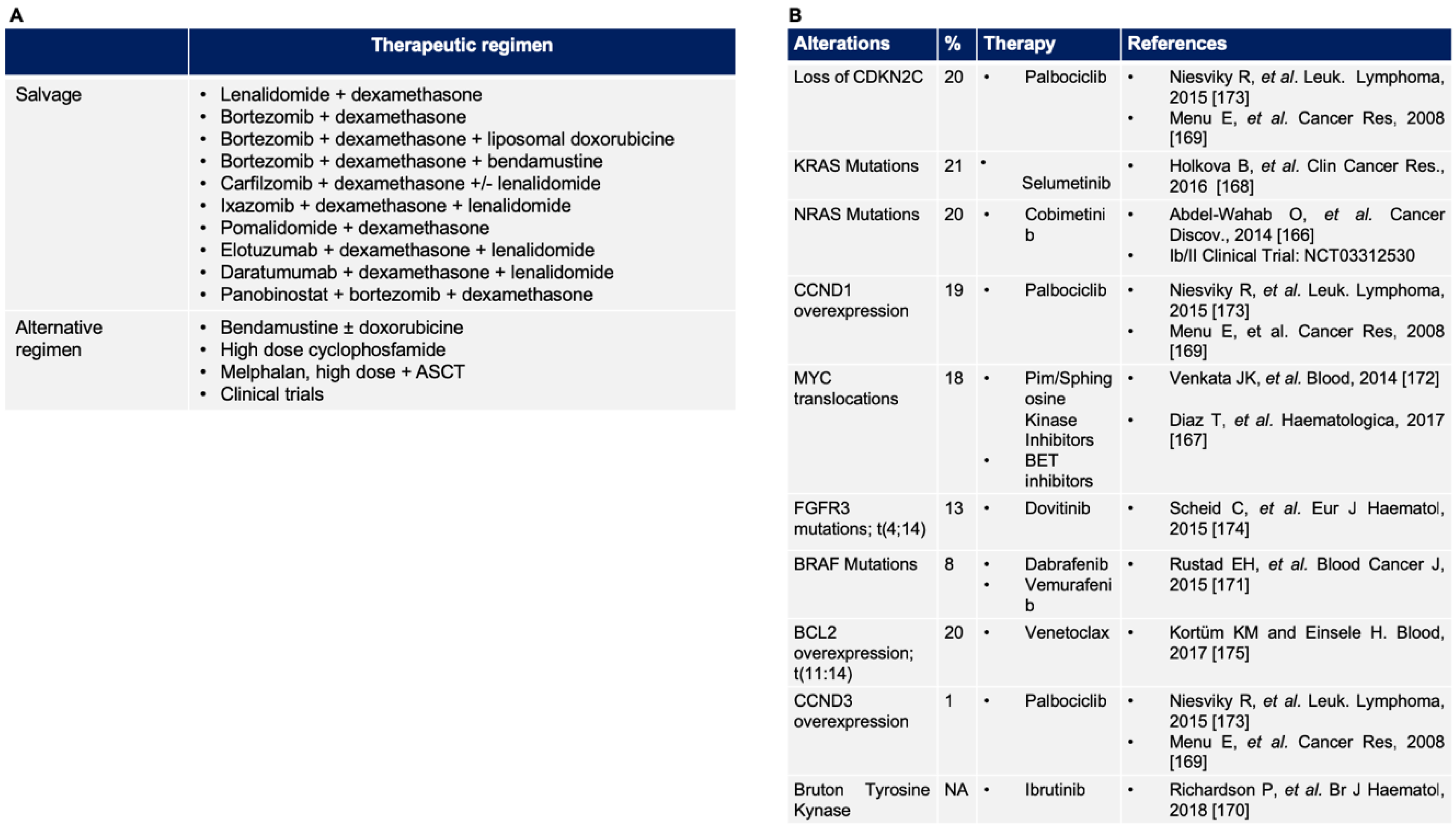
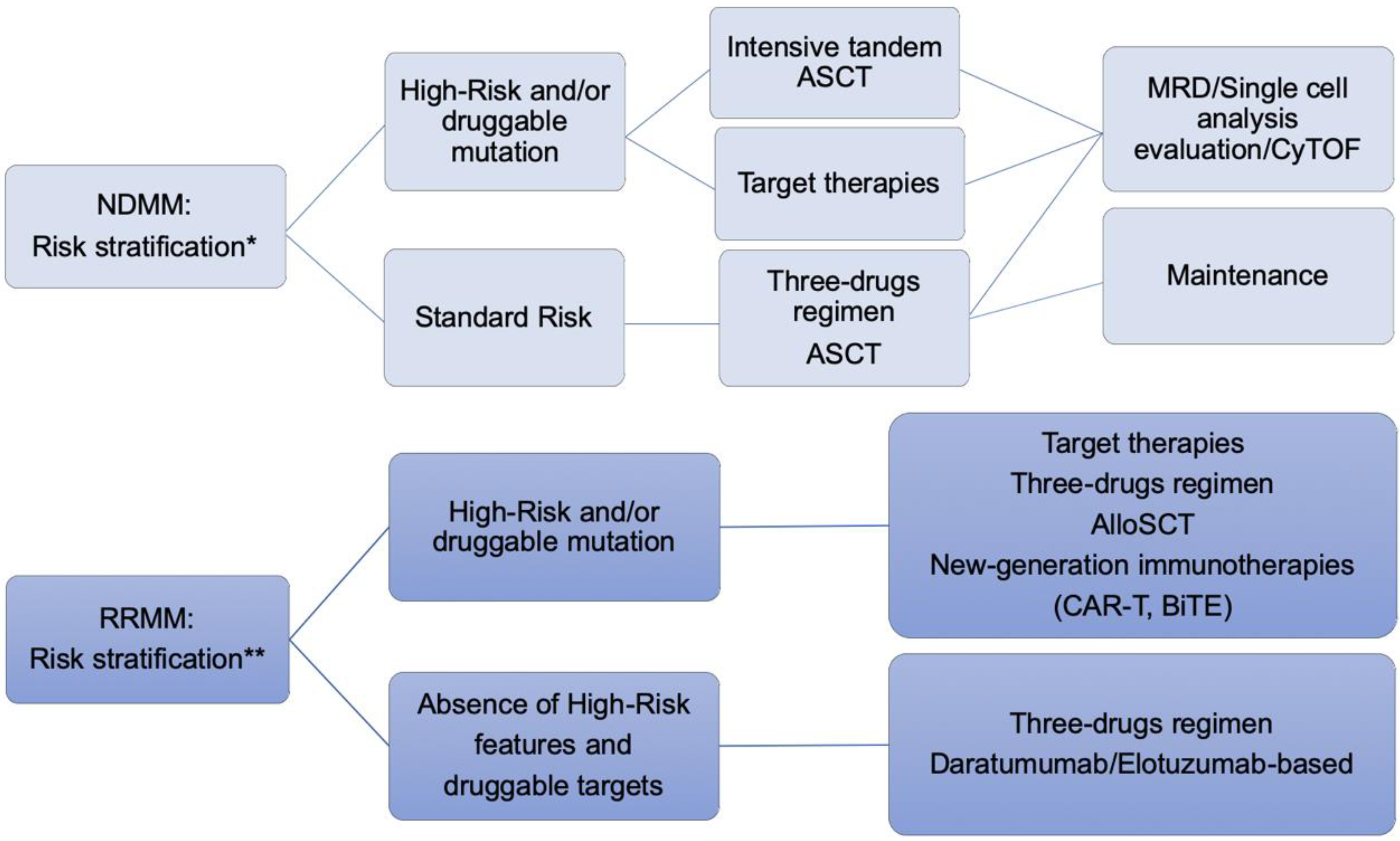
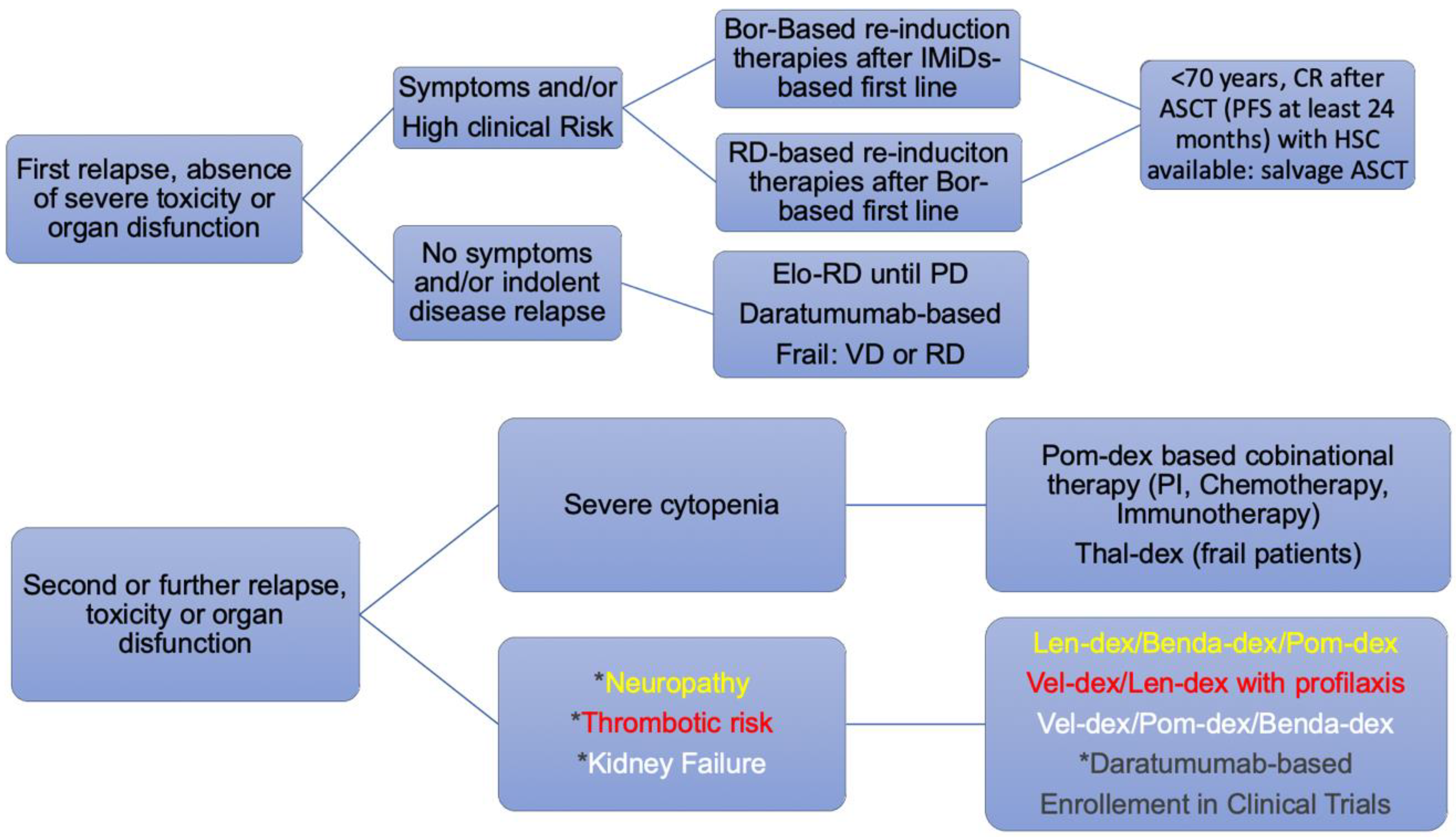
5. Approach to the Patient with High Risk Related to Relapsed/Refractory Multiple Myeloma
5.1. Validated Therapy for Relapsed/Refractory Multiple Myeloma
5.2. Novel Target in Relapsed/Refractory Multiple Myeloma
6. Future Perspectives
7. Envisioned Clinical Trial and Conclusive Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kumar, S.K.; Dispenzieri, A.; Lacy, M.Q.; Gertz, M.A.; Buadi, F.K.; Pandey, S.; Kapoor, P.; Dingli, D.; Hayman, S.R.; Leung, N.; et al. Continued improvement in survival in multiple myeloma: Changes in early mortality and outcomes in older patients. Leukemia 2014, 28, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Durie, B.G.; Salmon, S.E. A clinical staging system for multiple myeloma. Correlation of measured myeloma cell mass with presenting clinical features, response to treatment, and survival. Cancer 1975, 36, 842–854. [Google Scholar] [CrossRef]
- Kuehl, W.M.; Bergsagel, P.L. Multiple myeloma: Evolving genetic events and host interactions. Nat. Rev. Cancer 2002, 2, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Greipp, P.R.; San Miguel, J.; Durie, B.G.; Crowley, J.J.; Barlogie, B.; Blade, J.; Boccadoro, M.; Child, J.A.; Avet-Loiseau, H.; Kyle, R.A.; et al. International staging system for multiple myeloma. J. Clin. Oncol. 2005, 23, 3412–3420. [Google Scholar] [CrossRef] [PubMed]
- Sonneveld, P.; Avet-Loiseau, H.; Lonial, S.; Usmani, S.; Siegel, D.; Anderson, K.C.; Chng, W.J.; Moreau, P.; Attal, M.; Kyle, R.A.; et al. Treatment of multiple myeloma with high-risk cytogenetics: A consensus of the International Myeloma Working Group. Blood 2016, 127, 2955–2962. [Google Scholar] [CrossRef] [PubMed]
- Manier, S.; Salem, K.Z.; Park, J.; Landau, D.A.; Getz, G.; Ghobrial, I.M. Genomic complexity of multiple myeloma and its clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, R.; Bergsagel, P.L.; Drach, J.; Shaughnessy, J.; Gutierrez, N.; Stewart, A.K.; Morgan, G.; Van Ness, B.; Chesi, M.; Minvielle, S.; et al. International Myeloma Working Group molecular classification of multiple myeloma: Spotlight review. Leukemia 2009, 23, 2210–2221. [Google Scholar] [CrossRef]
- Rajan, A.M.; Rajkumar, S.V. Interpretation of cytogenetic results in multiple myeloma for clinical practice. Blood Cancer J. 2015, 5, e365. [Google Scholar] [CrossRef] [PubMed]
- Robiou du Pont, S.; Cleynen, A.; Fontan, C.; Attal, M.; Munshi, N.; Corre, J.; Avet-Loiseau, H. Genomics of Multiple Myeloma. J. Clin. Oncol. 2017, 35, 963–967. [Google Scholar] [CrossRef]
- Chretien, M.L.; Corre, J.; Lauwers-Cances, V.; Magrangeas, F.; Cleynen, A.; Yon, E.; Hulin, C.; Leleu, X.; Orsini-Piocelle, F.; Blade, J.S.; et al. Understanding the role of hyperdiploidy in myeloma prognosis: Which trisomies really matter? Blood 2015, 126, 2713–2719. [Google Scholar] [CrossRef]
- Barlogie, B.; Pineda-Roman, M.; van Rhee, F.; Haessler, J.; Anaissie, E.; Hollmig, K.; Alsayed, Y.; Waheed, S.; Petty, N.; Epstein, J.; et al. Thalidomide arm of Total Therapy 2 improves complete remission duration and survival in myeloma patients with metaphase cytogenetic abnormalities. Blood 2008, 112, 3115–3121. [Google Scholar] [CrossRef] [PubMed]
- Cavo, M.; Tacchetti, P.; Patriarca, F.; Petrucci, M.T.; Pantani, L.; Galli, M.; Di Raimondo, F.; Crippa, C.; Zamagni, E.; Palumbo, A.; et al. Bortezomib with thalidomide plus dexamethasone compared with thalidomide plus dexamethasone as induction therapy before, and consolidation therapy after, double autologous stem-cell transplantation in newly diagnosed multiple myeloma: A randomised phase 3 study. Lancet 2010, 376, 2075–2085. [Google Scholar] [CrossRef] [PubMed]
- Sonneveld, P.; Goldschmidt, H.; Rosinol, L.; Blade, J.; Lahuerta, J.J.; Cavo, M.; Tacchetti, P.; Zamagni, E.; Attal, M.; Lokhorst, H.M.; et al. Bortezomib-based versus nonbortezomib-based induction treatment before autologous stem-cell transplantation in patients with previously untreated multiple myeloma: A meta-analysis of phase III randomized, controlled trials. J. Clin. Oncol. 2013, 31, 3279–3287. [Google Scholar] [CrossRef] [PubMed]
- Sonneveld, P.; Schmidt-Wolf, I.G.; van der Holt, B.; El Jarari, L.; Bertsch, U.; Salwender, H.; Zweegman, S.; Vellenga, E.; Broyl, A.; Blau, I.W.; et al. Bortezomib induction and maintenance treatment in patients with newly diagnosed multiple myeloma: Results of the randomized phase III HOVON-65/ GMMG-HD4 trial. J. Clin. Oncol. 2012, 30, 2946–2955. [Google Scholar] [CrossRef] [PubMed]
- Boyd, K.D.; Ross, F.M.; Chiecchio, L.; Dagrada, G.P.; Konn, Z.J.; Tapper, W.J.; Walker, B.A.; Wardell, C.P.; Gregory, W.M.; Szubert, A.J.; et al. A novel prognostic model in myeloma based on co-segregating adverse FISH lesions and the ISS: Analysis of patients treated in the MRC Myeloma IX trial. Leukemia 2012, 26, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Decaux, O.; Lode, L.; Magrangeas, F.; Charbonnel, C.; Gouraud, W.; Jezequel, P.; Attal, M.; Harousseau, J.L.; Moreau, P.; Bataille, R.; et al. Prediction of survival in multiple myeloma based on gene expression profiles reveals cell cycle and chromosomal instability signatures in high-risk patients and hyperdiploid signatures in low-risk patients: A study of the Intergroupe Francophone du Myelome. J. Clin. Oncol. 2008, 26, 4798–4805. [Google Scholar] [CrossRef] [PubMed]
- van Beers, E.H.; van Vliet, M.H.; Kuiper, R.; de Best, L.; Anderson, K.C.; Chari, A.; Jagannath, S.; Jakubowiak, A.; Kumar, S.K.; Levy, J.B.; et al. Prognostic Validation of SKY92 and Its Combination With ISS in an Independent Cohort of Patients With Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2017, 17, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Shaughnessy, J.D., Jr.; Zhan, F.; Burington, B.E.; Huang, Y.; Colla, S.; Hanamura, I.; Stewart, J.P.; Kordsmeier, B.; Randolph, C.; Williams, D.R.; et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood 2007, 109, 2276–2284. [Google Scholar] [CrossRef]
- Kuiper, R.; Broyl, A.; de Knegt, Y.; van Vliet, M.H.; van Beers, E.H.; van der Holt, B.; el Jarari, L.; Mulligan, G.; Gregory, W.; Morgan, G.; et al. A gene expression signature for high-risk multiple myeloma. Leukemia 2012, 26, 2406–2413. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Cavo, M.; Sonneveld, P.; Rosinol, L.; Attal, M.; Pezzi, A.; Goldschmidt, H.; Lahuerta, J.J.; Marit, G.; Palumbo, A.; et al. Combination of international scoring system 3, high lactate dehydrogenase, and t(4;14) and/or del(17p) identifies patients with multiple myeloma (MM) treated with front-line autologous stem-cell transplantation at high risk of early MM progression-related death. J. Clin. Oncol. 2014, 32, 2173–2180. [Google Scholar] [CrossRef]
- Neben, K.; Jauch, A.; Bertsch, U.; Heiss, C.; Hielscher, T.; Seckinger, A.; Mors, T.; Muller, N.Z.; Hillengass, J.; Raab, M.S.; et al. Combining information regarding chromosomal aberrations t(4;14) and del(17p13) with the International Staging System classification allows stratification of myeloma patients undergoing autologous stem cell transplantation. Haematologica 2010, 95, 1150–1157. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, A.; Avet-Loiseau, H.; Oliva, S.; Lokhorst, H.M.; Goldschmidt, H.; Rosinol, L.; Richardson, P.; Caltagirone, S.; Lahuerta, J.J.; Facon, T.; et al. Revised International Staging System for Multiple Myeloma: A Report From International Myeloma Working Group. J. Clin. Oncol. 2015, 33, 2863–2869. [Google Scholar] [CrossRef] [PubMed]
- Bolli, N.; Biancon, G.; Moarii, M.; Gimondi, S.; Li, Y.; de Philippis, C.; Maura, F.; Sathiaseelan, V.; Tai, Y.T.; Mudie, L.; et al. Analysis of the genomic landscape of multiple myeloma highlights novel prognostic markers and disease subgroups. Leukemia 2017. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. A high-risk, Double-Hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia 2018. [Google Scholar] [CrossRef] [PubMed]
- Gay, F.; Larocca, A.; Wijermans, P.; Cavallo, F.; Rossi, D.; Schaafsma, R.; Genuardi, M.; Romano, A.; Liberati, A.M.; Siniscalchi, A.; et al. Complete response correlates with long-term progression-free and overall survival in elderly myeloma treated with novel agents: Analysis of 1175 patients. Blood 2011, 117, 3025–3031. [Google Scholar] [CrossRef]
- Lahuerta, J.J.; Mateos, M.V.; Martinez-Lopez, J.; Rosinol, L.; Sureda, A.; de la Rubia, J.; Garcia-Larana, J.; Martinez-Martinez, R.; Hernandez-Garcia, M.T.; Carrera, D.; et al. Influence of pre- and post-transplantation responses on outcome of patients with multiple myeloma: Sequential improvement of response and achievement of complete response are associated with longer survival. J. Clin. Oncol. 2008, 26, 5775–5782. [Google Scholar] [CrossRef]
- Miguel, J.S.; Weisel, K.; Moreau, P.; Lacy, M.; Song, K.; Delforge, M.; Karlin, L.; Goldschmidt, H.; Banos, A.; Oriol, A.; et al. Pomalidomide plus low-dose dexamethasone versus high-dose dexamethasone alone for patients with relapsed and refractory multiple myeloma (MM-003): A randomised, open-label, phase 3 trial. Lancet Oncol. 2013, 14, 1055–1066. [Google Scholar] [CrossRef]
- Barlogie, B.; Anaissie, E.; Haessler, J.; van Rhee, F.; Pineda-Roman, M.; Hollmig, K.; Alsayed, Y.; Epstein, J.; Shaughnessy, J.D., Jr.; Crowley, J. Complete remission sustained 3 years from treatment initiation is a powerful surrogate for extended survival in multiple myeloma. Cancer 2008, 113, 355–359. [Google Scholar] [CrossRef]
- Lahuerta, J.J.; Paiva, B.; Vidriales, M.B.; Cordon, L.; Cedena, M.T.; Puig, N.; Martinez-Lopez, J.; Rosinol, L.; Gutierrez, N.C.; Martin-Ramos, M.L.; et al. Depth of Response in Multiple Myeloma: A Pooled Analysis of Three PETHEMA/GEM Clinical Trials. J. Clin. Oncol. 2017, 35, 2900–2910. [Google Scholar] [CrossRef]
- Haessler, J.; Shaughnessy, J.D., Jr.; Zhan, F.; Crowley, J.; Epstein, J.; van Rhee, F.; Anaissie, E.; Pineda-Roman, M.; Zangari, M.; Hollmig, K.; et al. Benefit of complete response in multiple myeloma limited to high-risk subgroup identified by gene expression profiling. Clin. Cancer Res. 2007, 13, 7073–7079. [Google Scholar] [CrossRef]
- de Tute, R.M.; Rawstron, A.C.; Gregory, W.M.; Child, J.A.; Davies, F.E.; Bell, S.E.; Cook, G.; Szubert, A.J.; Drayson, M.T.; Jackson, G.H.; et al. Minimal residual disease following autologous stem cell transplant in myeloma: Impact on outcome is independent of induction regimen. Haematologica 2016, 101, e69-71. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Harousseau, J.L.; Durie, B.; Anderson, K.C.; Dimopoulos, M.; Kyle, R.; Blade, J.; Richardson, P.; Orlowski, R.; Siegel, D.; et al. Consensus recommendations for the uniform reporting of clinical trials: Report of the International Myeloma Workshop Consensus Panel 1. Blood 2011, 117, 4691–4695. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lopez, J.; Lahuerta, J.J.; Pepin, F.; Gonzalez, M.; Barrio, S.; Ayala, R.; Puig, N.; Montalban, M.A.; Paiva, B.; Weng, L.; et al. Prognostic value of deep sequencing method for minimal residual disease detection in multiple myeloma. Blood 2014, 123, 3073–3079. [Google Scholar] [CrossRef] [PubMed]
- Paiva, B.; van Dongen, J.J.; Orfao, A. New criteria for response assessment: Role of minimal residual disease in multiple myeloma. Blood 2015, 125, 3059–3068. [Google Scholar] [CrossRef] [PubMed]
- Avet-Loiseau, H. Minimal Residual Disease by Next-Generation Sequencing: Pros and Cons. Am. Soc. Clin. Oncol. Educ. Book 2016, 35, e425–e430. [Google Scholar] [CrossRef] [PubMed]
- Zappasodi, P.; Marbello, L.; Borlenghi, E.; Fumagalli, M.; Bernardi, M.; Fracchiolla, N.; Mancini, V.; Da Via, M.; Ravano, E.; Cerqui, E.; et al. Molecular remission at the end of treatment is a necessary goal for a good outcome in ELN favorable-risk acute myeloid leukemia: A real-life analysis on 201 patients by the Rete Ematologica Lombarda network. Ann. Hematol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Paiva, B.; Anderson, K.C.; Durie, B.; Landgren, O.; Moreau, P.; Munshi, N.; Lonial, S.; Blade, J.; Mateos, M.V.; et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016, 17, e328–e346. [Google Scholar] [CrossRef]
- Faham, M.; Zheng, J.; Moorhead, M.; Carlton, V.E.; Stow, P.; Coustan-Smith, E.; Pui, C.H.; Campana, D. Deep-sequencing approach for minimal residual disease detection in acute lymphoblastic leukemia. Blood 2012, 120, 5173–5180. [Google Scholar] [CrossRef]
- Martinez-Lopez, J.; Sanchez-Vega, B.; Barrio, S.; Cuenca, I.; Ruiz-Heredia, Y.; Alonso, R.; Rapado, I.; Marin, C.; Cedena, M.T.; Paiva, B.; et al. Analytical and clinical validation of a novel in-house deep-sequencing method for minimal residual disease monitoring in a phase II trial for multiple myeloma. Leukemia 2017, 31, 1446–1449. [Google Scholar] [CrossRef]
- Alaterre, E.; Raimbault, S.; Goldschmidt, H.; Bouhya, S.; Requirand, G.; Robert, N.; Boireau, S.; Seckinger, A.; Hose, D.; Klein, B.; et al. CD24, CD27, CD36 and CD302 gene expression for outcome prediction in patients with multiple myeloma. Oncotarget 2017, 8, 98931–98944. [Google Scholar] [CrossRef]
- Flores-Montero, J.; Sanoja-Flores, L.; Paiva, B.; Puig, N.; Garcia-Sanchez, O.; Bottcher, S.; van der Velden, V.H.J.; Perez-Moran, J.J.; Vidriales, M.B.; Garcia-Sanz, R.; et al. Next Generation Flow for highly sensitive and standardized detection of minimal residual disease in multiple myeloma. Leukemia 2017, 31, 2094–2103. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Zamagni, E. MRD in multiple myeloma: More questions than answers? Blood Cancer J. 2017, 7, 639. [Google Scholar] [CrossRef]
- Cavo, M.; Terpos, E.; Nanni, C.; Moreau, P.; Lentzsch, S.; Zweegman, S.; Hillengass, J.; Engelhardt, M.; Usmani, S.Z.; Vesole, D.H.; et al. Role of (18)F-FDG PET/CT in the diagnosis and management of multiple myeloma and other plasma cell disorders: A consensus statement by the International Myeloma Working Group. Lancet Oncol. 2017, 18, e206–e217. [Google Scholar] [CrossRef]
- Pandit-Taskar, N. Functional Imaging Methods for Assessment of Minimal Residual Disease in Multiple Myeloma: Current Status and Novel ImmunoPET Based Methods. Semin. Hematol. 2018, 55, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Rasche, L.; Alapat, D.; Kumar, M.; Gershner, G.; McDonald, J.; Wardell, C.P.; Samant, R.; Van Hemert, R.; Epstein, J.; Williams, A.F.; et al. Combination of flow cytometry and functional imaging for monitoring of residual disease in myeloma. Leukemia 2018. [Google Scholar] [CrossRef]
- Kircher, S.; Stolzenburg, A.; Kortuem, K.M.; Kircher, M.; Da Via, M.; Samnick, S.; Buck, A.; Einsele, H.; Rosenwald, A.; Lapa, C. Hexokinase-2 Expression in MET-positive FDG-negative Multiple Myeloma. J. Nucl. Med. 2018. [Google Scholar] [CrossRef]
- Rasche, L.; Angtuaco, E.; McDonald, J.E.; Buros, A.; Stein, C.; Pawlyn, C.; Thanendrarajan, S.; Schinke, C.; Samant, R.; Yaccoby, S.; et al. Low expression of hexokinase-2 is associated with false-negative FDG-positron emission tomography in multiple myeloma. Blood 2017, 130, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Lapa, C.; Knop, S.; Schreder, M.; Rudelius, M.; Knott, M.; Jorg, G.; Samnick, S.; Herrmann, K.; Buck, A.K.; Einsele, H.; et al. 11C-Methionine-PET in Multiple Myeloma: Correlation with Clinical Parameters and Bone Marrow Involvement. Theranostics 2016, 6, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Lapa, C.; Schreder, M.; Schirbel, A.; Samnick, S.; Kortum, K.M.; Herrmann, K.; Kropf, S.; Einsele, H.; Buck, A.K.; Wester, H.J.; et al. [(68)Ga]Pentixafor-PET/CT for imaging of chemokine receptor CXCR4 expression in multiple myeloma—Comparison to [(18)F]FDG and laboratory values. Theranostics 2017, 7, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Lapa, C.; Herrmann, K.; Schirbel, A.; Hanscheid, H.; Luckerath, K.; Schottelius, M.; Kircher, M.; Werner, R.A.; Schreder, M.; Samnick, S.; et al. CXCR4-directed endoradiotherapy induces high response rates in extramedullary relapsed Multiple Myeloma. Theranostics 2017, 7, 1589–1597. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Leong, T.; Roche, P.C.; Fonseca, R.; Dispenzieri, A.; Lacy, M.Q.; Lust, J.A.; Witzig, T.E.; Kyle, R.A.; Gertz, M.A.; et al. Prognostic value of bone marrow angiogenesis in multiple myeloma. Clin. Cancer Res. 2000, 6, 3111–3116. [Google Scholar] [PubMed]
- Yu, C.W.; Liang, X.; Lipsky, S.; Karaaslan, C.; Kozakewich, H.; Hotamisligil, G.S.; Bischoff, J.; Cataltepe, S. Dual role of fatty acid-binding protein 5 on endothelial cell fate: A potential link between lipid metabolism and angiogenic responses. Angiogenesis 2016, 19, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yang, Y.; Ding, Y.; Tang, X.; Sun, Z. Impacts of survivin and caspase-3 on apoptosis and angiogenesis in oral cancer. Oncol. Lett. 2017, 14, 3774–3779. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yan, Q.; Hu, M.; Qin, D.; Feng, Z. Effect of AURKA Gene Expression Knockdown on Angiogenesis and Tumorigenesis of Human Ovarian Cancer Cell Lines. Target Oncol. 2016, 11, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Migneco, G.; Whitaker-Menezes, D.; Chiavarina, B.; Castello-Cros, R.; Pavlides, S.; Pestell, R.G.; Fatatis, A.; Flomenberg, N.; Tsirigos, A.; Howell, A.; et al. Glycolytic cancer associated fibroblasts promote breast cancer tumor growth, without a measurable increase in angiogenesis: Evidence for stromal-epithelial metabolic coupling. Cell Cycle 2010, 9, 2412–2422. [Google Scholar] [CrossRef] [PubMed]
- Berardi, S.; Caivano, A.; Ria, R.; Nico, B.; Savino, R.; Terracciano, R.; De Tullio, G.; Ferrucci, A.; De Luisi, A.; Moschetta, M.; et al. Four proteins governing overangiogenic endothelial cell phenotype in patients with multiple myeloma are plausible therapeutic targets. Oncogene 2012, 31, 2258–2269. [Google Scholar] [CrossRef] [PubMed]
- Saltarella, I.; Morabito, F.; Giuliani, N.; Terragna, C.; Omede, P.; Palumbo, A.; Bringhen, S.; De Paoli, L.; Martino, E.; Larocca, A.; et al. Prognostic or predictive value of circulating cytokines and angiogenic factors for initial treatment of multiple myeloma in the GIMEMA MM0305 randomized controlled trial. J. Hematol. Oncol. 2019, 12, 4. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Witzig, T.E.; Timm, M.; Haug, J.; Wellik, L.; Kimlinger, T.K.; Greipp, P.R.; Rajkumar, S.V. Bone marrow angiogenic ability and expression of angiogenic cytokines in myeloma: Evidence favoring loss of marrow angiogenesis inhibitory activity with disease progression. Blood 2004, 104, 1159–1165. [Google Scholar] [CrossRef]
- Croucher, D.C.; Chesi, M.; Li, Z.; Garbitt, V.M.; Sharik, M.E.; Waller, D.; Sebag, M.; Bergsagel, P.L.; Pugh, T.J.; Trudel, S. A single-cell transcriptional analysis of tumour cells and the immune microenvironment during disease evolution in a transgenic mouse model of myeloma. In Proceedings of the Blood, American Society of Hematology Annual Meeting, San Diego, CA, USA, 1–4 December 2018. [Google Scholar]
- Seymour, F.; Cavenagh, J.; Gribben, J.G. Characterising the Immunological Microenvironment in Newly Diagnosed Multiple Myeloma Bone Marrow By Time of Flight Cytometry Reveals Abnormalities in Antigen Presenting and Effector Lymphocyte Populations with Prognostic Significance. In Proceedings of the Blood, American Society of Hematology Annual Meeting, San Diego, CA, USA, 1–4 December 2018. [Google Scholar]
- Hose, D.; Moreaux, J.; Meissner, T.; Seckinger, A.; Goldschmidt, H.; Benner, A.; Mahtouk, K.; Hillengass, J.; Reme, T.; De Vos, J.; et al. Induction of angiogenesis by normal and malignant plasma cells. Blood 2009, 114, 128–143. [Google Scholar] [CrossRef]
- Leone, P.; Di Lernia, G.; Solimando, A.G.; Cicco, S.; Saltarella, I.; Lamanuzzi, A.; Ria, R.; Frassanito, M.A.; Ponzoni, M.; Ditonno, P.; et al. Bone marrow endothelial cells sustain a tumor-specific CD8(+) T cell subset with suppressive function in myeloma patients. Oncoimmunology 2019, 8, e1486949. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Nico, B.; Vacca, A. Multiple myeloma as a model for the role of bone marrow niches in the control of angiogenesis. Int. Rev. Cell Mol. Biol. 2015, 314, 259–282. [Google Scholar] [CrossRef] [PubMed]
- Moschetta, M.; Mishima, Y.; Kawano, Y.; Manier, S.; Paiva, B.; Palomera, L.; Aljawai, Y.; Calcinotto, A.; Unitt, C.; Sahin, I.; et al. Targeting vasculogenesis to prevent progression in multiple myeloma. Leukemia 2016, 30, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Podar, K.; Anderson, K.C. The pathophysiologic role of VEGF in hematologic malignancies: Therapeutic implications. Blood 2005, 105, 1383–1395. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.M.; Figg, W.D. Angiogenesis inhibitors: Current strategies and future prospects. CA Cancer J. Clin. 2010, 60, 222–243. [Google Scholar] [CrossRef]
- Dimopoulos, M.; Spencer, A.; Attal, M.; Prince, H.M.; Harousseau, J.L.; Dmoszynska, A.; San Miguel, J.; Hellmann, A.; Facon, T.; Foa, R.; et al. Lenalidomide plus dexamethasone for relapsed or refractory multiple myeloma. N. Engl. J. Med. 2007, 357, 2123–2132. [Google Scholar] [CrossRef]
- Ghobrial, I.M.; Detappe, A.; Anderson, K.C.; Steensma, D.P. The bone-marrow niche in MDS and MGUS: Implications for AML and MM. Nat. Rev. Clin. Oncol. 2018, 15, 219–233. [Google Scholar] [CrossRef]
- Lopuch, S.; Kawalec, P.; Wisniewska, N. Effectiveness of targeted therapy as monotherapy or combined therapy in patients with relapsed or refractory multiple myeloma: A systematic review and meta-analysis. Hematology 2015, 20, 1–10. [Google Scholar] [CrossRef]
- Mateos, M.V.; Hernandez, M.T.; Giraldo, P.; de la Rubia, J.; de Arriba, F.; Lopez Corral, L.; Rosinol, L.; Paiva, B.; Palomera, L.; Bargay, J.; et al. Lenalidomide plus dexamethasone for high-risk smoldering multiple myeloma. N. Engl. J. Med. 2013, 369, 438–447. [Google Scholar] [CrossRef]
- Mo, H.N.; Liu, P. Targeting MET in cancer therapy. Chronic Dis. Transl. Med. 2017, 3, 148–153. [Google Scholar] [CrossRef]
- Singhal, S.; Mehta, J.; Desikan, R.; Ayers, D.; Roberson, P.; Eddlemon, P.; Munshi, N.; Anaissie, E.; Wilson, C.; Dhodapkar, M.; et al. Antitumor activity of thalidomide in refractory multiple myeloma. N. Engl. J. Med. 1999, 341, 1565–1571. [Google Scholar] [CrossRef]
- Zagouri, F.; Terpos, E.; Kastritis, E.; Dimopoulos, M.A. Emerging antibodies for the treatment of multiple myeloma. Expert Opin. Emerg. Drugs 2016, 21, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Ria, R.; Solimando, A.; Melaccio, A.; Sportelli, A.; Vacca, A. Angiogenesis and Antiangiogenesis in Multiple Myeloma. Available online: https://www.intechopen.com/books/update-on-multiple-myeloma/angiogenesis-and-antiangiogenesis-in-multiple-myeloma (accessed on 30 May 2019).
- Kawano, Y.; Moschetta, M.; Manier, S.; Glavey, S.; Gorgun, G.T.; Roccaro, A.M.; Anderson, K.C.; Ghobrial, I.M. Targeting the bone marrow microenvironment in multiple myeloma. Immunol. Rev. 2015, 263, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Ghobrial, I.M. Myeloma as a model for the process of metastasis: Implications for therapy. Blood 2012, 120, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The genetic architecture of multiple myeloma. Nat. Rev. Cancer 2012, 12, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Musto, P.; Simeon, V.; Todoerti, K.; Neri, A. Primary Plasma Cell Leukemia: Identity Card 2016. Curr. Treat. Options Oncol. 2016, 17, 19. [Google Scholar] [CrossRef] [PubMed]
- van de Donk, N.W.; Lokhorst, H.M.; Anderson, K.C.; Richardson, P.G. How I treat plasma cell leukemia. Blood 2012, 120, 2376–2389. [Google Scholar] [CrossRef] [PubMed]
- Blade, J.; Fernandez de Larrea, C.; Rosinol, L.; Cibeira, M.T.; Jimenez, R.; Powles, R. Soft-tissue plasmacytomas in multiple myeloma: Incidence, mechanisms of extramedullary spread, and treatment approach. J. Clin. Oncol. 2011, 29, 3805–3812. [Google Scholar] [CrossRef] [PubMed]
- Varettoni, M.; Corso, A.; Pica, G.; Mangiacavalli, S.; Pascutto, C.; Lazzarino, M. Incidence, presenting features and outcome of extramedullary disease in multiple myeloma: A longitudinal study on 1003 consecutive patients. Ann. Oncol. 2010, 21, 325–330. [Google Scholar] [CrossRef]
- Fernandez de Larrea, C.; Kyle, R.A.; Durie, B.G.; Ludwig, H.; Usmani, S.; Vesole, D.H.; Hajek, R.; San Miguel, J.F.; Sezer, O.; Sonneveld, P.; et al. Plasma cell leukemia: Consensus statement on diagnostic requirements, response criteria and treatment recommendations by the International Myeloma Working Group. Leukemia 2013, 27, 780–791. [Google Scholar] [CrossRef]
- Jurczyszyn, A.; Grzasko, N.; Gozzetti, A.; Czepiel, J.; Cerase, A.; Hungria, V.; Crusoe, E.; Silva Dias, A.L.; Vij, R.; Fiala, M.A.; et al. Central nervous system involvement by multiple myeloma: A multi-institutional retrospective study of 172 patients in daily clinical practice. Am. J. Hematol. 2016, 91, 575–580. [Google Scholar] [CrossRef]
- Varettoni, M.; Marchioni, E.; Bonfichi, M.; Picchiecchio, A.; Arcaini, L.; Arbasino, C.; Gotti, M.; Da Via, M.; Delmonte, M.; Sciarra, R.; et al. Successful treatment with Rituximab and Bendamustine in a patient with newly diagnosed Waldenstrom’s Macroglobulinemia complicated by Bing-Neel syndrome. Am. J. Hematol. 2015, 90, E152–E153. [Google Scholar] [CrossRef] [PubMed]
- Zamagni, E.; Patriarca, F.; Nanni, C.; Zannetti, B.; Englaro, E.; Pezzi, A.; Tacchetti, P.; Buttignol, S.; Perrone, G.; Brioli, A.; et al. Prognostic relevance of 18-F FDG PET/CT in newly diagnosed multiple myeloma patients treated with up-front autologous transplantation. Blood 2011, 118, 5989–5995. [Google Scholar] [CrossRef] [PubMed]
- Usmani, S.Z.; Heuck, C.; Mitchell, A.; Szymonifka, J.; Nair, B.; Hoering, A.; Alsayed, Y.; Waheed, S.; Haider, S.; Restrepo, A.; et al. Extramedullary disease portends poor prognosis in multiple myeloma and is over-represented in high-risk disease even in the era of novel agents. Haematologica 2012, 97, 1761–1767. [Google Scholar] [CrossRef] [PubMed]
- Pour, L.; Sevcikova, S.; Greslikova, H.; Kupska, R.; Majkova, P.; Zahradova, L.; Sandecka, V.; Adam, Z.; Krejci, M.; Kuglik, P.; et al. Soft-tissue extramedullary multiple myeloma prognosis is significantly worse in comparison to bone-related extramedullary relapse. Haematologica 2014, 99, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Touzeau, C.; Moreau, P. How I treat extramedullary myeloma. Blood 2016, 127, 971–976. [Google Scholar] [CrossRef]
- Andrulis, M.; Lehners, N.; Capper, D.; Penzel, R.; Heining, C.; Huellein, J.; Zenz, T.; von Deimling, A.; Schirmacher, P.; Ho, A.D.; et al. Targeting the BRAF V600E mutation in multiple myeloma. Cancer Discov. 2013, 3, 862–869. [Google Scholar] [CrossRef]
- Di Marzo, L.; Desantis, V.; Solimando, A.G.; Ruggieri, S.; Annese, T.; Nico, B.; Fumarulo, R.; Vacca, A.; Frassanito, M.A. Microenvironment drug resistance in multiple myeloma: Emerging new players. Oncotarget 2016, 7, 60698–60711. [Google Scholar] [CrossRef]
- Egan, J.B.; Kortuem, K.M.; Kurdoglu, A.; Izatt, T.; Aldrich, J.; Reiman, R.; Phillips, L.; Baker, A.; Shi, C.X.; Schmidt, J.; et al. Extramedullary myeloma whole genome sequencing reveals novel mutations in Cereblon, proteasome subunit G2 and the glucocorticoid receptor in multi drug resistant disease. Br. J. Haematol. 2013, 161, 748–751. [Google Scholar] [CrossRef]
- Roccaro, A.M.; Mishima, Y.; Sacco, A.; Moschetta, M.; Tai, Y.T.; Shi, J.; Zhang, Y.; Reagan, M.R.; Huynh, D.; Kawano, Y.; et al. CXCR4 Regulates Extra-Medullary Myeloma through Epithelial-Mesenchymal-Transition-like Transcriptional Activation. Cell Rep. 2015, 12, 622–635. [Google Scholar] [CrossRef]
- Fonseca, R.; Abouzaid, S.; Bonafede, M.; Cai, Q.; Parikh, K.; Cosler, L.; Richardson, P. Trends in overall survival and costs of multiple myeloma, 2000–2014. Leukemia 2017, 31, 1915–1921. [Google Scholar] [CrossRef]
- Barlogie, B.; Mitchell, A.; van Rhee, F.; Epstein, J.; Morgan, G.J.; Crowley, J. Curing myeloma at last: Defining criteria and providing the evidence. Blood 2014, 124, 3043–3051. [Google Scholar] [CrossRef] [PubMed]
- Pawlyn, C.; Morgan, G.J. Evolutionary biology of high-risk multiple myeloma. Nat. Rev. Cancer 2017, 17, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Rasche, L.; Chavan, S.S.; Stephens, O.W.; Patel, P.H.; Tytarenko, R.; Ashby, C.; Bauer, M.; Stein, C.; Deshpande, S.; Wardell, C.; et al. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Nat. Commun. 2017, 8, 268. [Google Scholar] [CrossRef] [PubMed]
- Kortuem, K.M.; Braggio, E.; Bruins, L.; Barrio, S.; Shi, C.S.; Zhu, Y.X.; Tibes, R.; Viswanatha, D.; Votruba, P.; Ahmann, G.; et al. Panel sequencing for clinically oriented variant screening and copy number detection in 142 untreated multiple myeloma patients. Blood Cancer J. 2016, 6, e397. [Google Scholar] [CrossRef] [PubMed]
- Melchor, L.; Brioli, A.; Wardell, C.P.; Murison, A.; Potter, N.E.; Kaiser, M.F.; Fryer, R.A.; Johnson, D.C.; Begum, D.B.; Hulkki Wilson, S.; et al. Single-cell genetic analysis reveals the composition of initiating clones and phylogenetic patterns of branching and parallel evolution in myeloma. Leukemia 2014, 28, 1705–1715. [Google Scholar] [CrossRef]
- Rasche, L.; Angtuaco, E.J.; Alpe, T.L.; Gershner, G.H.; McDonald, J.E.; Samant, R.S.; Kumar, M.; Van Hemert, R.; Epstein, J.; Deshpande, S.; et al. The presence of large focal lesions is a strong independent prognostic factor in multiple myeloma. Blood 2018, 132, 59–66. [Google Scholar] [CrossRef]
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.C.; Van Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 2014, 5, 2997. [Google Scholar] [CrossRef]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef]
- Walker, B.A.; Wardell, C.P.; Melchor, L.; Hulkki, S.; Potter, N.E.; Johnson, D.C.; Fenwick, K.; Kozarewa, I.; Gonzalez, D.; Lord, C.J.; et al. Intraclonal heterogeneity and distinct molecular mechanisms characterize the development of t(4;14) and t(11;14) myeloma. Blood 2012, 120, 1077–1086. [Google Scholar] [CrossRef]
- Dietrich, S.; Glimm, H.; Andrulis, M.; von Kalle, C.; Ho, A.D.; Zenz, T. BRAF inhibition in refractory hairy-cell leukemia. N. Engl. J. Med. 2012, 366, 2038–2040. [Google Scholar] [CrossRef]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Raab, M.S.; Lehners, N.; Xu, J.; Ho, A.D.; Schirmacher, P.; Goldschmidt, H.; Andrulis, M. Spatially divergent clonal evolution in multiple myeloma: Overcoming resistance to BRAF inhibition. Blood 2016, 127, 2155–2157. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Corral, L.; Gutierrez, N.C.; Vidriales, M.B.; Mateos, M.V.; Rasillo, A.; Garcia-Sanz, R.; Paiva, B.; San Miguel, J.F. The progression from MGUS to smoldering myeloma and eventually to multiple myeloma involves a clonal expansion of genetically abnormal plasma cells. Clin. Cancer Res. 2011, 17, 1692–1700. [Google Scholar] [CrossRef] [PubMed]
- Barrio, S.; DaVia, M.; Bruins, L.; Stuhmer, T.; Steinbrunn, T.; Bittrich, M.; Einsele, H.; Stewart, A.K.; Braggio, E.; Kortum, K.M. Protocol for M(3)P: A Comprehensive and Clinical Oriented Targeted Sequencing Panel for Routine Molecular Analysis in Multiple Myeloma. Methods Mol. Biol. 2018, 1792, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Kortum, K.M.; Mai, E.K.; Hanafiah, N.H.; Shi, C.X.; Zhu, Y.X.; Bruins, L.; Barrio, S.; Jedlowski, P.; Merz, M.; Xu, J.; et al. Targeted sequencing of refractory myeloma reveals a high incidence of mutations in CRBN and Ras pathway genes. Blood 2016, 128, 1226–1233. [Google Scholar] [CrossRef] [PubMed]
- Meads, M.B.; Hazlehurst, L.A.; Dalton, W.S. The bone marrow microenvironment as a tumor sanctuary and contributor to drug resistance. Clin. Cancer Res. 2008, 14, 2519–2526. [Google Scholar] [CrossRef] [PubMed]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.R.; Espitia, C.M.; Zhao, W.; Wendlandt, E.; Tricot, G.; Zhan, F.; Carew, J.S.; Nawrocki, S.T. Junctional adhesion molecule-A is overexpressed in advanced multiple myeloma and determines response to oncolytic reovirus. Oncotarget 2015, 6, 41275–41289. [Google Scholar] [CrossRef] [PubMed]
- Solimando, A.G.; Brandl, A.; Mattenheimer, K.; Graf, C.; Ritz, M.; Ruckdeschel, A.; Stuhmer, T.; Mokhtari, Z.; Rudelius, M.; Dotterweich, J.; et al. JAM-A as a prognostic factor and new therapeutic target in multiple myeloma. Leukemia 2018, 32, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Bjorklund, C.C.; Baladandayuthapani, V.; Lin, H.Y.; Jones, R.J.; Kuiatse, I.; Wang, H.; Yang, J.; Shah, J.J.; Thomas, S.K.; Wang, M.; et al. Evidence of a role for CD44 and cell adhesion in mediating resistance to lenalidomide in multiple myeloma: Therapeutic implications. Leukemia 2014, 28, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Podar, K.; Zimmerhackl, A.; Fulciniti, M.; Tonon, G.; Hainz, U.; Tai, Y.T.; Vallet, S.; Halama, N.; Jager, D.; Olson, D.L.; et al. The selective adhesion molecule inhibitor Natalizumab decreases multiple myeloma cell growth in the bone marrow microenvironment: Therapeutic implications. Br. J. Haematol. 2011, 155, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Nemkov, T.; Sun, K.; Reisz, J.A.; Song, A.; Yoshida, T.; Dunham, A.; Wither, M.J.; Francis, R.O.; Roach, R.C.; Dzieciatkowska, M.; et al. Hypoxia modulates the purine salvage pathway and decreases red blood cell and supernatant levels of hypoxanthine during refrigerated storage. Haematologica 2018, 103, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Rudelius, M.; Rosenfeldt, M.T.; Leich, E.; Rauert-Wunderlich, H.; Solimando, A.G.; Beilhack, A.; Ott, G.; Rosenwald, A. Inhibition of focal adhesion kinase overcomes resistance of mantle cell lymphoma to ibrutinib in the bone marrow microenvironment. Haematologica 2018, 103, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Andrade, V.C.; Vettore, A.L.; Panepucci, R.A.; Almeida, M.S.; Yamamoto, M.; De Carvalho, F.; Caballero, O.L.; Zago, M.A.; Colleoni, G.W. Number of expressed cancer/testis antigens identifies focal adhesion pathway genes as possible targets for multiple myeloma therapy. Leuk. Lymphoma 2010, 51, 1543–1549. [Google Scholar] [CrossRef] [PubMed]
- Barker, H.F.; Ball, J.; Drew, M.; Hamilton, M.S.; Franklin, I.M. The role of adhesion molecules in multiple myeloma. Leuk. Lymphoma 1992, 8, 189–196. [Google Scholar] [CrossRef]
- Hazlehurst, L.A.; Damiano, J.S.; Buyuksal, I.; Pledger, W.J.; Dalton, W.S. Adhesion to fibronectin via beta1 integrins regulates p27kip1 levels and contributes to cell adhesion mediated drug resistance (CAM-DR). Oncogene 2000, 19, 4319–4327. [Google Scholar] [CrossRef] [PubMed]
- Katz, B.Z. Adhesion molecules--The lifelines of multiple myeloma cells. Semin. Cancer Biol. 2010, 20, 186–195. [Google Scholar] [CrossRef]
- Muz, B.; Azab, F.; de la Puente, P.; Rollins, S.; Alvarez, R.; Kawar, Z.; Azab, A.K. Inhibition of P-Selectin and PSGL-1 Using Humanized Monoclonal Antibodies Increases the Sensitivity of Multiple Myeloma Cells to Bortezomib. Biomed. Res. Int. 2015, 2015, 417586. [Google Scholar] [CrossRef]
- Natoni, A.; Smith, T.A.G.; Keane, N.; McEllistrim, C.; Connolly, C.; Jha, A.; Andrulis, M.; Ellert, E.; Raab, M.S.; Glavey, S.V.; et al. E-selectin ligands recognised by HECA452 induce drug resistance in myeloma, which is overcome by the E-selectin antagonist, GMI-1271. Leukemia 2017, 31, 2642–2651. [Google Scholar] [CrossRef]
- Neri, P.; Ren, L.; Azab, A.K.; Brentnall, M.; Gratton, K.; Klimowicz, A.C.; Lin, C.; Duggan, P.; Tassone, P.; Mansoor, A.; et al. Integrin beta7-mediated regulation of multiple myeloma cell adhesion, migration, and invasion. Blood 2011, 117, 6202–6213. [Google Scholar] [CrossRef]
- Noborio-Hatano, K.; Kikuchi, J.; Takatoku, M.; Shimizu, R.; Wada, T.; Ueda, M.; Nobuyoshi, M.; Oh, I.; Sato, K.; Suzuki, T.; et al. Bortezomib overcomes cell-adhesion-mediated drug resistance through downregulation of VLA-4 expression in multiple myeloma. Oncogene 2009, 28, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Hawley, R.G.; Kodaka, M.; Okuno, H. Significance of VLA-4-VCAM-1 interaction and CD44 for transendothelial invasion in a bone marrow metastatic myeloma model. Clin. Exp. Metastasis 1999, 17, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Ria, R.; Vacca, A.; Ribatti, D.; Di Raimondo, F.; Merchionne, F.; Dammacco, F. Alpha(v)beta(3) integrin engagement enhances cell invasiveness in human multiple myeloma. Haematologica 2002, 87, 836–845. [Google Scholar] [PubMed]
- Schmidmaier, R.; Mandl-Weber, S.; Gaul, L.; Baumann, P.; Bumeder, I.; Straka, C.; Emmerich, B. Inhibition of lymphocyte function associated antigen 1 by LFA878 induces apoptosis in multiple myeloma cells and is associated with downregulation of the focal adhesion kinase/phosphatidylinositol 3 kinase/Akt pathway. Int. J. Oncol. 2007, 31, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Vacca, A.; Di Loreto, M.; Ribatti, D.; Di Stefano, R.; Gadaleta-Caldarola, G.; Iodice, G.; Caloro, D.; Dammacco, F. Bone marrow of patients with active multiple myeloma: Angiogenesis and plasma cell adhesion molecules LFA-1, VLA-4, LAM-1, and CD44. Am. J. Hematol. 1995, 50, 9–14. [Google Scholar] [CrossRef]
- Waldschmidt, J.M.; Simon, A.; Wider, D.; Muller, S.J.; Follo, M.; Ihorst, G.; Decker, S.; Lorenz, J.; Chatterjee, M.; Azab, A.K.; et al. CXCL12 and CXCR7 are relevant targets to reverse cell adhesion-mediated drug resistance in multiple myeloma. Br. J. Haematol. 2017, 179, 36–49. [Google Scholar] [CrossRef]
- Yao, Q.; Morgan, G.J.; Chim, C.S. Distinct promoter methylation profile reveals spatial epigenetic heterogeneity in 2 myeloma patients with multifocal extramedullary relapses. Clin. Epigenet. 2018, 10, 158. [Google Scholar] [CrossRef]
- Yin, L.; Tagde, A.; Gali, R.; Tai, Y.T.; Hideshima, T.; Anderson, K.; Avigan, D.; Kufe, D. MUC1-C is a target in lenalidomide resistant multiple myeloma. Br. J. Haematol. 2017, 178, 914–926. [Google Scholar] [CrossRef]
- Yoshida, T.; Ri, M.; Kinoshita, S.; Narita, T.; Totani, H.; Ashour, R.; Ito, A.; Kusumoto, S.; Ishida, T.; Komatsu, H.; et al. Low expression of neural cell adhesion molecule, CD56, is associated with low efficacy of bortezomib plus dexamethasone therapy in multiple myeloma. PLoS ONE 2018, 13, e0196780. [Google Scholar] [CrossRef]
- van de Donk, N.; Usmani, S.Z. CD38 Antibodies in Multiple Myeloma: Mechanisms of Action and Modes of Resistance. Front. Immunol. 2018, 9, 2134. [Google Scholar] [CrossRef]
- Kronke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Braggio, E.; Kortum, K.M.; Stewart, A.K. SnapShot: Multiple Myeloma. Cancer Cell 2015, 28, 678. [Google Scholar] [CrossRef] [PubMed]
- Kronke, J.; Kuchenbauer, F.; Kull, M.; Teleanu, V.; Bullinger, L.; Bunjes, D.; Greiner, A.; Kolmus, S.; Kopff, S.; Schreder, M.; et al. IKZF1 expression is a prognostic marker in newly diagnosed standard-risk multiple myeloma treated with lenalidomide and intensive chemotherapy: A study of the German Myeloma Study Group (DSMM). Leukemia 2017, 31, 1363–1367. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.X.; Braggio, E.; Shi, C.X.; Kortuem, K.M.; Bruins, L.A.; Schmidt, J.E.; Chang, X.B.; Langlais, P.; Luo, M.; Jedlowski, P.; et al. Identification of cereblon-binding proteins and relationship with response and survival after IMiDs in multiple myeloma. Blood 2014, 124, 536–545. [Google Scholar] [CrossRef]
- Eichner, R.; Heider, M.; Fernandez-Saiz, V.; van Bebber, F.; Garz, A.K.; Lemeer, S.; Rudelius, M.; Targosz, B.S.; Jacobs, L.; Knorn, A.M.; et al. Immunomodulatory drugs disrupt the cereblon-CD147-MCT1 axis to exert antitumor activity and teratogenicity. Nat. Med. 2016, 22, 735–743. [Google Scholar] [CrossRef]
- Herviou, L.; Kassambara, A.; Boireau, S.; Robert, N.; Requirand, G.; Muller-Tidow, C.; Vincent, L.; Seckinger, A.; Goldschmidt, H.; Cartron, G.; et al. PRC2 targeting is a therapeutic strategy for EZ score defined high-risk multiple myeloma patients and overcome resistance to IMiDs. Clin. Epigenet. 2018, 10, 121. [Google Scholar] [CrossRef]
- Ettari, R.; Zappala, M.; Grasso, S.; Musolino, C.; Innao, V.; Allegra, A. Immunoproteasome-selective and non-selective inhibitors: A promising approach for the treatment of multiple myeloma. Pharmacol. Ther. 2018, 182, 176–192. [Google Scholar] [CrossRef]
- Oerlemans, R.; Franke, N.E.; Assaraf, Y.G.; Cloos, J.; van Zantwijk, I.; Berkers, C.R.; Scheffer, G.L.; Debipersad, K.; Vojtekova, K.; Lemos, C.; et al. Molecular basis of bortezomib resistance: Proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood 2008, 112, 2489–2499. [Google Scholar] [CrossRef]
- Barrio, S.; Stuhmer, T.; Da-Via, M.; Barrio-Garcia, C.; Lehners, N.; Besse, A.; Cuenca, I.; Garitano-Trojaola, A.; Fink, S.; Leich, E.; et al. Spectrum and functional validation of PSMB5 mutations in multiple myeloma. Leukemia 2018. [Google Scholar] [CrossRef]
- Mitra, A.K.; Mukherjee, U.K.; Harding, T.; Jang, J.S.; Stessman, H.; Li, Y.; Abyzov, A.; Jen, J.; Kumar, S.; Rajkumar, V.; et al. Single-cell analysis of targeted transcriptome predicts drug sensitivity of single cells within human myeloma tumors. Leukemia 2016, 30, 1094–1102. [Google Scholar] [CrossRef]
- Shi, C.X.; Kortum, K.M.; Zhu, Y.X.; Bruins, L.A.; Jedlowski, P.; Votruba, P.G.; Luo, M.; Stewart, R.A.; Ahmann, J.; Braggio, E.; et al. CRISPR Genome-Wide Screening Identifies Dependence on the Proteasome Subunit PSMC6 for Bortezomib Sensitivity in Multiple Myeloma. Mol. Cancer Ther. 2017, 16, 2862–2870. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.D.; Baladandayuthapani, V.; Lin, H.; Mulligan, G.; Li, B.; Esseltine, D.W.; Qi, L.; Xu, J.; Hunziker, W.; Barlogie, B.; et al. Tight Junction Protein 1 Modulates Proteasome Capacity and Proteasome Inhibitor Sensitivity in Multiple Myeloma via EGFR/JAK1/STAT3 Signaling. Cancer Cell 2016, 29, 639–652. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, A.; Moschetta, M.; Frassanito, M.A.; Berardi, S.; Catacchio, I.; Ria, R.; Racanelli, V.; Caivano, A.; Solimando, A.G.; Vergara, D.; et al. A HGF/cMET autocrine loop is operative in multiple myeloma bone marrow endothelial cells and may represent a novel therapeutic target. Clin. Cancer Res. 2014, 20, 5796–5807. [Google Scholar] [CrossRef] [PubMed]
- Moschetta, M.; Basile, A.; Ferrucci, A.; Frassanito, M.A.; Rao, L.; Ria, R.; Solimando, A.G.; Giuliani, N.; Boccarelli, A.; Fumarola, F.; et al. Novel targeting of phospho-cMET overcomes drug resistance and induces antitumor activity in multiple myeloma. Clin. Cancer Res. 2013, 19, 4371–4382. [Google Scholar] [CrossRef] [PubMed]
- Rao, L.; De Veirman, K.; Giannico, D.; Saltarella, I.; Desantis, V.; Frassanito, M.A.; Solimando, A.G.; Ribatti, D.; Prete, M.; Harstrick, A.; et al. Targeting angiogenesis in multiple myeloma by the VEGF and HGF blocking DARPin((R)) protein MP0250: A preclinical study. Oncotarget 2018, 9, 13366–13381. [Google Scholar] [CrossRef] [PubMed]
- Desantis, V.; Frassanito, M.A.; Tamma, R.; Saltarella, I.; Di Marzo, L.; Lamanuzzi, A.; Solimando, A.G.; Ruggieri, S.; Annese, T.; Nico, B.; et al. Rhu-Epo down-regulates pro-tumorigenic activity of cancer-associated fibroblasts in multiple myeloma. Ann. Hematol. 2018, 97, 1251–1258. [Google Scholar] [CrossRef] [PubMed]
- Frassanito, M.A.; Desantis, V.; Di Marzo, L.; Craparotta, I.; Beltrame, L.; Marchini, S.; Annese, T.; Visino, F.; Arciuli, M.; Saltarella, I.; et al. Bone marrow fibroblasts overexpress miR-27b and miR-214 in step with multiple myeloma progression, dependent on tumour cell-derived exosomes. J. Pathol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Tibullo, D.; Di Rosa, M.; Giallongo, C.; La Cava, P.; Parrinello, N.L.; Romano, A.; Conticello, C.; Brundo, M.V.; Saccone, S.; Malaguarnera, L.; et al. Bortezomib modulates CHIT1 and YKL40 in monocyte-derived osteoclast and in myeloma cells. Front. Pharmacol. 2015, 6, 226. [Google Scholar] [CrossRef]
- Lamanuzzi, A.; Saltarella, I.; Desantis, V.; Frassanito, M.A.; Leone, P.; Racanelli, V.; Nico, B.; Ribatti, D.; Ditonno, P.; Prete, M.; et al. Inhibition of mTOR complex 2 restrains tumor angiogenesis in multiple myeloma. Oncotarget 2018, 9, 20563–20577. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.; Mitsiades, C.; Schlossman, R.; Ghobrial, I.; Hideshima, T.; Chauhan, D.; Munshi, N.; Anderson, K. The treatment of relapsed and refractory multiple myeloma. Hematol. Am. Soc. Hematol. Educ. Program 2007. [Google Scholar] [CrossRef] [PubMed]
- Sonneveld, P.; Broijl, A. Treatment of relapsed and refractory multiple myeloma. Haematologica 2016, 101, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Attal, M.; Lauwers-Cances, V.; Hulin, C.; Leleu, X.; Caillot, D.; Escoffre, M.; Arnulf, B.; Macro, M.; Belhadj, K.; Garderet, L.; et al. Lenalidomide, Bortezomib, and Dexamethasone with Transplantation for Myeloma. N. Engl. J. Med. 2017, 376, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.K.; Rajkumar, S.V.; Dimopoulos, M.A.; Masszi, T.; Spicka, I.; Oriol, A.; Hajek, R.; Rosinol, L.; Siegel, D.S.; Mihaylov, G.G.; et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N. Engl. J. Med. 2015, 372, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Sonneveld, P.; Schuster, M.W.; Irwin, D.; Stadtmauer, E.A.; Facon, T.; Harousseau, J.L.; Ben-Yehuda, D.; Lonial, S.; Goldschmidt, H.; et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N. Engl. J. Med. 2005, 352, 2487–2498. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, R.Z.; Nagler, A.; Sonneveld, P.; Blade, J.; Hajek, R.; Spencer, A.; San Miguel, J.; Robak, T.; Dmoszynska, A.; Horvath, N.; et al. Randomized phase III study of pegylated liposomal doxorubicin plus bortezomib compared with bortezomib alone in relapsed or refractory multiple myeloma: Combination therapy improves time to progression. J. Clin. Oncol. 2007, 25, 3892–3901. [Google Scholar] [CrossRef]
- Weber, D.M.; Chen, C.; Niesvizky, R.; Wang, M.; Belch, A.; Stadtmauer, E.A.; Siegel, D.; Borrello, I.; Rajkumar, S.V.; Chanan-Khan, A.A.; et al. Lenalidomide plus dexamethasone for relapsed multiple myeloma in North America. N. Engl. J. Med. 2007, 357, 2133–2142. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S.; Dimopoulos, M.; Palumbo, A.; White, D.; Grosicki, S.; Spicka, I.; Walter-Croneck, A.; Moreau, P.; Mateos, M.V.; Magen, H.; et al. Elotuzumab Therapy for Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2015, 373, 621–631. [Google Scholar] [CrossRef]
- Cook, G.; Williams, C.; Brown, J.M.; Cairns, D.A.; Cavenagh, J.; Snowden, J.A.; Ashcroft, A.J.; Fletcher, M.; Parrish, C.; Yong, K.; et al. High-dose chemotherapy plus autologous stem-cell transplantation as consolidation therapy in patients with relapsed multiple myeloma after previous autologous stem-cell transplantation (NCRI Myeloma X Relapse (Intensive trial)): A randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 874–885. [Google Scholar] [CrossRef]
- Maloney, D.G.; Molina, A.J.; Sahebi, F.; Stockerl-Goldstein, K.E.; Sandmaier, B.M.; Bensinger, W.; Storer, B.; Hegenbart, U.; Somlo, G.; Chauncey, T.; et al. Allografting with nonmyeloablative conditioning following cytoreductive autografts for the treatment of patients with multiple myeloma. Blood 2003, 102, 3447–3454. [Google Scholar] [CrossRef]
- Offidani, M.; Corvatta, L.; Maracci, L.; Liberati, A.M.; Ballanti, S.; Attolico, I.; Caraffa, P.; Alesiani, F.; Caravita di Toritto, T.; Gentili, S.; et al. Efficacy and tolerability of bendamustine, bortezomib and dexamethasone in patients with relapsed-refractory multiple myeloma: A phase II study. Blood Cancer J. 2013, 3, e162. [Google Scholar] [CrossRef]
- Lokhorst, H.M.; Plesner, T.; Laubach, J.P.; Nahi, H.; Gimsing, P.; Hansson, M.; Minnema, M.C.; Lassen, U.; Krejcik, J.; Palumbo, A.; et al. Targeting CD38 with Daratumumab Monotherapy in Multiple Myeloma. N. Engl. J. Med. 2015, 373, 1207–1219. [Google Scholar] [CrossRef] [PubMed]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, O.; Klimek, V.M.; Gaskell, A.A.; Viale, A.; Cheng, D.; Kim, E.; Rampal, R.; Bluth, M.; Harding, J.J.; Callahan, M.K.; et al. Efficacy of intermittent combined RAF and MEK inhibition in a patient with concurrent BRAF- and NRAS-mutant malignancies. Cancer Discov. 2014, 4, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Diaz, T.; Rodriguez, V.; Lozano, E.; Mena, M.P.; Calderon, M.; Rosinol, L.; Martinez, A.; Tovar, N.; Perez-Galan, P.; Blade, J.; et al. The BET bromodomain inhibitor CPI203 improves lenalidomide and dexamethasone activity in in vitro and in vivo models of multiple myeloma by blockade of Ikaros and MYC signaling. Haematologica 2017, 102, 1776–1784. [Google Scholar] [CrossRef] [PubMed]
- Holkova, B.; Zingone, A.; Kmieciak, M.; Bose, P.; Badros, A.Z.; Voorhees, P.M.; Baz, R.; Korde, N.; Lin, H.Y.; Chen, J.Q.; et al. A Phase II Trial of AZD6244 (Selumetinib, ARRY-142886), an Oral MEK1/2 Inhibitor, in Relapsed/Refractory Multiple Myeloma. Clin. Cancer Res. 2016, 22, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Menu, E.; Garcia, J.; Huang, X.; Di Liberto, M.; Toogood, P.L.; Chen, I.; Vanderkerken, K.; Chen-Kiang, S. A novel therapeutic combination using PD 0332991 and bortezomib: Study in the 5T33MM myeloma model. Cancer Res. 2008, 68, 5519–5523. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Bensinger, W.I.; Huff, C.A.; Costello, C.L.; Lendvai, N.; Berdeja, J.G.; Anderson, L.D., Jr.; Siegel, D.S.; Lebovic, D.; Jagannath, S.; et al. Ibrutinib alone or with dexamethasone for relapsed or relapsed and refractory multiple myeloma: Phase 2 trial results. Br. J. Haematol. 2018, 180, 821–830. [Google Scholar] [CrossRef]
- Rustad, E.H.; Dai, H.Y.; Hov, H.; Coward, E.; Beisvag, V.; Myklebost, O.; Hovig, E.; Nakken, S.; Vodak, D.; Meza-Zepeda, L.A.; et al. BRAF V600E mutation in early-stage multiple myeloma: Good response to broad acting drugs and no relation to prognosis. Blood Cancer J. 2015, 5, e299. [Google Scholar] [CrossRef] [PubMed]
- Venkata, J.K.; An, N.; Stuart, R.; Costa, L.J.; Cai, H.; Coker, W.; Song, J.H.; Gibbs, K.; Matson, T.; Garrett-Mayer, E.; et al. Inhibition of sphingosine kinase 2 downregulates the expression of c-Myc and Mcl-1 and induces apoptosis in multiple myeloma. Blood 2014, 124, 1915–1925. [Google Scholar] [CrossRef]
- Niesvizky, R.; Badros, A.Z.; Costa, L.J.; Ely, S.A.; Singhal, S.B.; Stadtmauer, E.A.; Haideri, N.A.; Yacoub, A.; Hess, G.; Lentzsch, S.; et al. Phase 1/2 study of cyclin-dependent kinase (CDK)4/6 inhibitor palbociclib (PD-0332991) with bortezomib and dexamethasone in relapsed/refractory multiple myeloma. Leuk. Lymphoma 2015, 56, 3320–3328. [Google Scholar] [CrossRef]
- Scheid, C.; Reece, D.; Beksac, M.; Spencer, A.; Callander, N.; Sonneveld, P.; Kalimi, G.; Cai, C.; Shi, M.; Scott, J.W.; et al. Phase 2 study of dovitinib in patients with relapsed or refractory multiple myeloma with or without t(4;14) translocation. Eur. J. Haematol. 2015, 95, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Kortum, K.M.; Einsele, H. First targeted therapy in multiple myeloma. Blood 2017, 130, 2359–2360. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jones, J.R.; Weinhold, N.; Ashby, C.; Walker, B.A.; Wardell, C.; Pawlyn, C.; Rasche, L.; Melchor, L.; Cairns, D.A.; Gregory, W.M.; et al. Clonal evolution in myeloma: The impact of maintenance lenalidomide and depth of response on the genetics and sub-clonal structure of relapsed disease in uniformly treated newly diagnosed patients. Haematologica 2019. [Google Scholar] [CrossRef] [PubMed]
- Heuck, C.J.; Jethava, Y.; Khan, R.; van Rhee, F.; Zangari, M.; Chavan, S.; Robbins, K.; Miller, S.E.; Matin, A.; Mohan, M.; et al. Inhibiting MEK in MAPK pathway-activated myeloma. Leukemia 2016, 30, 976–980. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kaufman, J.L.; Gasparetto, C.; Mikhael, J.; Vij, R.; Pegourie, B.; Benboubker, L.; Facon, T.; Amiot, M.; Moreau, P.; et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood 2017, 130, 2401–2409. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Chanan-Khan, A.; Roberts, A.W.; Agarwal, A.B.; Facon, T.; Kumar, S.; Touzeau, C.; Punnoose, E.A.; Cordero, J.; Munasinghe, W.; et al. Promising efficacy and acceptable safety of venetoclax plus bortezomib and dexamethasone in relapsed/refractory MM. Blood 2017, 130, 2392–2400. [Google Scholar] [CrossRef] [PubMed]
- Anwer, F.; Gee, K.M.; Iftikhar, A.; Baig, M.; Russ, A.D.; Saeed, S.; Zar, M.A.; Razzaq, F.; Carew, J.; Nawrocki, S.; et al. Future of Personalized Therapy Targeting Aberrant Signaling Pathways in Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2019. [Google Scholar] [CrossRef]
- Trudel, S.; Stewart, A.K.; Rom, E.; Wei, E.; Li, Z.H.; Kotzer, S.; Chumakov, I.; Singer, Y.; Chang, H.; Liang, S.B.; et al. The inhibitory anti-FGFR3 antibody, PRO-001, is cytotoxic to t(4;14) multiple myeloma cells. Blood 2006, 107, 4039–4046. [Google Scholar] [CrossRef]
- Walker, B.A.; Boyle, E.M.; Wardell, C.P.; Murison, A.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Johnson, D.C.; Kaiser, M.F.; Melchor, L.; et al. Mutational Spectrum, Copy Number Changes, and Outcome: Results of a Sequencing Study of Patients With Newly Diagnosed Myeloma. J. Clin. Oncol. 2015, 33, 3911–3920. [Google Scholar] [CrossRef]
- Kumar, L.; Gogi, R.; Patel, A.K.; Mookerjee, A.; Sahoo, R.K.; Malik, P.S.; Sharma, A.; Thulkar, S.; Kumar, R.; Biswas, A.; et al. Multiple myeloma with extramedullary disease: Impact of autologous stem cell transplantation on outcome. Bone Marrow Transplant. 2017, 52, 1473–1475. [Google Scholar] [CrossRef]
- Ria, R.; Reale, A.; Solimando, A.G.; Mangialardi, G.; Moschetta, M.; Gelao, L.; Iodice, G.; Vacca, A. Induction therapy and stem cell mobilization in patients with newly diagnosed multiple myeloma. Stem Cells Int. 2012, 2012, 607260. [Google Scholar] [CrossRef] [PubMed]
- Cavo, M.; Gay, F.M.; Patriarca, F.; Zamagni, E.; Montefusco, V.; Dozza, L.; Galli, M.; Bringhen, S.; Testoni, N.; Grasso, M.; et al. Double Autologous Stem Cell Transplantation Significantly Prolongs Progression-Free Survival and Overall Survival in Comparison with Single Autotransplantation in Newly Diagnosed Multiple Myeloma: An Analysis of Phase 3 EMN02/HO95 Study. Blood 2017, 130, 401. [Google Scholar]
- Vacca, A.; Melaccio, A.; Sportelli, A.; Solimando, A.G.; Dammacco, F.; Ria, R. Subcutaneous immunoglobulins in patients with multiple myeloma and secondary hypogammaglobulinemia: A randomized trial. Clin. Immunol. 2018, 191, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Barlogie, B.; Anaissie, E.; van Rhee, F.; Haessler, J.; Hollmig, K.; Pineda-Roman, M.; Cottler-Fox, M.; Mohiuddin, A.; Alsayed, Y.; Tricot, G.; et al. Incorporating bortezomib into upfront treatment for multiple myeloma: Early results of total therapy 3. Br. J. Haematol. 2007, 138, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Jakubowiak, A.J.; Dytfeld, D.; Griffith, K.A.; Lebovic, D.; Vesole, D.H.; Jagannath, S.; Al-Zoubi, A.; Anderson, T.; Nordgren, B.; Detweiler-Short, K.; et al. A phase 1/2 study of carfilzomib in combination with lenalidomide and low-dose dexamethasone as a frontline treatment for multiple myeloma. Blood 2012, 120, 1801–1809. [Google Scholar] [CrossRef]
- Rajkumar, S.V. Multiple myeloma: 2016 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2016, 91, 719–734. [Google Scholar] [CrossRef]
- van Rhee, F.; Szymonifka, J.; Anaissie, E.; Nair, B.; Waheed, S.; Alsayed, Y.; Petty, N.; Shaughnessy, J.D., Jr.; Hoering, A.; Crowley, J.; et al. Total Therapy 3 for multiple myeloma: Prognostic implications of cumulative dosing and premature discontinuation of VTD maintenance components, bortezomib, thalidomide, and dexamethasone, relevant to all phases of therapy. Blood 2010, 116, 1220–1227. [Google Scholar] [CrossRef]
- Jethava, Y.; Mitchell, A.; Zangari, M.; Waheed, S.; Schinke, C.; Thanendrarajan, S.; Sawyer, J.; Alapat, D.; Tian, E.; Stein, C.; et al. Dose-dense and less dose-intense Total Therapy 5 for gene expression profiling-defined high-risk multiple myeloma. Leukemia 2016, 6, e453. [Google Scholar] [CrossRef]
- Cavo, M.; Goldschmidt, H.; Rosinol, L.; Pantani, L.; Zweegman, S.; Salwender, H.J.; Lahuerta, J.J.; Lokhorst, H.M.; Petrucci, M.T.; Blau, I.; et al. Double Vs Single Autologous Stem Cell Transplantation for Newly Diagnosed Multiple Myeloma: Long-Term Follow-up (10-Years) Analysis of Randomized Phase 3 Studies. Blood 2018, 132, 124. [Google Scholar] [CrossRef]
- Smets, T.; Stevenaert, F.; Adams, H., 3rd; Vanhoof, G. Deep Profiling of the Immune System of Multiple Myeloma Patients Using Cytometry by Time-of-Flight (CyTOF). Methods Mol. Biol. 2018, 1792, 47–54. [Google Scholar] [CrossRef]
- Ledergor, G.; Weiner, A.; Zada, M.; Wang, S.Y.; Cohen, Y.C.; Gatt, M.E.; Snir, N.; Magen, H.; Koren-Michowitz, M.; Herzog-Tzarfati, K.; et al. Single cell dissection of plasma cell heterogeneity in symptomatic and asymptomatic myeloma. Nat. Med. 2018, 24, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Tikhonova, A.N.; Dolgalev, I.; Hu, H.; Sivaraj, K.K.; Hoxha, E.; Cuesta-Domínguez, Á.; Pinho, S.; Akhmetzyanova, I.; Gao, J.; Witkowski, M.; et al. The bone marrow microenvironment at single-cell resolution. Nature 2019, 569, 222–228. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solimando, A.G.; Da Vià, M.C.; Cicco, S.; Leone, P.; Di Lernia, G.; Giannico, D.; Desantis, V.; Frassanito, M.A.; Morizio, A.; Delgado Tascon, J.; et al. High-Risk Multiple Myeloma: Integrated Clinical and Omics Approach Dissects the Neoplastic Clone and the Tumor Microenvironment. J. Clin. Med. 2019, 8, 997. https://doi.org/10.3390/jcm8070997
Solimando AG, Da Vià MC, Cicco S, Leone P, Di Lernia G, Giannico D, Desantis V, Frassanito MA, Morizio A, Delgado Tascon J, et al. High-Risk Multiple Myeloma: Integrated Clinical and Omics Approach Dissects the Neoplastic Clone and the Tumor Microenvironment. Journal of Clinical Medicine. 2019; 8(7):997. https://doi.org/10.3390/jcm8070997
Chicago/Turabian StyleSolimando, Antonio Giovanni, Matteo Claudio Da Vià, Sebastiano Cicco, Patrizia Leone, Giuseppe Di Lernia, Donato Giannico, Vanessa Desantis, Maria Antonia Frassanito, Arcangelo Morizio, Julia Delgado Tascon, and et al. 2019. "High-Risk Multiple Myeloma: Integrated Clinical and Omics Approach Dissects the Neoplastic Clone and the Tumor Microenvironment" Journal of Clinical Medicine 8, no. 7: 997. https://doi.org/10.3390/jcm8070997
APA StyleSolimando, A. G., Da Vià, M. C., Cicco, S., Leone, P., Di Lernia, G., Giannico, D., Desantis, V., Frassanito, M. A., Morizio, A., Delgado Tascon, J., Melaccio, A., Saltarella, I., Ranieri, G., Ria, R., Rasche, L., Kortüm, K. M., Beilhack, A., Racanelli, V., Vacca, A., & Einsele, H. (2019). High-Risk Multiple Myeloma: Integrated Clinical and Omics Approach Dissects the Neoplastic Clone and the Tumor Microenvironment. Journal of Clinical Medicine, 8(7), 997. https://doi.org/10.3390/jcm8070997