A Clinical Efficacy of PRRT in Patients with Advanced, Nonresectable, Paraganglioma-Pheochromocytoma, Related to SDHx Gene Mutation

Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Study Parameters
2.3. Preparation of Radiotracer
2.4. Therapy–Administration Protocol
2.5. Biodistribution of the Radiotracer and Dosimetry
2.6. Radiology Proceedings
2.7. Assessment of Efficacy
2.8. Outcomes
2.9. Statistical Analysis
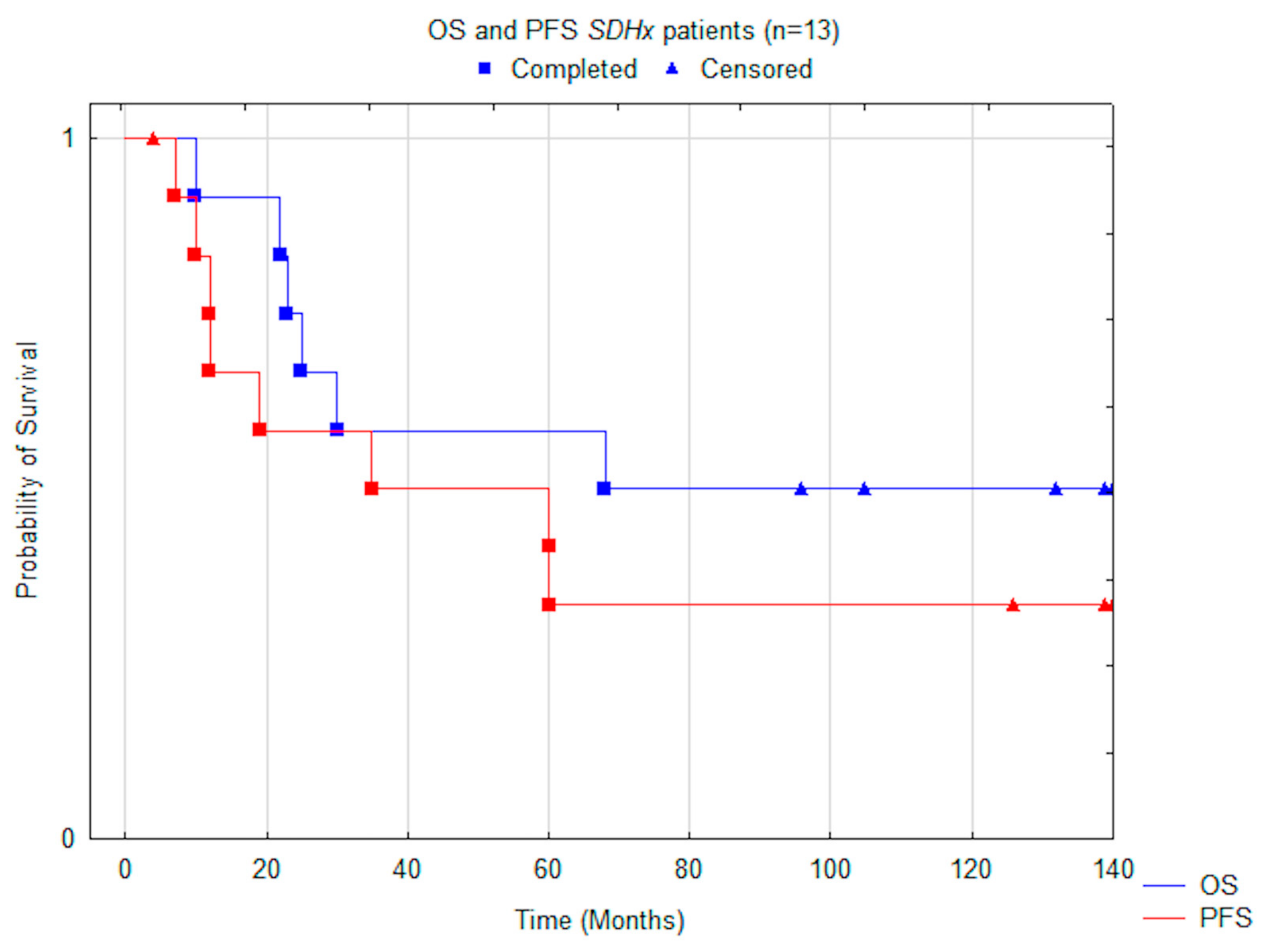
3. Results
3.1. Clinical Response
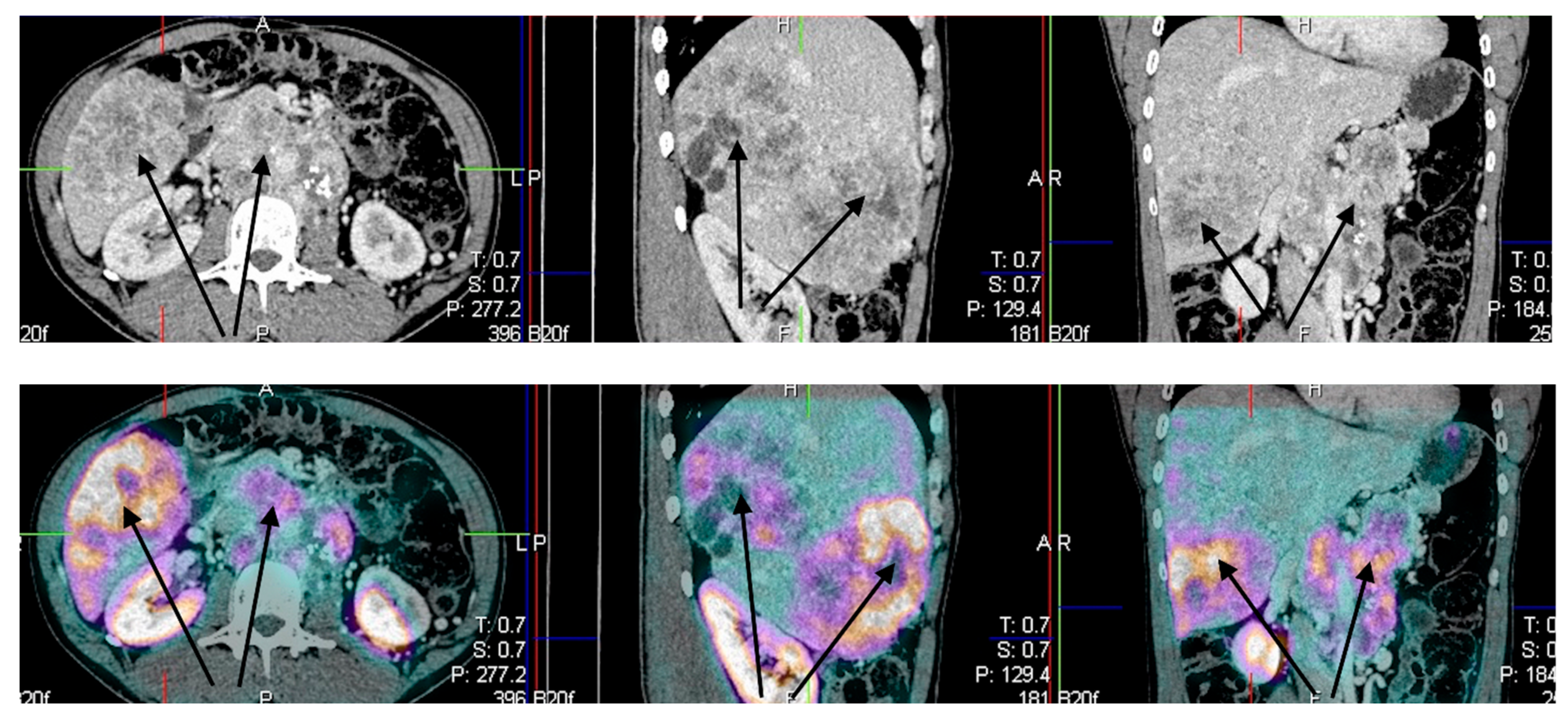
3.2. Radiological Response (ORR)
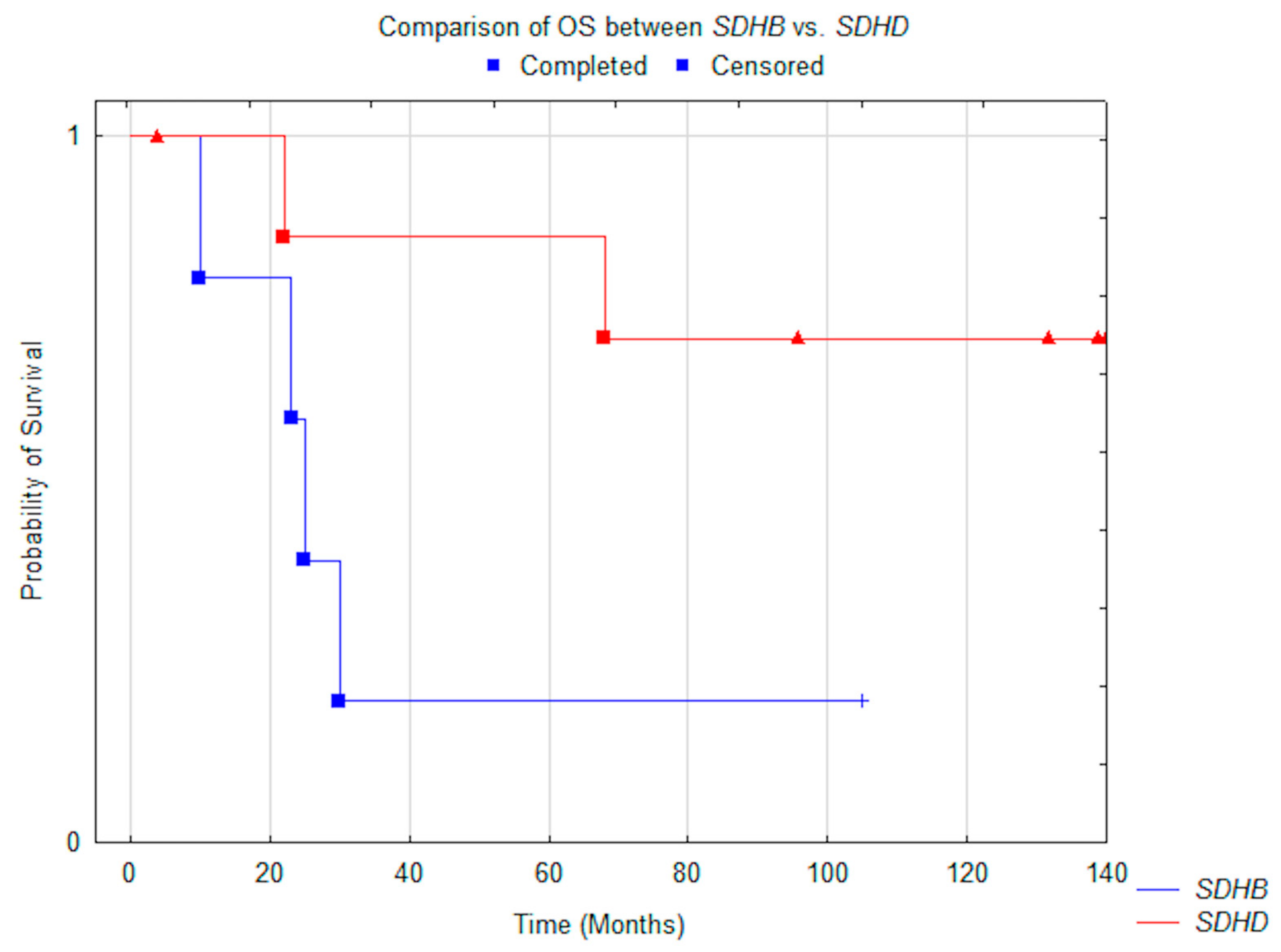
3.3. Outcome
3.4. Adverse Events CTC AEs NCI v. 4.0
3.5. Renal Toxicity
3.6. Hematological Toxicity
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Berends, A.M.A.; Buitenwerf, E.; de Krijger, R.R.; Veeger, N.J.G.M.; van der Horst-Schrivers, A.N.A.; Links, T.P.; Kerstens, M.N. Incidence of pheochromocytoma and sympathetic paraganglioma in the Netherlands: A nationwide study and systematic review. Eur. J. Intern. Med. 2018, 51, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Lenders, J.W.; Duh, Q.Y.; Eisenhofer, G.; Gimenez-Roqueplo, A.P.; Grebe, S.K.; Murad, M.H.; Naruse, M.; Pacak, K.; Young, W.F., Jr. Pheochromocytoma and paraganglioma: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2014, 99, 1915–1942. [Google Scholar] [CrossRef] [PubMed]
- Fishbein, L.; Orlowski, R.; Cohen, D. Pheochromocytoma/Paraganglioma: Review of perioperative management of blood pressure and update on genetic mutations associated with pheochromocytoma. J. Clin. Hypertens. (Greenwich) 2013, 15, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, C.; Rohren, E.; Habra, M.A.; Rich, T.; Jimenez, P.; Ayala-Ramirez, M.; Baudin, E. Current and future treatments for malignant pheochromocytoma and sympathetic paraganglioma. Curr. Oncol. Rep. 2013, 15, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Neumann, H.P.; Young, W.F., Jr.; Krauss, T.; Bayley, J.P.; Schiavi, F.; Opocher, G.; Boedeker, C.C.; Tirosh, A.; Castinetti, F.; Ruf, J.; et al. 65 Years of the double helix: Genetics informs precision practice in the diagnosis and management of pheochromocytoma. Endo. Relat. Cancer. 2018, 25, T201–T219. [Google Scholar] [CrossRef] [PubMed]
- Baudin, E.; Habra, M.A.; Deschamps, F.; Cote, G.; Dumont, F.; Cabanillas, M.; Arfi-Roufe, J.; Berdelou, A.; Moon, B.; Al Ghuzlan, A.; et al. Therapy of endocrine disease: Treatment of malignant pheochromocytoma and paraganglioma. Eur. J. Endocrinol. 2014, 171, R111–R122. [Google Scholar] [CrossRef] [PubMed]
- Baysal, B.E. Hereditary paraganglioma targets diverse paraganglia. J. Med. Genet. 2002, 39, 617–22. [Google Scholar] [CrossRef]
- Neumann, H.P.; Pawlu, C.; Peczkowska, M.; Bausch, B.; McWhinney, S.R.; Muresan, M.; Buchta, M.; Franke, G.; Klisch, J.; Bley, T.A.; et al. European-American Paraganglioma Study Group. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA 2004, 292, 943–951. [Google Scholar] [CrossRef]
- Crona, J.; Taïeb, D.; Pacak., K. New Perspectives on Pheochromocytoma and Paraganglioma: Towards a Molecular Classification. Endocr. Rev. 2017, 38, 489–515. [Google Scholar] [CrossRef]
- Fishbein, L.; Leshchiner, I.; Walter, V.; Danilova, L.; Robertson, G.; Johnson, A.R.; Lichtenberg, T.M.; Murray, B.A.; Ghayee, H.K.; Else, T.; et al. Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell 2017, 31, 181–193. [Google Scholar] [CrossRef]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Benn, D.E.; Robinson, B.G.; Clifton-Bligh, R.J. 15 Years of Paraganglioma: Clinical manifestations of paraganglioma syndromes types 1-5. Endocr. Relat. Cancer 2015, 22, T91–T103. [Google Scholar] [CrossRef] [PubMed]
- Pasini, B.; McWhinney, S.R.; Bei, T.; Matyakhina, L.; Stergiopoulos, S.; Muchow, M.; Boikos, S.A.; Ferrando, B.; Pacak, K.; Assie, G.; et al. Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur. J. Hum. Genet. 2008, 16, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Schiavi, F.; Boedeker, C.C.; Bausch, B.; Peczkowska, M.; Gomez, C.F.; Strassburg, T.; Pawlu, C.; Buchta, M.; Salzmann, M.; Hoffmann, M.M.; et al. Predictors and prevalence of paraganglioma syndrome associated with mutations of the SDHC gene. JAMA 2005, 294, 2057–2063. [Google Scholar] [CrossRef] [PubMed]
- Ricketts, C.J.; Forman, J.R.; Rattenberry, E.; Bradshaw, N.; Lalloo, F.; Izatt, L.; Cole, T.R.; Armstrong, R.; Kumar, V.K.; Morrison, P.J.; et al. Tumor risks and genotype–phenotype–proteotype analysis in 358 patients with germline mutations in SDHB and SDHD. Human Mutation 2010, 31, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Benn, D.E.; Gimenez-Roqueplo, A.P.; Reilly, J.R.; Bertherat, J.; Burgess, J.; Byth, K.; Croxson, M.; Dahia, P.L.; Elston, M.; Gimm, O.; et al. Clinical presentation and penetrance of pheochromocytoma/ paraganglioma syndromes. JCEM 2006, 91, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Malinoc, A.; Sullivan, M.; Wiech, T.; Schmid, K.W.; Jilg, C.; Straeter, J.; Deger, S.; Hoffmann, M.M.; Bosse, A.; Rasp, G.; et al. Biallelic inactivation of the SDHC gene in renal carcinoma associated with paraganglioma syndrome type 3. Endocr. Relat. Cancer 2012, 19, 283–290. [Google Scholar] [CrossRef]
- Ricketts, C.J.; Shuch, B.; Vocke, C.D.; Metwalli, A.R.; Bratslavsky, G.; Middelton, L.; Yang, Y.; Wei, M.H.; Pautler, S.E.; Peterson, J.; et al. Succinate Succinate dehydrogenase kidney cancer: An aggressive example of the Warburg effect in cancer. J. Urol. 2012, 188, 2063–2071. [Google Scholar] [CrossRef]
- Miettinen, M.; Lasota, J. Gastrointestinal stromal tumors. Gastroenterol Clin. North Am. 2013, 42, 399–415. [Google Scholar] [CrossRef]
- Haissaguerre, M.; Courel, M.; Caron, P.; Denost, S.; Dubessy, C.; Gosse, P.; Appavoupoulle, V.; Belleannee, G.; Jullie, M.L.; Montero-Hadjadje, M.; et al. Normotensive incidentally discovered pheochromocytomas display specific biochemical, cellular and molecular characteristics. J. Clin. Endocrinol. Metab. 2013, 3, 3. [Google Scholar] [CrossRef]
- Timmers, H.J.; Kozupa, A.; Eisenhofer, G.; Raygada, M.; Adams, K.T.; Solis, D.; Lenders, J.W.; Pacak, K. Clinical presentations, biochemical phenotypes, and genotype-phenotype correlations in patients with succinate dehydrogenase subunit B-associated pheochromocytomas and paragangliomas. J. Clin. Endocrinol. Metab. 2007, 92, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Turkova, H.; Prodanov, T.; Maly, M.; Martucci, V.; Adams, K.; Widimsky, J., Jr.; Chen, C.C.; Ling, A.; Kebebew, E.; Stratakis, C.; et al. Characteristics and outcomes of metastatic SDHB and sporadic pheochromocytoma/paraganglioma: An NIH study. Endocrine practice 2015, 22, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Lenders, J.W.M.; Eisenhofer, G. Update on Modern Management of Pheochromocytoma and Paraganglioma. Endocrinol. Metab. 2017, 32, 152–161. [Google Scholar] [CrossRef]
- Elston, M.S.; Meyer-Rochow, G.Y.; Conaglen, H.M.; Clarkson, A.; Clifton-Bligh, R.; Conaglen, J.V.; Gill, A.J. Increased SSTR2A and SSTR3 expression in succinate dehydrogenase-deficient pheochromocytomas and paragangliomas. Human Pathol. 2015, 46, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Epelbaum, J.; Bertherat, J.; Prevost, G.; Kordon, C.; Meyerhof, W.; Wulfsen, I.; Richter, D.; Plouin, P.F. Molecular and pharmacological characterization of somatostatin receptor subtypes in adrenal, extraadrenal, and malignant pheochromocytomas. J. Clin. Endocrinol. Metab. 1995, 80, 1837–1844. [Google Scholar] [PubMed]
- Reubi, J.C.; Waser, B.; Khosla, S.; Kvols, L.; Goellner, J.R.; Krenning, E.; Lamberts, S. In vitro and in vivo detection of somatostatin receptors in pheochromocytomas and paragangliomas. J. Clin. Endocrinol. Metab. 1992, 74, 1082–1089. [Google Scholar] [PubMed]
- Gonias, S.; Goldsby, R.; Matthay, K.K.; Hawkins, R.; Price, D.; Huberty, J.; Damon, L.; Linker, C.; Sznewajs, A.; Shiboski, S.; Fitzgerald, P. Phase II study of high-dose [131I] metaiodobenzylguanidine therapy for patients with metastatic pheochromocytoma and paraganglioma. J. Clin. Oncol. 2009, 27, 4162–4168. [Google Scholar] [CrossRef] [PubMed]
- Krempf, M.; Lumbroso, J.; Mornex, R.; Brendel, A.J.; Wemeau, J.L.; Delisle, M.J.; Aubert, B.; Carpentier, P.; Fleury-Goyon, M.C.; Gibold, C.; et al. Treatment of malignant pheochromocytoma with [131I] metaiodobenzylguanidine: A French multicenter study. J. Nucl. Med. Biol. Med. 1991, 35, 284–287. [Google Scholar]
- Jimenez, C.; Pryma, D.A.; Sullivan, D.C.; Schwarz, J.K.; Noto, R.B.; Stambler, N.; Armor, T.; Jensen, J.D.; Israel, R.J. Long Term Follow-up of a Pivotal Phase 2 Study of Ultratrace® Iobenguane I-131 (AZEDRA TM) in Patients with Malignant Relapsed/Refractory Pheochromocytoma /Paraganglioma. In Adrenal Tumors: Clinical Implications of the Recent Molecular and Genetic Findings; Endocrine Society’s 97th Annual Meeting and Expo, San Diego, CA, USA, 7 March 2015; Endocrine Society: Washington, DC, USA, 2015. [Google Scholar]
- Pryma, D.A.; Chin, B.B.; Noto, R.B.; Dillon, J.S.; Perkins, S.; Solnes, L.; Kostakoglu, L.; Serafini, A.N.; Pampaloni, M.H.; Jensen, J.; et al. Efficacy and Safety of High-Specific-Activity I-131 mIBG Therapy in Patients with Advanced Pheochromocytoma or Paraganglioma. JNM 2018, 60, 623–630. [Google Scholar] [CrossRef]
- Averbuch, S.D.; Steakley, C.S.; Young, R.C.; Gelmann, E.P.; Goldstein, D.S.; Stull, R.; Keiser, H.R. Malignant pheochromocytoma: Effective treatment with a combination of cyclophosphamide, vincristine, and dacarbazine. Ann. Internal Med. 1988, 109, 267–273. [Google Scholar] [CrossRef]
- Huang, H.; Abraham, J.; Hung, E.; Averbuch, S.; Merino, M.; Steinberg, S.M.; Pacak, K.; Fojo, T. Treatment of malignant pheochromocytoma/paraganglioma with cyclophosphamide, vincristine, and dacarbazine: Recommendation from a 22-year follow-up of 18 patients. Cancer 2008, 113, 2020–2028. [Google Scholar] [CrossRef] [PubMed]
- Niemeijer, N.D.; Alblas, G.; van Hulsteijn, L.T.; Dekkers, O.M.; Corssmit, E.P. Chemotherapy with cyclophosphamide, vincristine and dacarbazine for malignant paraganglioma and pheochromocytoma: Systematic review and meta-analysis. Clin. Endocrinol. 2014, 81, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Hadoux, J.; Favier, J.; Scoazec, J.Y.; Leboulleux, S.; Al Ghuzlan, A.; Caramella, C.; Deandreis, D.; Borget, I.; Loriot, C.; Chougnet, C.; et al. SDHB mutations are associated with response to temozolomide in patients with metastatic pheochromocytoma or paraganglioma. Int. J. Cancer 2014, 135, 2711–2720. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Ramirez, M.; Chougnet, C.N.; Habra, M.A.; Palmer, J.L.; Leboulleux, S.; Cabanillas, M.E.; Caramella, C.; Anderson, P.; Al Ghuzlan, A.; Waguespack, S.G.; et al. Treatment with sunitinib for patients with progressive metastatic pheochromocytomas and sympathetic paragangliomas. J. Clin. Endocrinol. Metab. 2012, 97, 4040–4050. [Google Scholar] [CrossRef] [PubMed]
- Jasim, S.; Suman, V.J.; Jimenez, C.; Harris, P.; Sideras, K.; Burton, J.K.; Worden, F.P.; Auchus, R.J.; Bible, K.C. Phase II trial of pazopanib in advanced/progressive malignant pheochromocytoma and paraganglioma. Endocrine 2017, 57, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Burotto Pichun, E.M.; Edgerly, M.; Velarde, M.; Bates, S.E.; Daerr, R.; Adams, K.; Pacak, K.; Fojo, T. Phase II clinical trial of axitinib in metastatic pheochromocytomas and paraganlgiomas (P/PG): Preliminary results. J. Clin. Oncol. 2015, 33, 457. [Google Scholar] [CrossRef]
- Zovato, S.; Kumanova, A.; Dematte, S.; Sansovini, M.; Bodei, L.; Di Sarra, D.; Casagranda, E.; Severi, S.; Ambrosetti, A.; Schiavi, F.; et al. Peptide receptor radionuclide therapy (PRRT) with 177Lu-DOTATATE in individuals with neck or mediastinal paraganglioma (PGL). Hormone Metab Res. 2012, 44, 411–414. [Google Scholar] [CrossRef]
- Forrer, F.; Riedweg, I.; Maecke, H.R.; Mueller-Brand, J. Radiolabeled DOTATOC in patients with advanced paraganglioma and pheochromocytoma. QJ Nucl. Med. Mol. Imaging 2008, 52, 334–340. [Google Scholar]
- Pinato, D.J.; Black, J.R.; Ramaswami, R.; Tan, T.M.; Adjogatse, D.; Sharma, R. Peptide receptor radionuclide therapy for metastatic paragangliomas. Med. Oncol. 2016, 33, 47. [Google Scholar] [CrossRef]
- Puranik, A.D.; Kulkarni, H.R.; Singh, A.; Baum, R.P. Peptide receptor radionuclide therapy with (90)Y/(177)Lu-labelled peptides for inoperable head and neck paragangliomas (glomus tumours). Eur. J. Nucl. Med. Mol. Imaging. 2015, 42, 1223–1230. [Google Scholar] [CrossRef]
- Van Essen, M.; Krenning, E.P.; Kooij, P.P.; Bakker, W.H.; Feelders, R.A.; de Herder, W.W.; Wolbers, J.G.; Kwekkeboom, D.J. Effects of therapy with [177Lu-DOTA0, Tyr3] octreotate in patients with paraganglioma, meningioma, small cell lung carcinoma, and melanoma. J. Nucl. Med. 2006, 47, 1599–1606. [Google Scholar] [PubMed]
- Michalowska, I.; Cwikla, J.B.; Peczkowska, M.; Furmanek, M.I.; Buscombe, J.R.; Michalski, W.; Prejbisz, A.; Szperl, M.; Malinoc, A.; Moczulski, D.; et al. Usefulness of Somatostatin Receptor Scintigraphy (Tc-[HYNIC, Tyr3]-Octreotide) and 123I-Metaiodobenzylguanidine Scintigraphy in Patients with SDHx Gene-Related Pheochromocytomas and Paragangliomas Detected by Computed Tomography. Neuroendocrinology 2015, 101, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Common Terminology Criteria for Adverse Events (CTCAE), Version 4.0. Available online: https://www.eortc.be/services/doc/ctc/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf (accessed on 26 May 2019).
- Ćwikła, J.B.; Sankowski, A.J.; Seklecka, N.; Buscombe, J.R.; Nasierowska-Guttmejer, A.; Jeziorski, K.G.; Mikołajczak, R.; Pawlak, D.; Walecki, J. Efficacy of radionuclide treatment 90Y-DOTATATE in patients with progressive metastatic gastroenteropancreatic neuroendocrine carcinomas (GEP-NET). A phase II study. Ann. Oncol. 2010, 21, 787–794. [Google Scholar] [CrossRef] [PubMed]
- De Jong, M.; Valkema, R.; Jamar, F.; Kvols, L.K.; Kwekkeboom, D.J.; Breeman, W.A.; Bakker, W.H.; Smith, C.; Pauwels, S.; Krenning, E.P. Somatostatin receptor-targeted radionuclide therapy of tumors: Preclinical and clinical findings. Semin. Nucl. Med. 2002, 32, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Breeman, W.A.; de Jong, M.; Kwekkeboom, D.J.; Valkema, R.; Bakker, WH.; Kooij, PP.; Visser, TJ.; Krenning, E.P. Somatostatin receptormediated imaging and therapy: Basic science, current knowledge, limitations and future perspectives. Eur. J. Nucl. Med. 2001, 28, 1421–1429. [Google Scholar] [CrossRef] [PubMed]
- Nieć, D.; Kunicki, P.K. Validation of an assay for quantification of free normetanephrine, metanephrine and methoxytyramine in plasma by high performance liquid chromatography with coulometric detection: Comparison of peak-area vs. peak-height measurements. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2015, 100, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Prasad, V.; Zachert, C.; Schuchardt, C.; Wortmann, R.; Baum, R.P. Peptide receptor radionuclide therapy (PRRT) for progressive, somatostatin receptor positive pheochromocytoma/paraganglioma. J. Nucl. Med. 2008, 49, 101P–101P. [Google Scholar]
- Cecchin, D.; Schiavi, F.; Fanti, S.; Favero, M.; Manara, R.; Fassina, A.; Briani, C.; Allegri, V.; Sansovini, M.; Bui, F.; et al. Peptide Receptor Radionuclide Therapy in a case of multiple spinal canal and cranial paragangliomas. J. Clin. Oncol. 2011, 29, e171–e174. [Google Scholar] [CrossRef]
- Makis, W.; McCann, K.; McEwan, A.J.B. The Challenges of Treating Paraganglioma Patients with 177Lu-DOTATATE PRRT: Catecholamine Crises, Tumor Lysis Sundrome and the Need for Modification of Treatment Protocols. Nucl. Med. Mol. Imaging. 2015, 49, 223–230. [Google Scholar] [CrossRef]
- Gupta, G.; Pacak, K. AACE Adrenal Scientific Committee. Recision Medicine an Update on Genetype/Biochemical Pheonotype Relationship in Pheochromocytoma/Paragnaglioma Patients. Endocr Pract. 2017, 23, 690–704. [Google Scholar] [CrossRef]
- Nastos, K.; Cheung, V.T.F.; Toumpanakis, C.; Navalkissoor, S.; Quigley, A.-M.; Caplin, M.; Khoo, B. Peptid Receptor Radionuclide Treatment and 131I mIBG in the management of patients with metastatic/progressive pheochromocytomas and paragangliomas. J. Surg. Oncol. 2017, 115, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Kong, G.; Grozinsky-Glasberg, S.; Hofman, M.S.; Callahan, J.; Meirovitz, A.; Maimon, O.; Pattison, D.A.; Gross, D.J.; Hicks, R.J. Efficacy of Peptide Receptor Radionuclide Therapyfor Functional Metastatic Paraganglioma and Pheochromocytoma. J. Clin. Endocrinol. Metab. 2017, 102, 3278–3287. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | All SDHx | SDHD n = 8 | SDHB n = 5 | p-value | |
|---|---|---|---|---|---|
| No. of patients, n (%) | 13 (100) | 8 (62) | 5 (38) | ||
| Mean age, years (range) | 41.9 (27–62) | 45.1 (31–62) | 36.6 (27–43) | 0.13 | |
| Gender, n (%) | |||||
| Female | 5 (38) | 4 (50) | 1 (20) | 0.42 | |
| Male | 8 (62) | 4 (50) | 4 (80) | ||
| Median time from initial diagnosis Months (range) | 48.0 (10–180) | 114.0 (10–180) | 30.0 (18–113) | 0.42 | |
| Initial performance status (PS), n (%) | |||||
| WHO/ECOG = 1 | 11 (85) | 7 (88) | 4 (80) | 0.09 | |
| WHO/ECOG = 2 | 2 (15) | 1 (12) | 1 (20) | ||
| Grading of primary tumor, n (%) | |||||
| G1 | 7 (54) | 6 (75) | 1 (25) | ||
| G2 | 6 (46) | 2 (25) | 4 (75) | ||
| Median Ki-67 index, % (95% CI) | 2.2 (1.9–6.4) | 2.0 (1.6–3.4) | 5.0 (0.8–13) | 0.048 | |
| Liver involvement n (%) | 6 (46) | 3 (38) | 3 (60) | 0.83 | |
| Presence of bone mts n (%) | 9 (41) | 4 (50) | 5 (100) | 0.86 | |
| Secretor tumors, n (%) | 4 (31) | 2 (25) | 2 (40) | 0.18 | |
| Mean initial CgA x ULN (Range) | 5.1 (0.8–26.4) | 4.7 (0.78–26.0) | 5.8 (1.4–12.0) | 0.12 | |
| SRS Krenning uptake scale 3/4 | 2/11 | 1/7 | 1/4 | 0.97 | |
| No | Gender | Age *, | Type of Mutation | Primary Tumor Localization | Grading (Ki-67 in %) | Secretion before PRRT pg/mL | Secretion after PRRT pg/mL (Three Months) |
|---|---|---|---|---|---|---|---|
| 1 | M | 62 | SDHD C11X, ex 1 33TGC-TGC | Right PPC | 2 (5) | normal | normal |
| 2 | F | 31 | SDHD C11X, ex 1 33TGC-TGC | Chest & abdominal PGLs | 1 (2) | normal | normal |
| 3 | M | 52 | SDHD C11X, ex 1 33TGC-TGC | Right HNP | 1 (1) | normal | normal |
| 4 | F | 51 | SDHD C11X, ex 1 33TGC-TGC | Left HNP | 2 (3) | MTY = 1890 NMN = 126 | MTY = 1060 NMN = 143 |
| 5 | M | 42 | SDHD C11X, ex 1 33TGC-TGC | Right HNP | 1 (2) | NMN = 183,6 | NMN = normal |
| 6 | F | 47 | SDHD C11X, ex 1 33TGC-TGC | Bilateral PPC | 1 (2) | normal | normal |
| 7 | M | 31 | SDHD C11X, ex 1 33TGC-TGC | Left PPC | 1 (1) | normal | normal |
| 8 | F | 45 | SDHD C11X, ex 1 33TGC-TGC | Left HNP | 1 (2) | normal | normal |
| 9 | M | 36 | SDHB ex.3 p.R90X | Left PPC | 1 (2) | MTY = 9189 NMN = 6911 | MTY = 7235 NMN = 6430 |
| 10 | M | 38 | SDHB exon 1 deletion | Bladder PGL | 2 (5) | normal | normal |
| 11 | M | 27 | SDHB c. 708 T > C (int. 574 T > C) heterozygotic | Paraspinal PGL | 2 (8) | MTY = 3344 NMN = 2622 | MTY = 2570 NMN = 1987 |
| 12 | F | 39 | SDHB exon 1 deletion | Abdominal PGL | 2 (15) | normal | normal |
| 13 | M | 43 | SDHB R230L, exon7 | Left HNP | 2 (5) | normal | normal |
| Variable | All patients (n = 13) | SDHD (n = 8) | SDHB (n = 5) | p-Value |
|---|---|---|---|---|
| Previous surgery, n (%) | 13 (100) | 8 (100) | 5(100) | |
| Initial Previous surgery with ITT (intention to treat) n (%) | 5 (38) | 3 (38) | 2 (40) | 0.68 |
| Previous SST analogues, n (%) | 8 (62) | 6 (75) | 2 (40) | 0.56 |
| Previous any other systemic or local therapy (%) # | 9 (69) | 5 (63) | 4 (80) | 0.42 |
| All (n = 13) | PGL-1 (n = 8) | PGL-4 (n = 5) | |
|---|---|---|---|
| Mean therapy sessions | 2.5 | 2.4 | 3.0 |
| Activity per session 90Y in GBq mean (range) | 3.4 | 2.9 | 3.3 |
| Cumulative Activity 90Y in GBq, mean (range) | 8.3 | 7.3 | 9.9 |
| Variable | Subjects | Median OS Months (+/−95% CI) | P-value | Median PFS Months (+/−95% CI) | p-Value |
|---|---|---|---|---|---|
| All subjects | 13 | 68.0 (38.6–105.1) | 35.0 (24.4–93.1) | ||
| Mutation | 0.05 | 0.014 | |||
| SDHD | 8 | N.R. (not reached) | N.R. (not reached) | ||
| SDHB | 5 | 25.0 (3.2–85.6) | 12.0 (2.3–48.8) | ||
| Clinical response | 0.005 | 0.0004 | |||
| PR | 8 | N.R. (not reached) | N.R. (not reached) | ||
| SD/DP | 5 | 22.0 (6.5–27.9) | 7.0 (3.8–11.8) | ||
| Liver mts | 0.033 | 0.005 | |||
| present | 7 | 25.0 (6.0–67.7) | 10.0 (1.3–39.0) | ||
| absent | 6 | N.R. (not reached) | N.R. (not reached) | ||
| Bone mts | 0.029 | 0.0027 | |||
| present | 9 | 25.0 (14.1–71.9) | 12 (6.3–41.0) | ||
| absent | 4 | N.R. (not reached) | N.R. (not reached) | ||
| Hormonal status | 0.496 | 0.84 | |||
| secretor | 4 | 49.0 (0–152.4) | 27.0 (0–148.2) | ||
| non-secretor | 9 | 97.0 (30.0–120.1) | 60.0 (15.9–109.4) |
| Variable | All n (%) | SDHDn (%) | SDHB n (%) |
|---|---|---|---|
| RECIST six months, | n = 12 | n = 8 | n = 4 |
| PR | 1 (8) | 0 | 1 (25) |
| SD | 9 (75) | 6 (75) | 3 (75) |
| DP | 2 (17) | 2 (25) | 0 |
| RECIST 12 months, | n = 11 | n = 8 | n = 4 |
| PR | 0 | 0 | 0 |
| SD | 9 (82) | 6 (75) | 3 (75) |
| DP | 2 (18) | 2 (25) | 1 (25) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolasinska-Ćwikła, A.; Pęczkowska, M.; Ćwikła, J.B.; Michałowska, I.; Pałucki, J.M.; Bodei, L.; Lewczuk-Myślicka, A.; Januszewicz, A. A Clinical Efficacy of PRRT in Patients with Advanced, Nonresectable, Paraganglioma-Pheochromocytoma, Related to SDHx Gene Mutation. J. Clin. Med. 2019, 8, 952. https://doi.org/10.3390/jcm8070952
Kolasinska-Ćwikła A, Pęczkowska M, Ćwikła JB, Michałowska I, Pałucki JM, Bodei L, Lewczuk-Myślicka A, Januszewicz A. A Clinical Efficacy of PRRT in Patients with Advanced, Nonresectable, Paraganglioma-Pheochromocytoma, Related to SDHx Gene Mutation. Journal of Clinical Medicine. 2019; 8(7):952. https://doi.org/10.3390/jcm8070952
Chicago/Turabian StyleKolasinska-Ćwikła, Agnieszka, Mariola Pęczkowska, Jarosław B. Ćwikła, Ilona Michałowska, Jakub M. Pałucki, Lisa Bodei, Anna Lewczuk-Myślicka, and Andrzej Januszewicz. 2019. "A Clinical Efficacy of PRRT in Patients with Advanced, Nonresectable, Paraganglioma-Pheochromocytoma, Related to SDHx Gene Mutation" Journal of Clinical Medicine 8, no. 7: 952. https://doi.org/10.3390/jcm8070952
APA StyleKolasinska-Ćwikła, A., Pęczkowska, M., Ćwikła, J. B., Michałowska, I., Pałucki, J. M., Bodei, L., Lewczuk-Myślicka, A., & Januszewicz, A. (2019). A Clinical Efficacy of PRRT in Patients with Advanced, Nonresectable, Paraganglioma-Pheochromocytoma, Related to SDHx Gene Mutation. Journal of Clinical Medicine, 8(7), 952. https://doi.org/10.3390/jcm8070952