TNF-α/TNF-R System May Represent a Crucial Mediator of Proliferative Synovitis in Hemophilia A

 ,
, 
 ,
,  ,
,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Clinical and Imaging Score
2.3. Serum TNF-α Measurements
2.4. Synovial Biopsy Samples
2.5. Cell Isolation and Culture
2.6. Immunohistochemistry
2.7. Western Blotting
2.8. Cell Viability Assay
2.9. Active Caspase-3 Assay
2.10. Statistical Analysis
3. Results
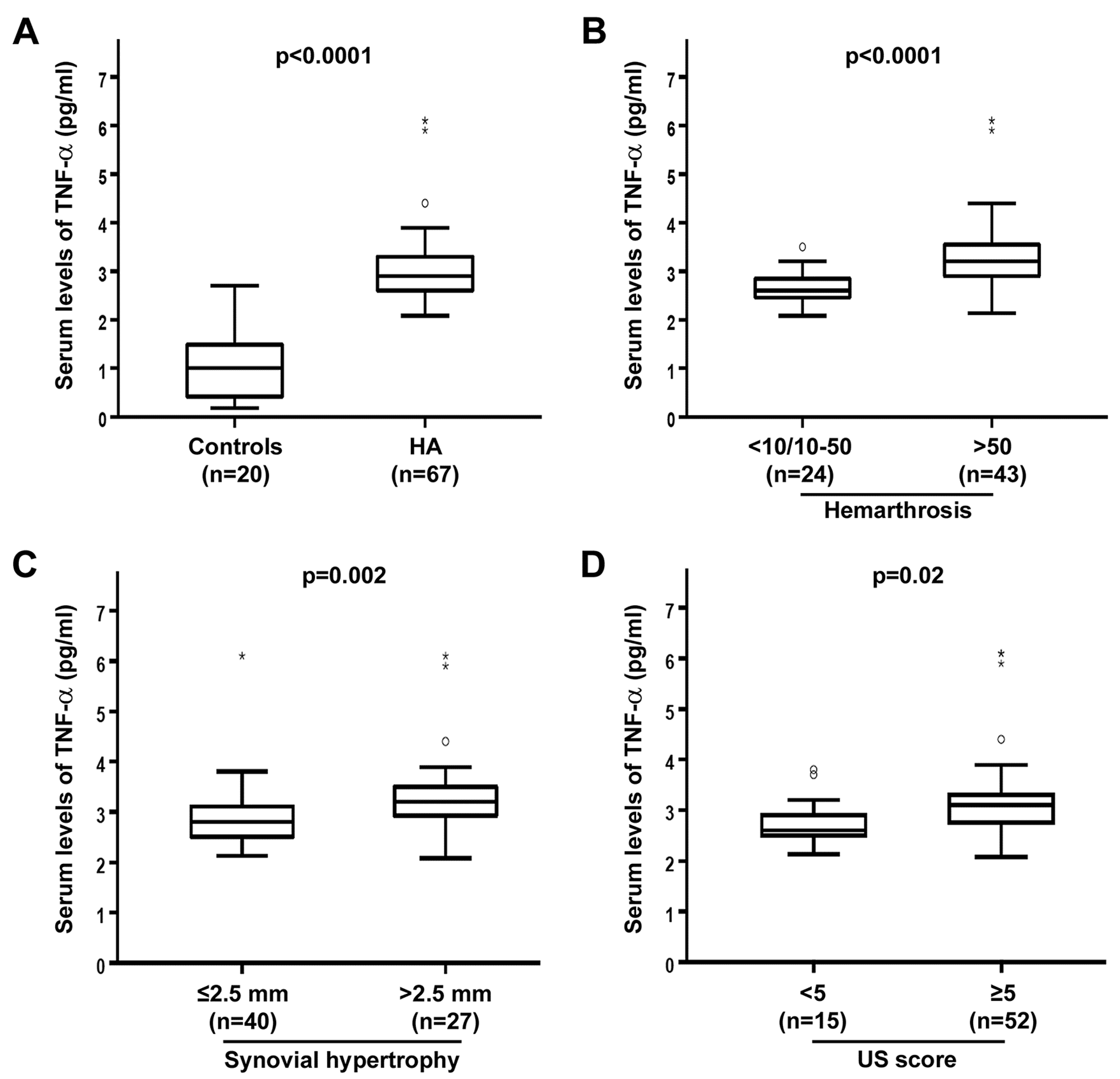
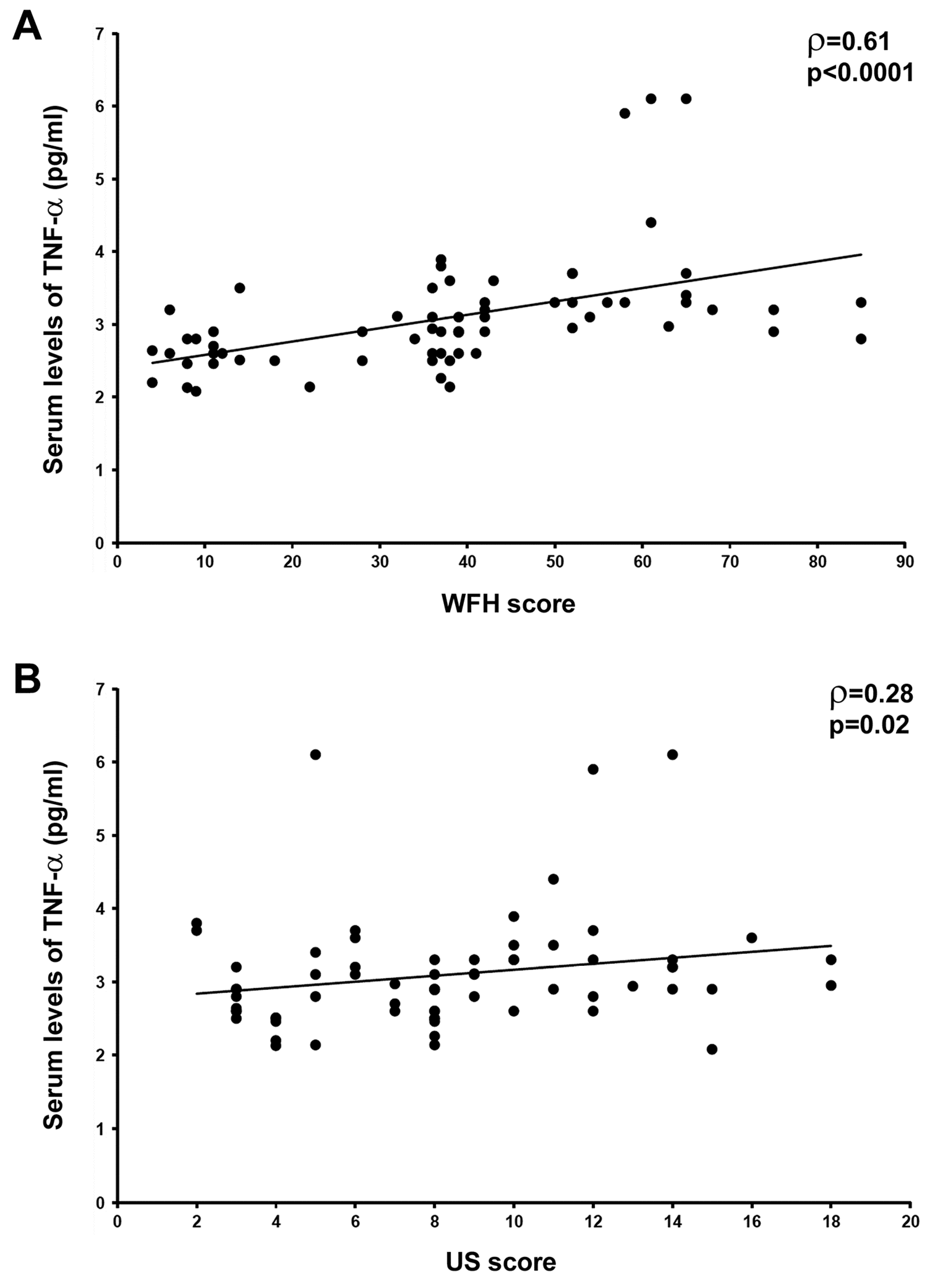
3.1. Circulating Levels of TNF-α Are Raised and Correlate with Joint Disease Severity in HA Patients
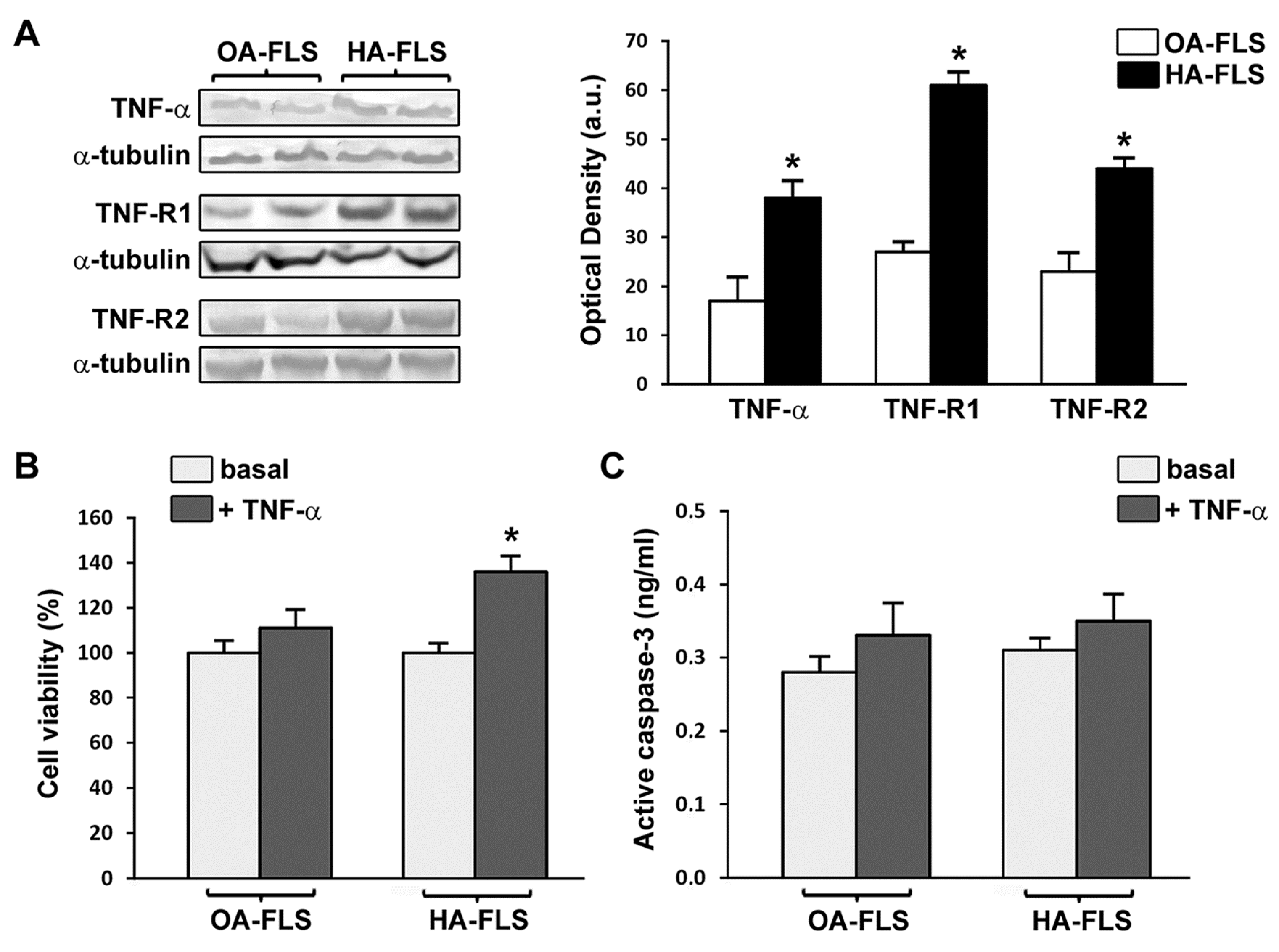
3.2. The Expression of TNF-α, TNF-R1, and TNF-R2 Is Strongly Increased in HA Synovial Tissue and Cultured HA Fibroblast-Like Synoviocytes
3.3. TNF-α Fosters HA Fibroblast-Like Synoviocyte Proliferation
4. Discussion
Author Contributions
Conflicts of Interest
References
- Peyvandi, F.; Garagiola, I.; Young, G. The past and future of haemophilia: Diagnosis, treatments, and its complications. Lancet 2016, 388, 187–197. [Google Scholar] [CrossRef]
- Van Vulpen, L.F.D.; Mastbergen, S.C.; Lafeber, F.P.J.G.; Schutgens, R.E.G. Differential effects of bleeds on the development of arthropathy-basic and applied issues. Haemophilia 2017, 23, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Valentino, L.A. Blood-induced joint disease: The pathophysiology of hemophilic arthropathy. J. Thromb. Haemost. 2010, 8, 1895–1902. [Google Scholar] [CrossRef] [PubMed]
- Valentino, L.A.; Hakobyan, N.; Enockson, C.; Simpson, M.L.; Kakodkar, N.C.; Cong, L.; Song, X. Exploring the biological basis of haemophilic joint disease: Experimental studies. Haemophilia 2012, 18, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Blobel, C.P.; Haxaire, C.; Kalliolias, G.D.; DiCarlo, E.; Salmon, J.; Srivastava, A. Blood-induced arthropathy in hemophilia: Mechanisms and heterogeneity. Semin. Thromb. Hemost. 2015, 41, 832–837. [Google Scholar]
- Lee, A.; Boyd, S.K.; Kline, G.; Poon, M.C. Premature changes in trabecular and cortical microarchitecture result in decreased bone strength in hemophilia. Blood 2015, 125, 2160–2163. [Google Scholar] [CrossRef]
- Kovacs, C.S. Hemophilia, low bone mass, and osteopenia/osteoporosis. Transfus. Apheresis Sci. 2008, 38, 33–40. [Google Scholar] [CrossRef]
- Melchiorre, D.; Manetti, M.; Matucci-Cerinic, M. Pathophysiology of hemophilic arthropathy. J. Clin. Med. 2017, 6, 63. [Google Scholar] [CrossRef]
- Pulles, A.E.; Mastbergen, S.C.; Schutgens, R.E.; Lafeber, F.P.; van Vulpen, L.F. Pathophysiology of hemophilic arthropathy and potential targets for therapy. Pharmacol. Res. 2017, 115, 192–199. [Google Scholar] [CrossRef]
- Hanley, J.; McKernan, A.; Creagh, M.D.; Classey, S.; McLaughlin, P.; Goddard, N.; Briggs, P.J.; Frostick, S.; Giangrande, P.; Wilde, J.; et al. Musculoskeletal working party of the UKHCDO. Guidelines for the management of acute joint bleeds and chronic synovitis in haemophilia: A United Kingdom Haemophilia Centre Doctors’ Organisation (UKHCDO) guideline. Haemophilia 2017, 23, 511–520. [Google Scholar] [CrossRef]
- Hua, B.; Olsen, E.H.N.; Sun, S.; Gudme, C.N.; Wang, L.; Vandahl, B.; Roepstorff, K.; Kjelgaard-Hansen, M.; Sørensen, B.B.; Zhao, Y.; et al. Serological biomarkers detect active joint destruction and inflammation in patients with haemophilic arthropathy. Haemophilia 2017, 23, e294–e300. [Google Scholar] [CrossRef] [PubMed]
- Prince, R.; Bologna, L.; Manetti, M.; Melchiorre, D.; Rosa, I.; Dewarrat, N.; Suardi, S.; Amini, P.; Fernández, J.A.; Burnier, L.; et al. Targeting anticoagulant protein S to improve hemostasis in hemophilia. Blood 2018, 131, 1360–1371. [Google Scholar] [CrossRef] [PubMed]
- Ehrenfeld, M.; Gur, H.; Shoenfeld, Y. Rheumatologic features of hematologic disorders. Curr. Opin. Rheumatol. 1999, 11, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Lövgren, K.M.; Christensen, K.R.; Majewski, W.; Østrup, O.; Skov, S.; Wiinberg, B. Acute haemarthrosis in the haemophilia a rat generates a local and systemic proinflammatory response. Thromb. Haemost. 2017, 117, 2092–2104. [Google Scholar] [CrossRef] [PubMed]
- Christensen, K.R.; Kjelgaard-Hansen, M.; Nielsen, L.N.; Wiinberg, B.; Alexander Althoehn, F.; Bloksgaard Poulsen, N.; Kryger Vøls, K.; Popp Thyme, A.; Maria Lövgren, K.; Kornerup Hansen, A.; et al. Rapid inflammation and early degeneration of bone and cartilage revealed in a time-course study of induced haemarthrosis in haemophilic rats. Rheumatology 2019, 58, 588–599. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, M.; Maini, R.N. Anti-TNF therapy, from rationale to standard of care: What lessons has it taught us? J. Immunol. 2010, 185, 791–794. [Google Scholar] [CrossRef]
- Zhao, B. TNF and bone remodeling. Curr. Osteoporos. Rep. 2017, 15, 126–134. [Google Scholar] [CrossRef]
- Sakthiswary, R.; Das, S. The effects of TNF α antagonist therapy on bone metabolism in rheumatoid arthritis: A systematic review. Curr. Drug Targets 2013, 14, 1552–1557. [Google Scholar] [CrossRef]
- Haxaire, C.; Hakobyan, N.; Pannellini, T.; Carballo, C.; McIlwain, D.; Mak, T.W.; Rodeo, S.; Acharya, S.; Li, D.; Szymonifka, J.; et al. Blood-induced bone loss in murine hemophilic arthropathy is prevented by blocking the iRhom2/ADAM17/TNF-α pathway. Blood 2018, 132, 1064–1074. [Google Scholar] [CrossRef]
- Roosendaal, G.; Vianen, M.E.; Wenting, M.J.; van Rinsum, A.C.; van den Berg, H.M.; Lafeber, F.P.; Bijlsma, J.W. Iron deposits and catabolic properties of synovial tissue from patients with haemophilia. J. Bone Joint Surg. Br. 1998, 80, 540–545. [Google Scholar] [CrossRef]
- Roosendaal, G.; van Rinsum, A.C.; Vianen, M.E.; van den Berg, H.M.; Lafeber, F.P.; Bijlsma, J.W. Haemophilic arthropathy resembles degenerative rather than inflammatory joint disease. Histopathology 1999, 34, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Brewer, A.K.; Mauser-Bunschoten, E.P.; Key, N.S.; Kitchen, S.; Llinas, A.; Ludlam, C.A.; Mahlangu, J.N.; Mulder, K.; Poon, M.C.; et al. Guidelines for the management of hemophilia. Haemophilia 2013, 19, e1–e47. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.S. Prophylaxis: Musculoskeletal evaluation. Semin. Hematol. 1993, 30 (Suppl. 2), 3–6. [Google Scholar] [PubMed]
- Pettersson, H.; Ahlberg, A.; Nilsson, I.M. A radiologic classification of the haemophilic arthropathy. Clin. Orthop. Relat. Res. 1980, 149, 153–159. [Google Scholar]
- Melchiorre, D.; Linari, S.; Innocenti, M.; Biscoglio, I.; Toigo, M.; Cerinic, M.M.; Morfini, M. Ultrasound detects joint damage and bleeding in haemophilic arthropathy: A proposal of a score. Haemophilia 2011, 17, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Melchiorre, D.; Linari, S.; Manetti, M.; Romano, E.; Sofi, F.; Matucci-Cerinic, M.; Carulli, C.; Innocenti, M.; Ibba-Manneschi, L.; Castaman, G. Clinical, instrumental, serological and histological findings suggest that hemophilia B may be less severe than hemophilia A. Haematologica 2016, 101, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Querol, F.; Rodriguez-Merchan, E.C. The role of ultrasonography in the diagnosis of the musculo-skeletal problems of haemophilia. Haemophilia 2012, 18, e215–e226. [Google Scholar] [CrossRef]
- Melchiorre, D.; Milia, A.F.; Linari, S.; Romano, E.; Benelli, G.; Manetti, M.; Guiducci, S.; Ceccarelli, C.; Innocenti, M.; Carulli, C.; et al. RANK-RANKL-OPG in hemophilic arthropathy: From clinical and imaging diagnosis to histopathology. J. Rheumatol. 2012, 39, 1678–1686. [Google Scholar] [CrossRef]
- Romano, E.; Manetti, M.; Peruzzi, F.; Melchiorre, D.; Milia, A.F.; Bellando-Randone, S.; Nishioka, K.; Innocenti, M.; Carulli, C.; Linari, S.; et al. Agonistic anti-human Fas monoclonal antibody induces fibroblast-like synoviocyte apoptosis in haemophilic arthropathy: Potential therapeutic implications. Haemophilia 2014, 20, e32–e39. [Google Scholar] [CrossRef]
- Aggarwal, B.B. Signalling pathways of the TNF superfamily: A double-edged sword. Nat. Rev. Immunol. 2003, 3, 745–756. [Google Scholar] [CrossRef]
- Vanamee, É.S.; Faustman, D.L. Structural principles of tumor necrosis factor superfamily signaling. Sci. Signal. 2018, 11, eaao4910. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Qiao, Y.; Grigoriev, G.; Chen, J.; Park-Min, K.H.; Park, S.H.; Ivashkiv, L.B.; Kalliolias, G.D. Tumor necrosis factor α induces sustained signaling and a prolonged and unremitting inflammatory response in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2013, 65, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Radner, H.; Aletaha, D. Anti-TNF in rheumatoid arthritis: An overview. Wien. Med. Wochenschr. 2015, 165, 3–9. [Google Scholar]
- Cantini, F.; Niccoli, L.; Nannini, C.; Cassarà, E.; Kaloudi, O.; Giulio Favalli, E.; Becciolini, A.; Biggioggero, M.; Benucci, M.; Li Gobbi, F.; et al. Tailored first-line biologic therapy in patients with rheumatoid arthritis, spondyloarthritis, and psoriatic arthritis. Semin. Arthritis Rheum. 2016, 45, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Melchiorre, D.; Morfini, M.; Linari, S.; Zignego, A.L.; Innocenti, M.; Matucci Cerinic, M. Anti-TNF-α therapy prevents the recurrence of joint bleeding in haemophilia and arthritis. Rheumatology (Oxford) 2014, 53, 576–578. [Google Scholar] [CrossRef] [PubMed]
- Brennan, F.M.; McInnes, I.B. Evidence that cytokines play a role in rheumatoid arthritis. J. Clin. Invest. 2008, 118, 3537–3545. [Google Scholar] [CrossRef] [PubMed]
- Ogata, A.; Hirano, T.; Hishitani, Y.; Tanaka, T. Safety and efficacy of tocilizumab for the treatment of rheumatoid arthritis. Clin. Med. Insights Arthritis Musculoskelet. Disord. 2012, 5, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.W.; Lee, N.R.; Pi, R.H.; Lim, Y.S.; Lee, Y.M.; Lee, J.M.; Jeong, H.S.; Chung, S.H. IL-6 inhibitors for treatment of rheumatoid arthritis: Past, present, and future. Arch. Pharm. Res. 2015, 38, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Narkbunnam, N.; Sun, J.; Hu, G.; Lin, F.C.; Bateman, T.A.; Mihara, M.; Monahan, P.E. IL-6 receptor antagonist as adjunctive therapy with clotting factor replacement to protect against bleeding-induced arthropathy in hemophilia. J. Thromb. Haemost. 2013, 11, 881–893. [Google Scholar] [CrossRef]
- Van Vulpen, L.F.; Schutgens, R.E.; Coeleveld, K.; Alsema, E.C.; Roosendaal, G.; Mastbergen, S.C.; Lafeber, F.P. IL-1β, in contrast to TNFα, is pivotal in blood-induced cartilage damage and is a potential target for therapy. Blood 2015, 126, 2239–2246. [Google Scholar] [CrossRef]
- Acharya, S.S.; Kaplan, R.N.; Macdonald, D.; Fabiyi, O.T.; DiMichele, D.; Lyden, D. Neoangiogenesis contributes to the development of hemophilic synovitis. Blood 2011, 117, 2484–2493. [Google Scholar] [CrossRef]
- Zetterberg, E.; Palmblad, J.; Wallensten, R.; Morfini, M.; Melchiorre, D.; Holmström, M. Angiogenesis is increased in advanced haemophilic joint disease and characterised by normal pericyte coverage. Eur. J. Haematol. 2014, 92, 256–262. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical characteristics/Imaging Findings | Patients |
|---|---|
| Median age (range), years | 36.3 (16–69) |
| Primary and secondary prophylaxis treatment, n (%) | 8 (12.0%) |
| Tertiary prophylaxis treatment, n (%) | 22 (32.8%) |
| On demand treatment, n (%) | 37 (55.2%) |
| Viral infections,n (%) | |
| HCV-RNA | 29 (43.3%) |
| Anti-HCV | 43 (64.2%) |
| HIV | 14 (20.9%) * |
| Hemarthroses,n (%) | |
| <10 | 7 (10.4%) |
| 10–50 | 17 (25.4%) |
| >50 | 43 (64.2%) |
| Synovial hypertrophy,n (%) | |
| ≤2.5 mm | 40 (59.7%) |
| >2.5 mm | 27 (40.3%) |
| Clinical WFH score, mean ± SD | 37.6 ± 21.2 |
| RadiographicPettersson score, mean ± SD | 8.46 ± 7.62 |
| US score, mean ± SD | 8.32 ± 4.09 |
| n (%) | |
| <5 | 15 (22.4%) |
| ≥5 | 52 (77.6%) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manetti, M.; Linari, S.; Romano, E.; Rosa, I.; Carulli, C.; Innocenti, M.; Matucci-Cerinic, M.; Ibba-Manneschi, L.; Castaman, G.; Melchiorre, D. TNF-α/TNF-R System May Represent a Crucial Mediator of Proliferative Synovitis in Hemophilia A. J. Clin. Med. 2019, 8, 939. https://doi.org/10.3390/jcm8070939
Manetti M, Linari S, Romano E, Rosa I, Carulli C, Innocenti M, Matucci-Cerinic M, Ibba-Manneschi L, Castaman G, Melchiorre D. TNF-α/TNF-R System May Represent a Crucial Mediator of Proliferative Synovitis in Hemophilia A. Journal of Clinical Medicine. 2019; 8(7):939. https://doi.org/10.3390/jcm8070939
Chicago/Turabian StyleManetti, Mirko, Silvia Linari, Eloisa Romano, Irene Rosa, Christian Carulli, Massimo Innocenti, Marco Matucci-Cerinic, Lidia Ibba-Manneschi, Giancarlo Castaman, and Daniela Melchiorre. 2019. "TNF-α/TNF-R System May Represent a Crucial Mediator of Proliferative Synovitis in Hemophilia A" Journal of Clinical Medicine 8, no. 7: 939. https://doi.org/10.3390/jcm8070939
APA StyleManetti, M., Linari, S., Romano, E., Rosa, I., Carulli, C., Innocenti, M., Matucci-Cerinic, M., Ibba-Manneschi, L., Castaman, G., & Melchiorre, D. (2019). TNF-α/TNF-R System May Represent a Crucial Mediator of Proliferative Synovitis in Hemophilia A. Journal of Clinical Medicine, 8(7), 939. https://doi.org/10.3390/jcm8070939