Current Therapeutic Options in the Treatment of Rheumatoid Arthritis

Abstract
1. Introduction
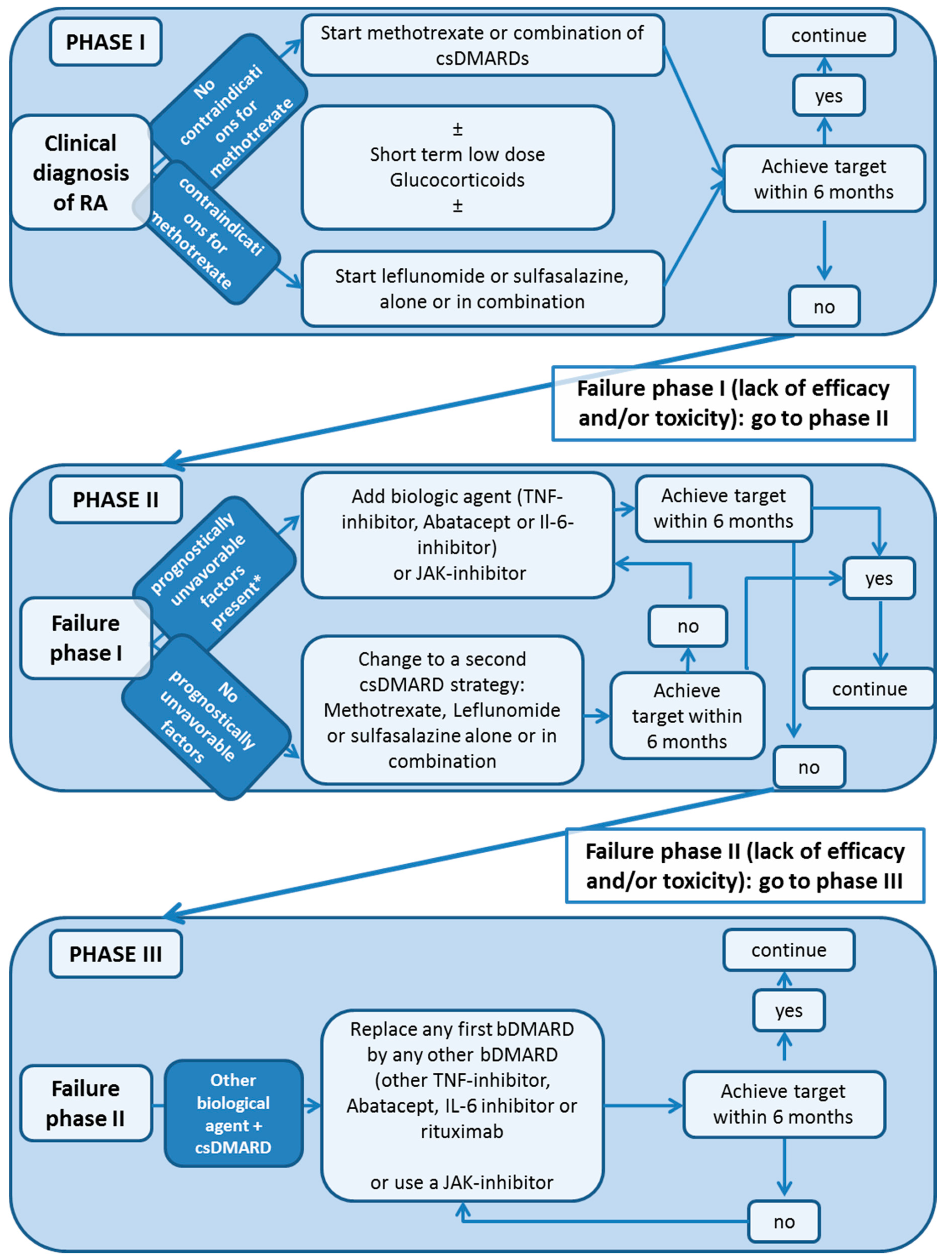
2. Treatment Guidelines: Conventional Synthetic DMARD (csDMARD)
2.1. MTX
2.2. Leflunomide
2.3. Sulfasalazine
3. Biologic DMARDs (bDMARD)
3.1. Tumor Necrosis Factor-Alpha Inhibitors (TNFi)
Pregnancy
3.2. Interleukin 1 Inhibitor
Pregnancy
4. Interleukin 6 and Interleukin 6 Receptor Inhibitors
4.1. Pregnancy
4.2. CD80/86-CD 28 Inhibitor
Pregnancy
5. Anti-CD 20 Antibody
Pregnancy
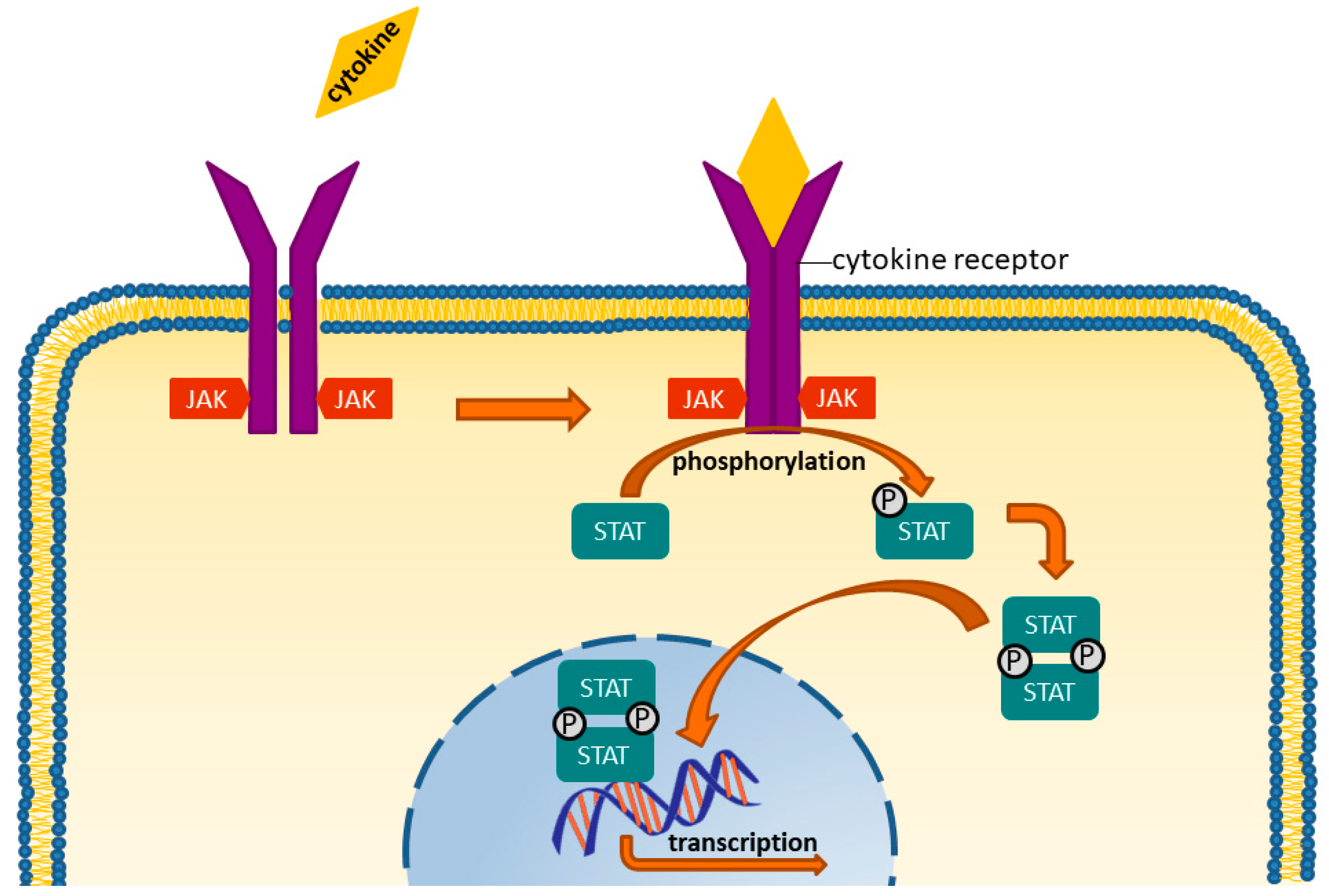
6. Targeted Synthetic DMARDS: JAK-Inhibitors
Side Effects
7. Conclusions
Author Contributions
Conflicts of Interest
References
- Hoes, J.N.; Jacobs, J.W.G.; Boers, M.; Boumpas, D.; Buttgereit, F.; Caeyers, N.; Choy, E.H.; Cutolo, M.; Silva, J.A.P.D.; Esselens, G.; et al. EULAR evidence-based recommendations on the management of systemic glucocorticoid therapy in rheumatic diseases. Ann. Rheum. Dis. 2007, 66, 1560–1567. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Landewe, R.; Breedveld, F.C.; Buch, M.; Burmester, G.; Dougados, M.; Emery, P.; Gaujoux-Viala, C.; Gossec, L.; Nam, J.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2013 update. Ann. Rheum. Dis. 2014, 73, 492–509. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Breedveld, F.C.; Burmester, G.R.; Bykerk, V.; Dougados, M.; Emery, P.; Kvien, T.K.; Navarro-Compan, M.V.; Oliver, S.; Schoels, M.; et al. Treating rheumatoid arthritis to target: 2014 update of the recommendations of an international task force. Ann. Rheum. Dis. 2016, 75, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Aletaha, D.; Smolen, J.S. Diagnosis and management of rheumatoid arthritis: A review. JAMA 2018, 320, 1360–1372. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, C.; Holle, J.; Iking-Konert, C.; Leipe, J.; Weseloh, C.; Frerix, M.; Alten, R.; Behrens, F.; Baerwald, C.; Braun, J.; et al. S2e guideline: Treatment of rheumatoid arthritis with disease-modifying drugs. Z. Rheumatol. 2018, 77, 3553. [Google Scholar]
- Verschueren, P.; De Cock, D.; Corluy, L.; Joos, R.; Langenaken, C.; Taelman, V.; Raeman, F.; Ravelingien, I.; Vandevyvere, K.; Lenaerts, J.; et al. Effectiveness of methotrexate with step-down glucocorticoid remission induction (COBRA Slim) versus other intensive treatment strategies for early rheumatoid arthritis in a treat-to-target approach: 1-year results of CareRA, a randomised pragmatic open-label superiority trial. Ann. Rheum. Dis. 2017, 76, 511–520. [Google Scholar] [PubMed]
- Smolen, J.S.; Kalden, J.R.; Scott, D.L.; Rozman, B.; Kvien, T.K.; Larsen, A.; Loew-Friedrich, I.; Oed, C.; Rosenburg, R. Efficacy and safety of leflunomide compared with placebo and sulphasalazine in active rheumatoid arthritis: A double-blind, randomised, multicentre trial. Lancet 1999, 353, 259–266. [Google Scholar] [CrossRef]
- Brown, P.M.; Pratt, A.G.; Isaacs, J.D. Mechanism of action of methotrexate in rheumatoid arthritis, and the search for biomarkers. Nat. Rev. Rheumatol. 2016, 12, 731–742. [Google Scholar] [CrossRef]
- Kremer, J.M. Major Side Effects of Low-Dose Methotrexate. Available online: www.UpToDate.com (accessed on 2 April 2019).
- Shea, B.; Swinden, M.V.; Ghogomu, E.T.; Ortiz, Z.; Katchamart, W.; Rader, T.; Bombardier, C.; A Wells, G.; Tugwell, P. Folic acid and folinic acid for reducing side effects in patients receiving methotrexate for rheumatoid arthritis. Cochrane Database Syst. Rev. 2013, CD000951. [Google Scholar] [CrossRef]
- Kruger, K.; Albrecht, K.; Rehart, S.; Scholz, R. Recommendations of the German society for rheumatology on the perioperative approach under therapy with DMARDs and biologicals in inflammatory rheumatic diseases. Z. Rheumatol. 2014, 73, 77–84. [Google Scholar] [CrossRef]
- Østensen, M.; Khamashta, M.; Lockshin, M.; Parke, A.; Brucato, A.; Carp, H.; Doria, A.; Rai, R.; Meroni, P.L.; Cetin, I.; et al. Anti-inflammatory and immunosuppressive drugs and reproduction. Arthritis Res. Ther. 2006, 8, 209. [Google Scholar] [CrossRef] [PubMed]
- Breedveld, F.C.; Dayer, J. Leflunomide: Mode of action in the treatment of rheumatoid arthritis. Ann. Rheum. Dis. 2000, 59, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Kruger, K.; Bolten, W. Treatment with leflunomide in rheumatoid arthritis. Z. Rheumatol. 2005, 64, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Fox, R.; Helfgott, S.M. Pharmacology, Dosing, and Adverse Effects of Leflunomide in the Treatment of Rheumatoid Arthritis. Available online: www.UpToDate.com (accessed on 2 April 2019).
- Smedegård, G.; Björk, J. Sulphasalazine: Mechanism of Action in Rheumatoid Arthritis. Rheumatology 1995, 34, 7–15. [Google Scholar] [CrossRef]
- Michael, H.; Weisman, R.Z.R. Sulfasalazine: Pharmacology, Administration, and Adverse Effects in the Treatment of Rheumatoid Arthritis. Available online: www.UpToDate.com (accessed on 2 April 2019).
- Pullar, T.; A Box, S. Sulphasalazine in the treatment of rheumatoid arthritis. Rheumatology 1997, 36, 382–386. [Google Scholar]
- Fachinformation Sulfasalazin HEXAL. Available online: https://s3.eu-central-1.amazonaws.com/prod-cerebro-ifap/media_all/72466.pdf (accessed on 2 April 2019).
- Keffer, J.; Probert, L.; Cazlaris, H.; Georgopoulos, S.; Kaslaris, E.; Kioussis, D.; Kollias, G. Transgenic mice expressing human tumour necrosis factor: A predictive genetic model of arthritis. EMBO J. 1991, 10, 4025–4031. [Google Scholar] [CrossRef] [PubMed]
- Emery, P. Infliximab: A new treatment for rheumatoid arthritis. Hosp. Med. 2001, 62, 150–152. [Google Scholar] [CrossRef]
- Moreland, L.W. Recent advances in anti-tumour necrosis factor (TNF) therapy in rheumatoid arthritis: Focus on the soluble TNF receptor p75 fusion protein, etanercept. BioDrugs 1999, 11, 201–210. [Google Scholar] [CrossRef]
- Mikuls, T.R.; Moreland, L.W. TNF blockade in the treatment of rheumatoid arthritis: Infliximab versus etanercept. Expert Opin. Pharmacother. 2001, 2, 75–84. [Google Scholar] [CrossRef]
- Rau, R. Adalimumab (a fully human anti-tumour necrosis factor alpha monoclonal antibody) in the treatment of active rheumatoid arthritis: The initial results of five trials. Ann. Rheum. Dis. 2002, 61, 70–73. [Google Scholar] [CrossRef]
- Kay, J.; Matteson, E.L.; Dasgupta, B.; Nash, P.; Durez, P.; Hall, S.; Hsia, E.C.; Han, J.; Wagner, C.; Xu, Z.; et al. Golimumab in patients with active rheumatoid arthritis despite treatment with methotrexate: A randomized, double-blind, placebo-controlled, dose-ranging study. Arthritis Rheum. 2008, 58, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.; Landewe, R.B.; Mease, P.; Brzezicki, J.; Mason, D.; Luijtens, K.; van Vollenhoven, R.F.; Kavanaugh, A.; Schiff, M.; Burmester, G.R.; et al. Efficacy and safety of certolizumab pegol plus methotrexate in active rheumatoid arthritis: The RAPID 2 study. A randomised controlled trial. Ann. Rheum. Dis. 2009, 68, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, R.; Vencovsky, J.; van Vollenhoven, R.F.; Borenstein, D.; Box, J.; Coteur, G.; Goel, N.; Brezinschek, H.P.; Innes, A.; Strand, V. Efficacy and safety of certolizumab pegol monotherapy every 4 weeks in patients with rheumatoid arthritis failing previous disease-modifying antirheumatic therapy: The FAST4WARD study. Ann. Rheum. Dis. 2009, 68, 805–811. [Google Scholar] [CrossRef]
- Fleischmann, R.; van Vollenhoven, R.F.; Vencovsky, J.; Alten, R.; Davies, O.; Mountian, I.; de Longueville, M.; Carter, D.; Choy, E. Long-Term Maintenance of Certolizumab Pegol Safety and Efficacy, in Combination with Methotrexate and as Monotherapy, in Rheumatoid Arthritis Patients. Rheumatol. Ther. 2017, 4, 57–69. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Keystone, E.C.; Breedveld, F.C.; Kupper, H.; Li, Y.; Florentinus, S.; Sainsbury, I. Long-term use of adalimumab as monotherapy after attainment of low disease activity with adalimumab plus methotrexate in patients with rheumatoid arthritis. RMD Open 2018, 4, e000637. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, S.; Lu, Y.; Hutchings, R.; Baynton, E.; Hautamaki, E. Comparison of Disease Status And Outcomes of Patients With Rheumatoid Arthritis (Ra) Receiving Adalimumab or Etanercept Monotherapy In Europe. Value Heal. 2014, 17, A374. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gaubitz, M.; Göttl, K.-H.; Behmer, O.; Lippe, R.; Meng, T.; Löschmann, P.-A. Etanercept is effective as monotherapy or in combination with methotrexate in rheumatoid arthritis: Subanalysis of an observational study. Clin. Rheumatol. 2017, 36, 1989–1996. [Google Scholar] [CrossRef] [PubMed]
- Cheifetz, A.; Mayer, L. Monoclonal antibodies, immunogenicity, and associated infusion reactions. Mount Sinai J. Med. 2005, 72, 250–256. [Google Scholar]
- Weinblatt, M.E.; Schiff, M.; Valente, R.; van der Heijde, D.; Citera, G.; Zhao, C.; Maldonado, M.; Fleischmann, R. Head-to-head comparison of subcutaneous abatacept versus adalimumab for rheumatoid arthritis: Findings of a phase IIIb, multinational, prospective, randomized study. Arthritis Rheum. 2013, 65, 28–38. [Google Scholar] [CrossRef]
- Cheifetz, A.; Smedley, M.; Martín, S.; Reiter, M.; Leone, G.; Mayer, L.; Plevy, S. The incidence and management of infusion reactions to infliximab: A large center experience. Am. J. Gastroenterol. 2003, 98, 1315–1324. [Google Scholar] [CrossRef]
- A Wells, G.; Christensen, R.; Ghogomu, E.T.; Maxwell, L.; Macdonald, J.K.; Filippini, G.; Skoetz, N.; Francis, D.K.; Lopes, L.C.; Guyatt, G.H.; et al. Adverse effects of biologics: A network meta-analysis and Cochrane overview. Cochrane Database Syst. Rev. 2011. [Google Scholar]
- Calabrese, L.H.; Calabrese, C.; Kirchner, E. The 2015 American College of Rheumatology Guideline for the Treatment of Rheumatoid Arthritis Should Include New Standards for Hepatitis B Screening: Comment on the Article by Singh et al. Arthritis Rheum. 2016, 68, 723–724. [Google Scholar] [CrossRef] [PubMed]
- Hastings, R.; Ding, T.; Butt, S.; Gadsby, K.; Zhang, W.; Moots, R.J.; Deighton, C. Neutropenia in patients receiving anti-tumor necrosis factor therapy. Arthritis Rheum. 2010, 62, 764–769. [Google Scholar] [CrossRef] [PubMed]
- van Oosten, B.W.; Barkhof, F.; Truyen, L.; Boringa, J.B.; Bertelsmann, F.W.; von Blomberg, B.M.; Woody, J.N.; Hartung, H.P.; Polman, C.H. Increased MRI activity and immune activation in two multiple sclerosis patients treated with the monoclonal anti-tumor necrosis factor antibody cA2. Neurology 1996, 47, 1531–1534. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.J.; Cote, T.R.; Cuffe, M.S.; Kramer, J.M.; Braun, M.M. Case reports of heart failure after therapy with a tumor necrosis factor antagonist. Ann. Intern. Med. 2003, 138, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, S.E. Tumor necrosis factor inhibition: A part of the solution or a part of the problem of heart failure in rheumatoid arthritis? Arthritis Rheum. 2008, 58, 637–640. [Google Scholar] [CrossRef] [PubMed]
- Marchioni, R.M.; Lichtenstein, G.R. Tumor necrosis factor-alpha inhibitor therapy and fetal risk: A systematic literature review. World J. Gastroenterol. 2013, 19, 2591–2602. [Google Scholar] [CrossRef]
- Malek, A.; Sager, R.; Schneider, H. Maternal-fetal transport of immunoglobulin-G and its subclasses during the 3rd trimester of human-pregnancy. Am. J. Reprod. Immunol. 1994, 32, 8–14. [Google Scholar] [CrossRef]
- Mahadevan, U.; Wolf, D.C.; Dubinsky, M.; Cortot, A.; Lee, S.D.; Siegel, C.A.; Ullman, T.; Glover, S.; Valentine, J.F.; Rubin, D.T.; et al. Placental transfer of anti-tumor necrosis factor agents in pregnant patients with inflammatory bowel disease. Clin. Gastroenterol. Hepatol. 2013, 11, 286–292. [Google Scholar] [CrossRef]
- Clowse, M.E.B.; Scheuerle, A.E.; Chambers, C.; Afzali, A.; Kimball, A.B.; Cush, J.J.; Cooney, M.; Shaughnessy, L.; Vanderkelen, M.; Forger, F. Pregnancy Outcomes After Exposure to Certolizumab Pegol: Updated Results From a Pharmacovigilance Safety Database. Arthritis Rheumatol. 2018, 70, 1399–1407. [Google Scholar] [CrossRef]
- Konttinen, L.; Kankaanpää, E.; Luosujärvi, R.; Blåfield, H.; Vuori, K.; Hakala, M.; Rantalaiho, V.; Savolainen, E.; Uutela, T.; Nordström, D.; et al. Effectiveness of anakinra in rheumatic disease in patients naive to biological drugs or previously on TNF blocking drugs: An observational study. Clin. Rheumatol. 2006, 25, 882–884. [Google Scholar] [CrossRef] [PubMed]
- Mertens, M.; Singh, J.A. Anakinra for Rheumatoid Arthritis: A Systematic Review. J. Rheumatol. 2009, 36, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Eungdamrong, J.; Boyd, K.P.; A Meehan, S.; Latkowski, J.-A. Muckle-Wells treatment with anakinra. Dermatol. Online J. 2013, 19. [Google Scholar]
- Kohler, B.M.; Lorenz, H.-M.; Blank, N. IL1-blocking therapy in colchicine-resistant familial Mediterranean fever. Eur. J. Rheumatol. 2018, 5, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Vounotrypidis, P.; Sakellariou, G.T.; Zisopoulos, D.; Berberidis, C. Refractory relapsing polychondritis: Rapid and sustained response in the treatment with an IL-1 receptor antagonist (anakinra). Rheumatology 2006, 45, 491–492. [Google Scholar] [CrossRef] [PubMed]
- Gratton, S.B.; Scalapino, K.J.; Fye, K.H. Case of anakinra as a steroid-sparing agent for gout inflammation. Arthritis Rheum. 2009, 61, 1268–1270. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, R.M.; Tesser, J.; Schiff, M.H.; Schechtman, J.; Burmester, G.-R.; Bennett, R.; Modafferi, D.; Zhou, L.; Bell, D.; Appleton, B. Safety of extended treatment with anakinra in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2006, 65, 1006–1012. [Google Scholar] [CrossRef] [PubMed]
- Chang, Z.; Spong, C.Y.; Jesus, A.A.; Davis, M.A.; Plass, N.; Stone, D.L.; Chapelle, D.; Hoffmann, P.; Kastner, D.L.; Barron, K.; et al. Anakinra Use During Pregnancy in Patients with Cryopyrin-Associated Periodic Syndromes (CAPS). Arthritis Rheumatol. 2014, 66, 3227–3232. [Google Scholar] [CrossRef] [PubMed]
- Gabay, C.; Emery, P.; van Vollenhoven, R.; Dikranian, A.; Alten, R.; Pavelka, K.; Klearman, M.; Musselman, D.; Agarwal, S.; Green, J.; et al. Tocilizumab monotherapy versus adalimumab monotherapy for treatment of rheumatoid arthritis (ADACTA): A randomised, double-blind, controlled phase 4 trial. Lancet 2013, 381, 1541–1550. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, Y.-C.; Fettner, S.; Rowell, L.; Gott, T.; Grimsey, P.; Unsworth, A. Pharmacokinetics and pharmacodynamics of tocilizumab after subcutaneous administration in patients with rheumatoid arthritis. Int. J. Clin. Pharmacol. Ther. 2013, 51, 620–630. [Google Scholar] [CrossRef]
- Burmester, G.R.; Lin, Y.; Patel, R.; van Adelsberg, J.; Mangan, E.K.; Graham, N.M.; van Hoogstraten, H.; Bauer, D.; Ignacio Vargas, J.; Lee, E.B. Efficacy and safety of sarilumab monotherapy versus adalimumab monotherapy for the treatment of patients with active rheumatoid arthritis (MONARCH): A randomised, double-blind, parallel-group phase III trial. Ann. Rheum. Dis. 2017, 76, 840–847. [Google Scholar] [CrossRef] [PubMed]
- Genovese, M.C.; Rubbert-Roth, A.; Smolen, J.S.; Kremer, J.; Khraishi, M.; Gómez-Reino, J.; Sebba, A.; Pilson, R.; Williams, S.; Van Vollenhoven, R. Longterm Safety and Efficacy of Tocilizumab in Patients with Rheumatoid Arthritis: A Cumulative Analysis of Up to 4.6 Years of Exposure. J. Rheumatol. 2013, 40, 768–780. [Google Scholar] [CrossRef] [PubMed]
- Saito, J.; Yakuwa, N.; Kaneko, K.; Takai, C.; Goto, M.; Nakajima, K.; Yamatani, A.; Murashima, A. Tocilizumab during pregnancy and lactation: Drug levels in maternal serum, cord blood, breast milk and infant serum. Rheumatology 2019. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, K.; Watanabe, O.; Mochizuki, M.; Nakasone, A.; Ishizuka, N.; Murashima, A. Pregnancy outcomes after exposure to tocilizumab: A retrospective analysis of 61 patients in Japan. Mod. Rheumatol. 2016, 26, 667–671. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, L.; Singh, J.A. Abatacept for rheumatoid arthritis. Cochrane Database Syst. Rev. 2009, CD007277. [Google Scholar] [CrossRef] [PubMed]
- Herrero-Beaumont, G.; Calatrava, M.J.M.; Castañeda, S. Abatacept Mechanism of Action: Concordance With Its Clinical Profile. Reumatol. Clínica (Engl. Ed.) 2012, 8, 78–83. [Google Scholar] [CrossRef]
- Atzeni, F.; Sarzi-Puttini, P.; Mutti, A.; Bugatti, S.; Cavagna, L.; Caporali, R. Long-term safety of abatacept in patients with rheumatoid arthritis. Autoimmun. Rev. 2013, 12, 1115–1117. [Google Scholar] [CrossRef]
- Smolen, J.S.; Keystone, E.C.; Emery, P.; Breedveld, F.C.; Betteridge, N.; Burmester, G.R.; Dougados, M.; Ferraccioli, G.; Jaeger, U.; Klareskog, L.; et al. Consensus statement on the use of rituximab in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2007, 66, 143–150. [Google Scholar] [CrossRef]
- Harrold, L.R.; John, A.; Best, J.; Zlotnick, S.; Karki, C.; Li, Y.; Greenberg, J.D.; Kremer, J.M. Impact of rituximab on patient-reported outcomes in patients with rheumatoid arthritis from the US Corrona Registry. Clin. Rheumatol. 2017, 36, 2135–2140. [Google Scholar] [CrossRef]
- Chatzidionysiou, K.; Lie, E.; Nasonov, E.; Lukina, G.; Hetland, M.L.; Tarp, U.; Gabay, C.; van Riel, P.L.; Nordstrom, D.C.; Gomez-Reino, J.; et al. Highest clinical effectiveness of rituximab in autoantibody-positive patients with rheumatoid arthritis and in those for whom no more than one previous TNF antagonist has failed: Pooled data from 10 European registries. Ann. Rheum. Dis. 2011, 70, 1575–1580. [Google Scholar] [CrossRef]
- Lee, Y.H.; Bae, S.C.; Song, G.G. The efficacy and safety of rituximab for the treatment of active rheumatoid arthritis: A systematic review and meta-analysis of randomized controlled trials. Rheumatol. Int. 2011, 31, 1493–1499. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, E.F.; Murray, E.R.; Kelman, A.; Farmer, P. Pregnancy outcomes after maternal exposure to rituximab. Blood 2011, 117, 1499–1506. [Google Scholar] [CrossRef] [PubMed]
- Iwata, S.; Tanaka, Y. Progress in understanding the safety and efficacy of Janus kinase inhibitors for treatment of rheumatoid arthritis. Expert Rev. Clin. Immunol. 2016, 12, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Migita, K.; Izumi, Y.; Torigoshi, T.; Satomura, K.; Izumi, M.; Nishino, Y.; Jiuchi, Y.; Nakamura, M.; Kozuru, H.; Nonaka, F.; et al. Inhibition of Janus kinase/signal transducer and activator of transcription (JAK/STAT) signalling pathway in rheumatoid synovial fibroblasts using small molecule compounds. Clin. Exp. Immunol. 2013, 174, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Migita, K.; Koga, T.; Komori, A.; Torigoshi, T.; Maeda, Y.; Izumi, Y.; Sato, J.; Jiuchi, Y.; Miyashita, T.; Yamasaki, S.; et al. Influence of Janus Kinase Inhibition on Interleukin 6-mediated Induction of Acute-phase Serum Amyloid A in Rheumatoid Synovium. J. Rheumatol. 2011, 38, 2309–2317. [Google Scholar] [CrossRef] [PubMed]
- Arkin, M.R.; Wells, J.A. Small-molecule inhibitors of protein–protein interactions: Progressing towards the dream. Nat. Rev. Drug Discov. 2004, 3, 301–317. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Tofacitinib: A Review in Rheumatoid Arthritis. Drugs 2017, 77, 1987–2001. [Google Scholar] [CrossRef]
- Kremer, J.; Li, Z.G.; Hall, S.; Fleischmann, R.; Genovese, M.; Martin-Mola, E.; Isaacs, J.D.; Gruben, D.; Wallenstein, G.; Krishnaswami, S.; et al. Tofacitinib in combination with nonbiologic disease-modifying antirheumatic drugs in patients with active rheumatoid arthritis: A randomized trial. Ann. Intern. Med. 2013, 159, 253–261. [Google Scholar] [CrossRef]
- Taylor, P.C.; Keystone, E.C.; van der Heijde, D.; Weinblatt, M.E.; Del Carmen Morales, L.; Reyes Gonzaga, J.; Yakushin, S.; Ishii, T.; Emoto, K.; Beattie, S.; et al. Baricitinib versus Placebo or Adalimumab in Rheumatoid Arthritis. N. Engl. J. Med. 2017, 376, 652–662. [Google Scholar] [CrossRef]
- Fleischmann, R.; Schiff, M.; van der Heijde, D.; Ramos-Remus, C.; Spindler, A.; Stanislav, M.; Zerbini, C.A.; Gurbuz, S.; Dickson, C.; de Bono, S.; et al. Baricitinib, Methotrexate, or Combination in Patients With Rheumatoid Arthritis and No or Limited Prior Disease-Modifying Antirheumatic Drug Treatment. Arthritis Rheumatol. 2017, 69, 506–517. [Google Scholar] [CrossRef]
- Dougados, M.; van der Heijde, D.; Chen, Y.C.; Greenwald, M.; Drescher, E.; Liu, J.; Beattie, S.; Witt, S.; de la Torre, I.; Gaich, C.; et al. Baricitinib in patients with inadequate response or intolerance to conventional synthetic DMARDs: Results from the RA-BUILD study. Ann. Rheum. Dis. 2017, 76, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, K. Benefit and Risk of Tofacitinib in the Treatment of Rheumatoid Arthritis: A Focus on Herpes Zoster. Drug Saf. 2016, 39, 823–840. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.B.; Koenig, A.; Wang, L.; Kwok, K.; Mebus, C.A.; Riese, R.; Fleischmann, R. Efficacy and safety of tofacitinib in US and non-US rheumatoid arthritis patients: Pooled analyses of phase II and III. Clin. Exp. Rheumatol. 2016, 34, 32–36. [Google Scholar] [PubMed]
- Keystone, E.C.; Taylor, P.C.; Drescher, E.; Schlichting, D.E.; Beattie, S.D.; Berclaz, P.Y.; Lee, C.H.; Fidelus-Gort, R.K.; Luchi, M.E.; Rooney, T.P.; et al. Safety and efficacy of baricitinib at 24 weeks in patients with rheumatoid arthritis who have had an inadequate response to methotrexate. Ann. Rheum. Dis. 2015, 74, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Genovese, M.C.; Takeuchi, T.; Hyslop, D.L.; Macias, W.L.; Rooney, T.; Chen, L.; Dickson, C.L.; Riddle Camp, J.; Cardillo, T.E.; et al. Safety Profile of Baricitinib in Patients with Active Rheumatoid Arthritis with over 2 Years Median Time in Treatment. J. Rheumatol. 2019, 46, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Westhovens, R. Clinical efficacy of new JAK inhibitors under development. Just more of the same? Rheumatology 2019, 58 (Suppl. 1), i27–i33. [Google Scholar] [CrossRef]
- Serhal, L.; Edwards, C.J. Upadacitinib for the treatment of rheumatoid arthritis. Expert Rev. Clin. Immunol. 2018, 15, 1–13. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Overarching Principles of the T2T Strategy | |
|---|---|
| 1 | Basis for the treatment is a shared decision making between patient and doctor |
| 2 | Major treatment goals are: maximization of quality of life, normalisation of function and participation in social and professional life |
| 3 | The elimination of inflammation is essential to achieve the treatment goals |
| 4 | Outcomes in rheumatoid arthritis are improved by implementing T2T |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Köhler, B.M.; Günther, J.; Kaudewitz, D.; Lorenz, H.-M. Current Therapeutic Options in the Treatment of Rheumatoid Arthritis. J. Clin. Med. 2019, 8, 938. https://doi.org/10.3390/jcm8070938
Köhler BM, Günther J, Kaudewitz D, Lorenz H-M. Current Therapeutic Options in the Treatment of Rheumatoid Arthritis. Journal of Clinical Medicine. 2019; 8(7):938. https://doi.org/10.3390/jcm8070938
Chicago/Turabian StyleKöhler, Birgit M., Janine Günther, Dorothee Kaudewitz, and Hanns-Martin Lorenz. 2019. "Current Therapeutic Options in the Treatment of Rheumatoid Arthritis" Journal of Clinical Medicine 8, no. 7: 938. https://doi.org/10.3390/jcm8070938
APA StyleKöhler, B. M., Günther, J., Kaudewitz, D., & Lorenz, H.-M. (2019). Current Therapeutic Options in the Treatment of Rheumatoid Arthritis. Journal of Clinical Medicine, 8(7), 938. https://doi.org/10.3390/jcm8070938