Molecular Genetics and Interferon Signature in the Italian Aicardi Goutières Syndrome Cohort: Report of 12 New Cases and Literature Review

 , ,
, , 
 , add
Show full author list
, add
Show full author list
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Genetic Tests and Data Analysis
2.3. Multiplex Ligation-Dependent Probe Amplification (MLPA)
2.4. Interferon Signature
3. Results
3.1. RNASEH2B Variants
3.2. TREX1 Variants
3.3. RNASEH2A Variants
3.4. RNASEH2C Variants
3.5. SAMHD1 Variants
3.6. ADAR1 Variants
3.7. IFIH1 variants
3.8. AGS Patients without Mutation in AGS-Related Genes
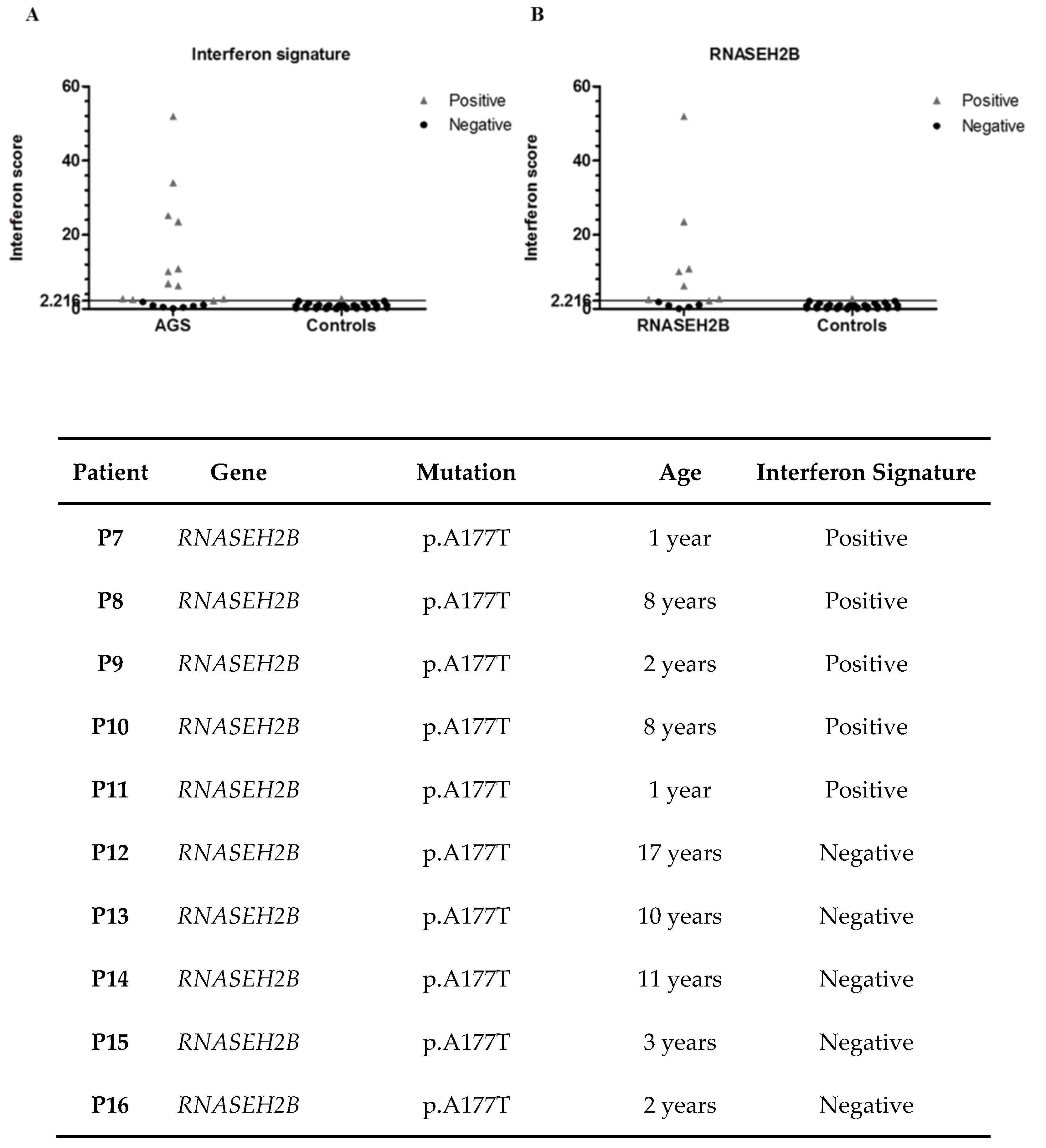
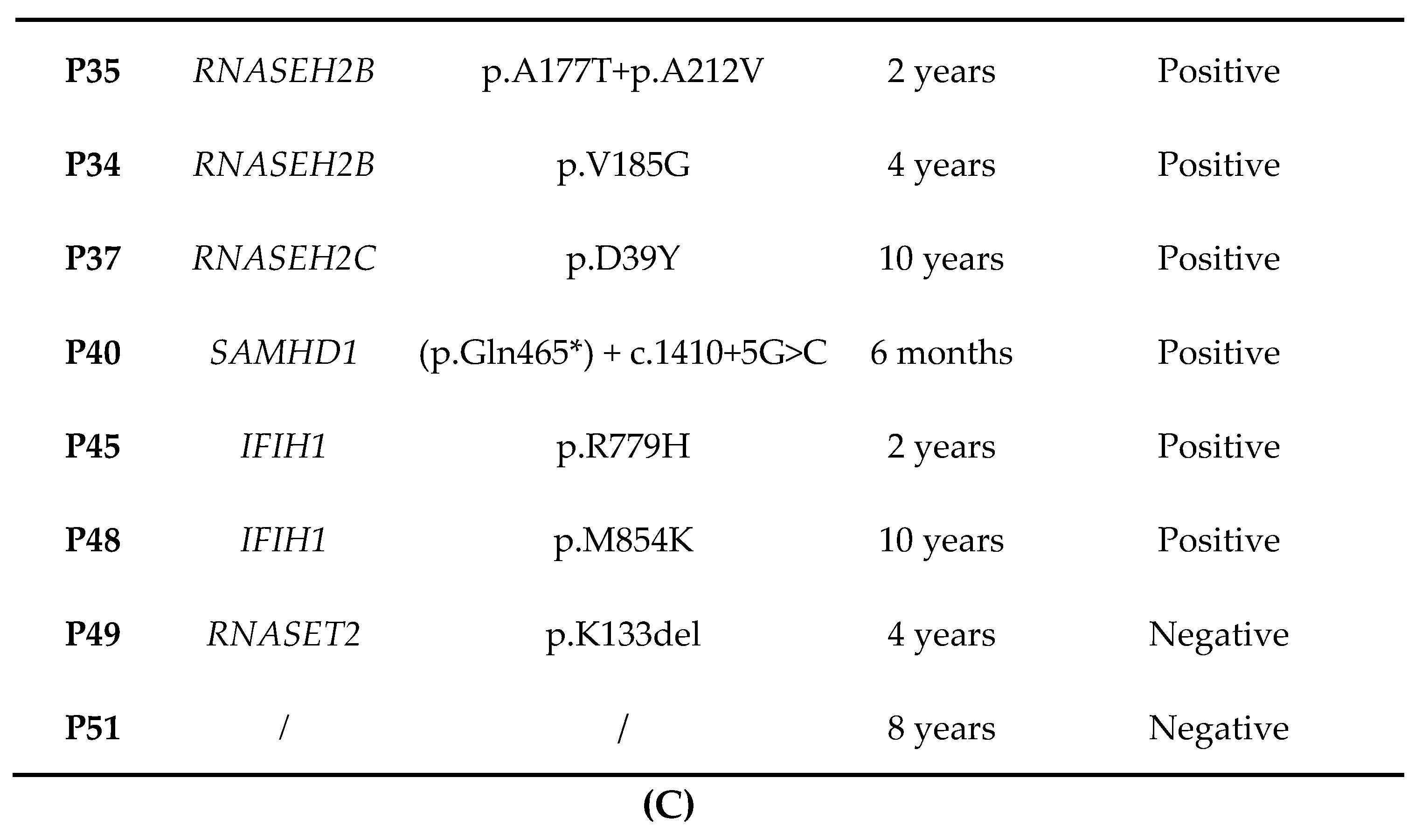
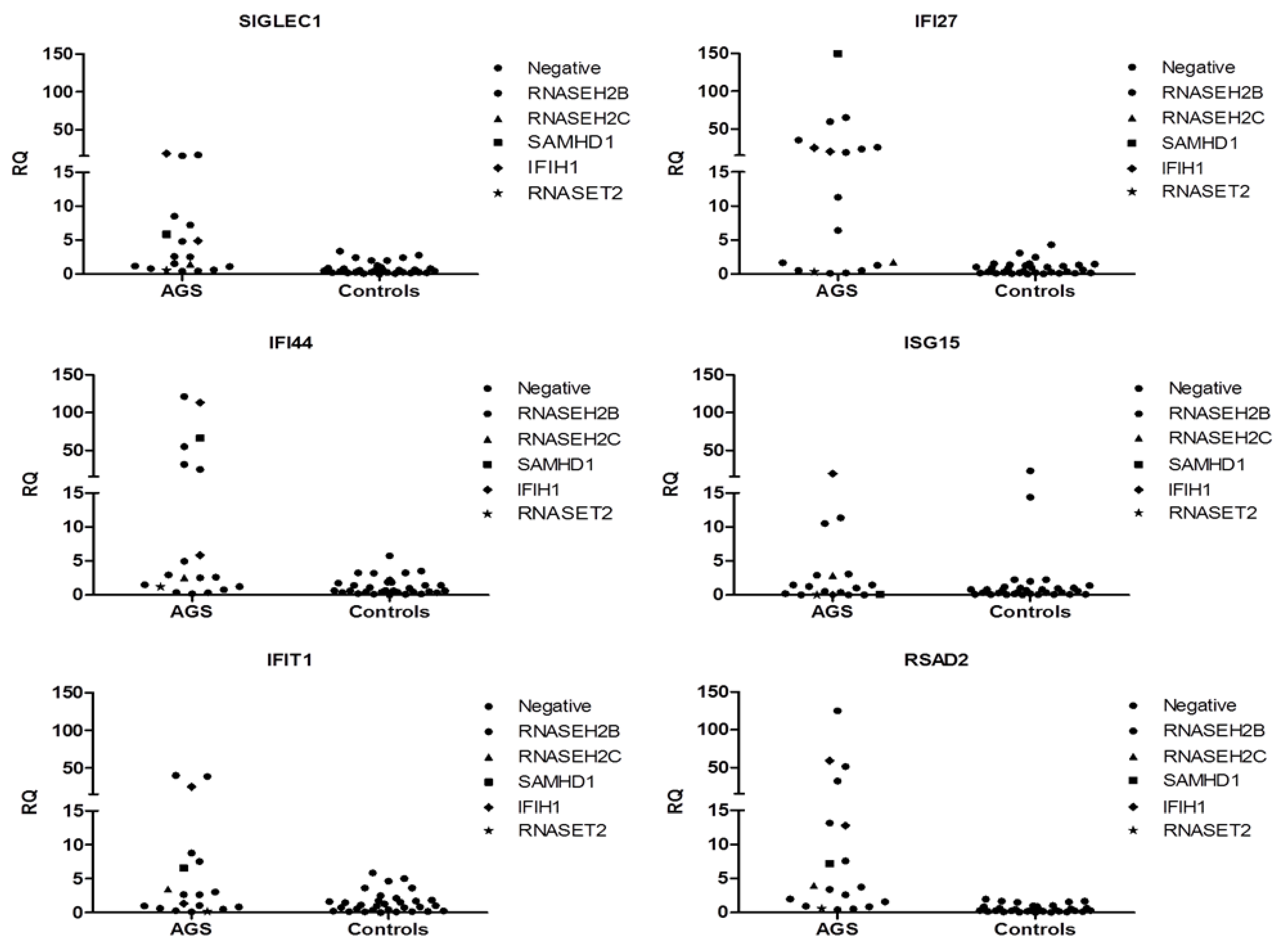
3.9. Interferon Signature
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Crow, Y.J.; Chase, D.S.; Lowenstein Schmidt, J.; Szynkiewicz, M.; Forte, G.M.; Gornall, H.L.; Oojageer, A.; Anderson, B.; Pizzino, A.; Helman, G.; et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am. J. Med. Genet. A. 2015, 167, 296–312. [Google Scholar] [CrossRef] [PubMed]
- Rice, G.; Patrick, T.; Parmar, R.; Taylor, C.F.; Aeby, A.; Aicardi, J.; Artuch, R.; Montalto, S.A.; Bacino, C.A.; Barroso, B.; et al. Clinical and molecular phenotype of Aicardi-Goutieres syndrome. Am. J. Hum. Genet. 2007, 81, 713–725. [Google Scholar] [CrossRef]
- Vanderver, A.; Prust, M.; Kadom, N.; Demarest, S.; Crow, Y.J.; Helman, G.; Orcesi, S.; La Piana, R.; Uggetti, C.; Wang, J.; et al. Early-Onset Aicardi-Goutières Syndrome: Magnetic Resonance Imaging (MRI) Pattern Recognition. J. Child. Neurol. 2015, 30, 1343–1348. [Google Scholar] [CrossRef] [PubMed]
- Fazzi, E.; Cattalini, M.; Orcesi, S.; Tincani, A.; Andreoli, L.; Balottin, U.; De Simone, M.; Fredi, M.; Facchetti, F.; Galli, J.; et al. Aicardi-Goutieres syndrome, a rare neurological disease in children: A new autoimmune disorder? Autoimmun. Rev. 2013, 12, 506–509. [Google Scholar] [CrossRef] [PubMed]
- Livingston, J.H.; Crow, Y.J. Neurologic Phenotypes Associated with Mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1, and IFIH1: Aicardi-Goutières Syndrome and Beyond. Neuropediatrics 2016, 47, 355–360. [Google Scholar] [PubMed]
- Orcesi, S.; Pessagno, A.; Biancheri, R.; La Piana, R.; Mascaretti, M.; Rossi, A.; Rice, G.I.; Crow, Y.J.; Fazzi, E.; Veneselli, E. Aicardi-Goutières syndrome presenting atypically as a sub-acute leukoencephalopathy. Eur. J. Paediatr. Neurol. 2008, 12, 408–411. [Google Scholar] [CrossRef] [PubMed]
- Orcesi, S.; La Piana, R.; Fazzi, E. Aicardi-Goutières syndrome. Br. Med. Bull. 2009, 89, 183–201. [Google Scholar] [CrossRef]
- Goutières, F. Aicardi-Goutières syndrome. Brain Dev. 2005, 27, 201–206. [Google Scholar] [CrossRef]
- Lebon, P.; Badoual, J.; Ponsot, G.; Goutières, F.; Hémeury-Cukier, F.; Aicardi, J. Intrathecal synthesis of interferon-alpha in infants with progressive familial encephalopathy. J. Neurol. Sci. 1988, 84, 201–208. [Google Scholar] [CrossRef]
- van Heteren, J.T.; Rozenberg, F.; Aronica, E.; Troost, D.; Lebon, P.; Kuijpers, T.W. Astrocytes produce interferon-alpha and CXCL10, but not IL-6 or CXCL8, in Aicardi-Goutières syndrome. Glia 2008, 56, 568–578. [Google Scholar] [CrossRef] [PubMed]
- Baechler, E.C.; Batliwalla, F.M.; Karypis, G.; Gaffney, P.M.; Ortmann, W.A.; Espe, K.J.; Shark, K.B.; Grande, W.J.; Hughes, K.M.; Kapur, V.; et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. USA 2003, 100, 2610–2615. [Google Scholar] [CrossRef]
- Bennett, L.; Palucka, A.K.; Arce, E.; Cantrell, V.; Borvak, J.; Banchereau, J.; Pascual, V. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 2003, 197, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Rice, G.I.; Forte, G.M.; Szynkiewicz, M.; Chase, D.S.; Aeby, A.; Abdel-Hamid, M.S.; Ackroyd, S.; Allcock, R.; Bailey, K.M.; Balottin, U.; et al. Assessment of interferon-related biomarkers in Aicardi-Goutières syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: A case-control study. Lancet Neurol. 2013, 12, 1159–1169. [Google Scholar] [CrossRef]
- Höss, M.; Robins, P.; Naven, T.J.; Pappin, D.J.; Sgouros, J.; Lindahl, T. A human DNA editing enzyme homologous to the Escherichia coli DnaQ/MutD protein. EMBO J. 1999, 18, 3868–3875. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.; Backlund, P.S.; Chen, H.; Karavanov, A.A.; Crouch, R.J. RNase H2 of Saccharomyces cerevisiae is a complex of three proteins. Nucleic Acids Res. 2004, 32, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Keegan, L.P.; Leroy, A.; Sproul, D.; O’Connell, M.A. Adenosine deaminases acting on RNA (ADARs): RNA-editing enzymes. Genome Biol. 2004, 5, 209. [Google Scholar] [CrossRef] [PubMed]
- Goldstone, D.C.; Ennis-Adeniran, V.; Hedden, J.J.; Groom, H.C.; Rice, G.I.; Christodoulou, E.; Walker, P.A.; Kelly, G.; Haire, L.F.; Yap, M.W.; et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 2011, 480, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Diamond, J. Autosomal dominant IFIH1 gain-of-function mutations cause Aicardi-Goutières syndrome. Clin. Genet. 2014, 86, 473–474. [Google Scholar] [CrossRef]
- Livingston, J.H.; Stivaros, S.; van der Knaap, M.S.; Crow, Y.J. Recognizable phenotypes associated with intracranial calcification. Dev. Med. Child. Neurol. 2013, 55, 46–57. [Google Scholar] [CrossRef]
- Tonduti, D.; Panteghini, C.; Pichiecchio, A.; Decio, A.; Carecchio, M.; Reale, C.; Moroni, I.; Nardocci, N.; Campistol, J.; Garcia-Cazorla, A.; et al. Encephalopathies with intracranial calcification in children: Clinical and genetic characterization. Orphanet. J. Rare Dis. 2018, 13, 135. [Google Scholar] [CrossRef]
- Palisano, R.; Rosenbaum, P.; Walter, S.; Russell, D.; Wood, E.; Galuppi, B. Development and reliability of a system to classify gross motor function in children with cerebral palsy. Dev. Med. Child. Neurol. 1997, 39, 214–223. [Google Scholar] [CrossRef]
- Eliasson, A.C.; Krumlinde-Sundholm, L.; Rösblad, B.; Beckung, E.; Arner, M.; Ohrvall, A.M.; Rosenbaum, P. The Manual Ability Classification System (MACS) for children with cerebral palsy: Scale development and evidence of validity and reliability. Dev. Med. Child. Neurol. 2006, 48, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Hidecker, M.J.; Paneth, N.; Rosenbaum, P.L.; Kent, R.D.; Lillie, J.; Eulenberg, J.B.; Chester, K., Jr.; Johnson, B.; Michalsen, L.; Evatt, M.; et al. Developing and validating the Communication Function Classification System for individuals with cerebral palsy. Dev. Med. Child. Neurol. 2011, 53, 704–710. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, I.; Cattalini, M.; Tonduti, D.; La Piana, R.; Uggetti, C.; Galli, J.; Meini, A.; Tincani, A.; Moratto, D.; Fazzi, E.; et al. Dysregulation of the immune system in Aicardi-Goutières syndrome: Another example in a TREX1-mutated patient. Lupus 2013, 22, 1064–1069. [Google Scholar] [CrossRef]
- Rice, G.I.; Reijns, M.A.; Coffin, S.R.; Forte, G.M.; Anderson, B.H.; Szynkiewicz, M.; Gornall, H.; Gent, D.; Leitch, A.; Botella, M.P.; et al. Synonymous mutations in RNASEH2A create cryptic splice sites impairing RNase H2 enzyme function in Aicardi-Goutières syndrome. Hum. Mutat. 2013, 34, 1066–1070. [Google Scholar] [CrossRef]
- D’Arrigo, S.; Riva, D.; Bulgheroni, S.; Chiapparini, L.; Lebon, P.; Rice, G.; Crow, Y.J.; Pantaleoni, C. Aicardi-Goutières syndrome: Description of a late onset case. Dev. Med. Child. Neurol. 2008, 50, 631–634. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, V.; Bernardi, B.; Stafa, A.; Garone, C.; Franzoni, E.; Abinun, M.; Mitchell, P.; Mitra, D.; Friswell, M.; Nelson, J.; et al. Intracerebral large artery disease in Aicardi-Goutières syndrome implicates SAMHD1 in vascular homeostasis. Dev. Med. Child. Neurol. 2010, 52, 725–732. [Google Scholar] [CrossRef]
- La Piana, R.; Uggetti, C.; Olivieri, I.; Tonduti, D.; Balottin, U.; Fazzi, E.; Orcesi, S. Bilateral striatal necrosis in two subjects with Aicardi-Goutières syndrome due to mutations in ADAR1 (AGS6). Am. J. Med. Genet. A. 2014, 164, 815–819. [Google Scholar] [CrossRef]
- Rice, G.I.; Del Toro Duany, Y.; Jenkinson, E.M.; Forte, G.M.; Anderson, B.H.; Ariaudo, G.; Bader-Meunier, B.; Baildam, E.M.; Battini, R.; Beresford, M.W.; et al. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat. Genet. 2014, 46, 503–509. [Google Scholar] [CrossRef]
- Galli, J.; Gavazzi, F.; De Simone, M.; Giliani, S.; Garau, J.; Valente, M.; Vairo, D.; Cattalini, M.; Mortilla, M.; Andreoli, L.; et al. Sine causa tetraparesis: A pilot study on its possible relationship with interferon signature analysis and Aicardi Goutières syndrome related genes analysis. Medicine 2018, 97, e13893. [Google Scholar] [CrossRef]
- Tonduti, D.; Orcesi, S.; Jenkinson, E.M.; Dorboz, I.; Renaldo, F.; Panteghini, C.; Rice, G.I.; Henneke, M.; Livingston, J.H.; Elmaleh, M.; et al. Clinical, radiological and possible pathological overlap of cystic leukoencephalopathy without megalencephaly and Aicardi-Goutières syndrome. Eur. J. Paediatr. Neurol. 2016, 20, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, R. The Griffiths Mental Development Scales from birth to 2 years; National Testing Agency: Henley, UK, 1996. [Google Scholar]
- Wechsler, D. Wechsler Preschool and Primary Scale of Intelligence -Revised (WPPSI-R): Short Form Vocabulary and Block Design; The Psychological Corporation: Amersham, UK, 1989. [Google Scholar]
- Wechsler, D. Wechsler Intelligence Scale for Children, 3rd ed.; Psychological Corporation: San Antonio, CA, USA, 1992. [Google Scholar]
- Zucca, S.; Villaraggia, M.; Gagliardi, S.; Grieco, G.S.; Valente, M.; Cereda, C.; Magni, P. Analysis of amplicon-based NGS data from neurological disease gene panels: A new method for allele drop-out management. BMC Bioinformatics. 2016, 17, 339. [Google Scholar] [CrossRef] [PubMed]
- Rice, G.I.; Melki, I.; Frémond, M.L.; Briggs, T.A.; Rodero, M.P.; Kitabayashi, N.; Oojageer, A.; Bader-Meunier, B.; Belot, A.; Bodemer, C.; et al. Assessment of Type I Interferon Signaling in Pediatric Inflammatory Disease. J. Clin. Immunol. 2017, 37, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Haud, N.; Kara, F.; Diekmann, S.; Henneke, M.; Willer, J.R.; Hillwig, M.S.; Gregg, R.G.; Macintosh, G.C.; Gärtner, J.; Alia, A.; et al. Rnaset2 mutant zebrafish model familial cystic leukoencephalopathy and reveal a role for RNase T2 in degrading ribosomal RNA. Proc. Natl. Acad. Sci. USA 2011, 108, 1099–1103. [Google Scholar] [CrossRef]
- Al Mutairi, F.; Alfadhel, M.; Nashabat, M.; El-Hattab, A.W.; Ben-Omran, T.; Hertecant, J.; Eyaid, W.; Ali, R.; Alasmari, A.; Kara, M.; et al. Phenotypic and Molecular Spectrum of Aicardi-Goutières Syndrome: A Study of 24 Patients. Pediatr. Neurol. 2018, 78, 35–40. [Google Scholar] [CrossRef]
- Abe, J.; Nakamura, K.; Nishikomori, R.; Kato, M.; Mitsuiki, N.; Izawa, K.; Awaya, T.; Kawai, T.; Yasumi, T.; Toyoshima, I.; et al. A nationwide survey of Aicardi-Goutières syndrome patients identifies a strong association between dominant TREX1 mutations and chilblain lesions: Japanese cohort study. Rheumatology 2014, 53, 448–458. [Google Scholar] [CrossRef]
- Figiel, M.; Chon, H.; Cerritelli, S.M.; Cybulska, M.; Crouch, R.J.; Nowotny, M. The structural and biochemical characterization of human RNase H2 complex reveals the molecular basis for substrate recognition and Aicardi-Goutières syndrome defects. J. Biol. Chem. 2011, 286, 10540–10550. [Google Scholar] [CrossRef]
- Reijns, M.A.; Bubeck, D.; Gibson, L.C.; Graham, S.C.; Baillie, G.S.; Jones, E.Y.; Jackson, A.P. The structure of the human RNase H2 complex defines key interaction interfaces relevant to enzyme function and human disease. J. Biol. Chem. 2011, 286, 10530–10539. [Google Scholar] [CrossRef]
- Mackenzie, K.J.; Carroll, P.; Lettice, L.; Tarnauskaitė, Ž.; Reddy, K.; Dix, F.; Revuelta, A.; Abbondati, E.; Rigby, R.E.; Rabe, B.; et al. Ribonuclease H2 mutations induce a cGAS/STING-dependent innate immune response. EMBO J. 2016, 35, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Tüngler, V.; Schmidt, F.; Hieronimus, S.; Reyes-Velasco, C.; Lee-Kirsch, M.A. Phenotypic variability in a family with aicardi-goutières syndrome due to the common A177T RNASEH2B mutation. Case Rep. Clin. Med. 2014, 3, 153–156. [Google Scholar] [CrossRef]
- Pizzi, S.; Sertic, S.; Orcesi, S.; Cereda, C.; Bianchi, M.; Jackson, A.P.; Lazzaro, F.; Plevani, P.; Muzi-Falconi, M. Reduction of hRNase H2 activity in Aicardi-Goutières syndrome cells leads to replication stress and genome instability. Hum. Mol. Genet. 2015, 24, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Chon, H.; Vassilev, A.; DePamphilis, M.L.; Zhao, Y.; Zhang, J.; Burgers, P.M.; Crouch, R.J.; Cerritelli, S.M. Contributions of the two accessory subunits, RNASEH2B and RNASEH2C, to the activity and properties of the human RNase H2 complex. Nucleic Acids Res. 2009, 37, 96–110. [Google Scholar] [CrossRef]
- Baranzini, N.; Pedrini, E.; Girardello, R.; Tettamanti, G.; de Eguileor, M.; Taramelli, R.; Acquati, F.; Grimaldi, A. Human recombinant RNASET2-induced inflammatory response and connective tissue remodeling in the medicinal leech. Cell Tissue Res. 2017, 368, 337–351. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Gene | Variant | Amino Acidic Substitution | Number of Patients (Gender) | Annotation * | Onset ** | Clinical Score # | Clinical Phenotype | Epilepsy | GQ/IQ ° | Chilblains and/or Recurrent Fevers | Other |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | TREX1 | c.341G>A | p.R114H | 1 (Male) | Described [1] | Prenatal/neonatal | ≥12 | Spastic tetraparesis | Yes | Not evaluable | No | s- |
| P2 | TREX1 | c.262 ins AG + c.290G>A | p.S88Kfs* + p.R97H | 1(Male) | Described [25] | Prenatal/neonatal | ≥12 | Spastic-dystonic tetraparesis | No | Not evaluable | No | Antiphospholipid syndrome, thyroiditis, cerebral ischemia |
| P3 | TREX1 | c.150_151del | p.N51Gfs*50 | 1 (Male) | Described [1] | Prenatal/neonatal | ≥12 | Spastic tetraparesis | No | Not evaluable | Yes | Cardiomyopathy |
| P4 | TREX1 | c.868_885del + c.341G>A | p.P290_A295del + p.R114H | 1(Male) | Described [1] | Prenatal/neonatal | ≥12 | Spastic-dystonic tetraparesis | Yes | Not evaluable | Yes | Cardiomyopathy, pulmonary hypertension, sensoryneural hearing loss |
| P5 | RNASEH2A | c.322C>T + c.690C>A | p.R108W + p.F231L | 1(Female) | Described [1] | Prenatal/neonatal | ≥12 | Spastic-dystonic tetraparesis | No | Not evaluable | No | Celiac disease |
| P6 | RNASEH2A | c.556C>T + c.69G>A | p.R186W + p.V23V | 1 (Male) | Described [26] | Prenatal/neonatal | ≥12 | Spastic tetraparesis | Yes | Not evaluable | No | - |
| P7-P23 | RNASEH2B | c.529G>A | p.A177T | 17 (9 Males, 8 Females) | Described [1,21] | Infantile to later onset | 4 patients ≤ 6 2 patients 6–12 11 patients≥ 12 | Variable (Hemiparesis/Spastic diplegia/hypotonic-dystonic syndrome/Spastic-dystonic tetraparesis) | Yes (4 patients) No (13 patients) | Variable (from notevaluable to 90) | Yes/No | Variable neuroradiological features (brain calcification/no calcification/diffuse microcalcification/MRI normalization at follow up) |
| P24-P30 | RNASEH2B | c.529G>A + c.488C>T | p.A177T + p.T163I | 7 (2 Males, 5 Females) | Described [1] | Prenatal to infantile | ≥12 | Spastic-dystonic tetraparesis | Yes (3 patients) No (4 patients) | Variable (from not evaluable to >50) | No | Variable (no other features to celiac disease) |
| P31-P32† | RNASEH2B | c.529G>A + c.218G>T | p.A177T + p.W73L | 2 (1 Male, 1 Female) | Described [27] | Infantile | 6–12 (1 patient 11 1 patient 8) | Spastic-dystonic tetraparesis | No | 1 patient <501 patient not evaluable | Yes | Variable neuroradiological features (brain calcification/ no calcification) |
| P33 | RNASEH2B | c.529G>A +c.Ex9_Ex11del | p.A177T + p.Ex9_Ex11del | 1 (Male) | Novel | Prenatal/neonatal | ≥12 | Spastic-dystonic tetraparesis | Yes | Not evaluable | Yes | - |
| P34 | RNASEH2B | c.554T>G | p.V185G | 1 (Male) | Described [1] | Prenatal/neonatal | ≥12 | Spastic-dystonic tetraparesis | Yes | Not evaluable | Yes | - |
| P35 | RNASEH2B | c.529G>A + c.635C>T | p.A177T + p.A212V | 1 (Female) | Novel | Infantile | ≥12 | Spastic-dystonic tetraparesis | No | Not evaluable | Yes | - |
| P36 | RNASEH2B | c.529G>A + c.64+1G>A | p.A177T | 1 (Male) | Described [1] | Infantile | ≥12 | Spastic tetraparesis | Yes | Not evaluable | Yes | - |
| P37 | RNASEH2C | c.115G>T + c.173-1G>C | p.D39Y | 1 (Male) | Described [1] | Infantile | ≥12 | Spastic-dystonic tetraparesis | No | Not evaluable | Yes | - |
| P38-P39† | SAMHD1 | c.410A>G | p.D137G | 2 (1 Male, 1 Female) | Novel | Infantile | 1patient 6 1 patient ≥12 | Spastic paraparesis/spastic-dystonic tetraparesis | No | 1 patient 60 1 patient <50 | Yes | - |
| P40 | SAMHD1 | c.1393C>T +c.1410+5G>C | p.Q465* | 1 (Female) | Novel | Infantile | ≥12 | Spastic-dystonic tetraparesis | No | <50 | Yes | - |
| P41 | SAMHD1 | Ex12_Ex16del | p.Ex12_Ex16del | 1 (Male) | Described [28] | Infantile | ≥12 | Spastic-dystonic tetraparesis | Yes | <50 | No | Cerebral vasculitis, three intracranial aneurysms |
| P42 | ADAR1 | c.577C>G + c.2608G>A | p.P193A + p.A870T | 1 (Male) | Described [1,29] | Infantile | ≥12 | Spastic-dystonic tetraparesis | No | Not evaluable | No | Striatal necrosis |
| P43 | IFIH1 | c.1178A>T | p.D393V | 1 (Male) | Described [1,30] | Later onset | ≥12 | Spastic-dystonic tetraparesis | Yes | Not evaluable | No | - |
| P44 | IFIH1 | c.2471G>A | p.R824K | 1 (Male) | Described [31] | Infantile | ≥12 | Spastic-dystonic tetraparesis | No | Not evaluable | No | - |
| P45-46 | IFIH1 | c.2336G>A | p.R779H | 2 (2 Males) | Described [1] | Infantile | ≥12 | Spastic-dystonic tetraparesis | No | Not evaluable | Yes | - |
| P47 | IFIH1 | c.2159G>A | p.R720Q | 1 (Male) | Described [1] | Infantile | ≥12 | Spastic tetraparesis | Yes | Not evaluable | Yes | |
| P48 | IFIH1 | c.2561T>A | p.M854K | 1 (Male) | Novel | Later onset | <6 | Spastic paraparesis | Yes | 97 | No | Erythematous cheeks, lentiges, hyperkeratotic lesions, glaucoma, abnormal dentition, demyelinating sensory-motor polyneuropathy |
| P49 | RNASET2 | c.397_399delAAG + c.145G > T | p.K133del +p.E49* | 1 (Male) | Described [32] | Infantile | ≥12 | Spastic-dystonic tetraparesis | No | Not evaluable | No | Cerebellar atrophy |
| P50 | / | / | / | 1 (Male) | / | Infantile | ≥12 | Spastic-dystonic tetraparesis | No | < 50 | No | Hypopigmented lesions, thyroiditis |
| P51 | / | / | / | 1 (Female) | / | Infantile | <6 | Spastic diplegia | Yes | <50 | Yes | Hypochromic lesions on trunk |
| Gene | Mutation | Homozygous/Heterozygous % | MAF £ (ExAC) |
|---|---|---|---|
| TREX1 | p.S88Kfs* | 0/1 | NA |
| p.R97H | 0/1 | T = 0.000008/1 | |
| p.R114H | 1/1 | A = 0.0002/19 | |
| p.N51Gfs*50 | 1/0 | NA | |
| p.P290_A295del | 0/1 | - = 0.00007/8 | |
| p.R169H | 0/1 | A = 0.0002/19 | |
| RNASEH2A | p.R108W | 0/1 | A = 0.000008/1 |
| p.F230L | 0/1 | A = 0.000008/1 | |
| p.R186W | 0/1 | NA | |
| p.V23V | 0/1 | A = 0.00002/3 | |
| RNASEH2B | p.W73L | 0/2 | NA |
| p.T163I | 0/7 | NA | |
| p.A177T | 17/12 | A = 0.0013/158 | |
| p.V185G | 1/0 | NA | |
| p.A212V | 0/1 | NA | |
| p.Ex9_Ex11del | 0/1 | NA | |
| c.64+1G>A | 0/1 | NA | |
| RNASEH2C | p.D39Y | 0/1 | NA |
| c.173-1G>C | 0/1 | ||
| SAMHD1 | p.D137G | 2/0 | T = 0.000008/1 |
| p.Q465* | 0/1 | NA | |
| c.1410+5G>C | 0/1 | NA | |
| p.Ex12_Ex16del | 1/0 | NA | |
| ADAR1 | p.P193A | 0/1 | C = 0.0021/260 |
| p.A870T | 0/1 | NA | |
| IFIH1 | p.D393V | 0/1 | C = 0.000008/1 |
| p.R720Q | 0/1 | NA | |
| p.R824K | 0/1 | NA | |
| p.R779H | 0/2 | NA | |
| p.M854K | 0/1 | NA | |
| RNASET2 | p.K133del | 0/1 | - = 0.00002/2 |
| p.E49* | 0/1 | NA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garau, J.; Cavallera, V.; Valente, M.; Tonduti, D.; Sproviero, D.; Zucca, S.; Battaglia, D.; Battini, R.; Bertini, E.; Cappanera, S.; et al. Molecular Genetics and Interferon Signature in the Italian Aicardi Goutières Syndrome Cohort: Report of 12 New Cases and Literature Review. J. Clin. Med. 2019, 8, 750. https://doi.org/10.3390/jcm8050750
Garau J, Cavallera V, Valente M, Tonduti D, Sproviero D, Zucca S, Battaglia D, Battini R, Bertini E, Cappanera S, et al. Molecular Genetics and Interferon Signature in the Italian Aicardi Goutières Syndrome Cohort: Report of 12 New Cases and Literature Review. Journal of Clinical Medicine. 2019; 8(5):750. https://doi.org/10.3390/jcm8050750
Chicago/Turabian StyleGarau, Jessica, Vanessa Cavallera, Marialuisa Valente, Davide Tonduti, Daisy Sproviero, Susanna Zucca, Domenica Battaglia, Roberta Battini, Enrico Bertini, Silvia Cappanera, and et al. 2019. "Molecular Genetics and Interferon Signature in the Italian Aicardi Goutières Syndrome Cohort: Report of 12 New Cases and Literature Review" Journal of Clinical Medicine 8, no. 5: 750. https://doi.org/10.3390/jcm8050750
APA StyleGarau, J., Cavallera, V., Valente, M., Tonduti, D., Sproviero, D., Zucca, S., Battaglia, D., Battini, R., Bertini, E., Cappanera, S., Chiapparini, L., Crasà, C., Crichiutti, G., Dalla Giustina, E., D’Arrigo, S., De Giorgis, V., De Simone, M., Galli, J., La Piana, R., ... Cereda, C. (2019). Molecular Genetics and Interferon Signature in the Italian Aicardi Goutières Syndrome Cohort: Report of 12 New Cases and Literature Review. Journal of Clinical Medicine, 8(5), 750. https://doi.org/10.3390/jcm8050750