A Novel Heterozygous Deletion Variant in KLOTHO Gene Leading to Haploinsufficiency and Impairment of Fibroblast Growth Factor 23 Signaling Pathway

 , , ,
, , ,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement and Human Samples
2.2. Gene Sequencing and Variant Analysis
2.3. Laboratory Measurements
2.4. Expression Vector
2.5. Cell Culture Experiments
2.6. RNA Extraction and Quantitative PCR
2.7. Protein Extraction and Western Blot
2.8. Statistical Analysis
3. Results
3.1. Case Report
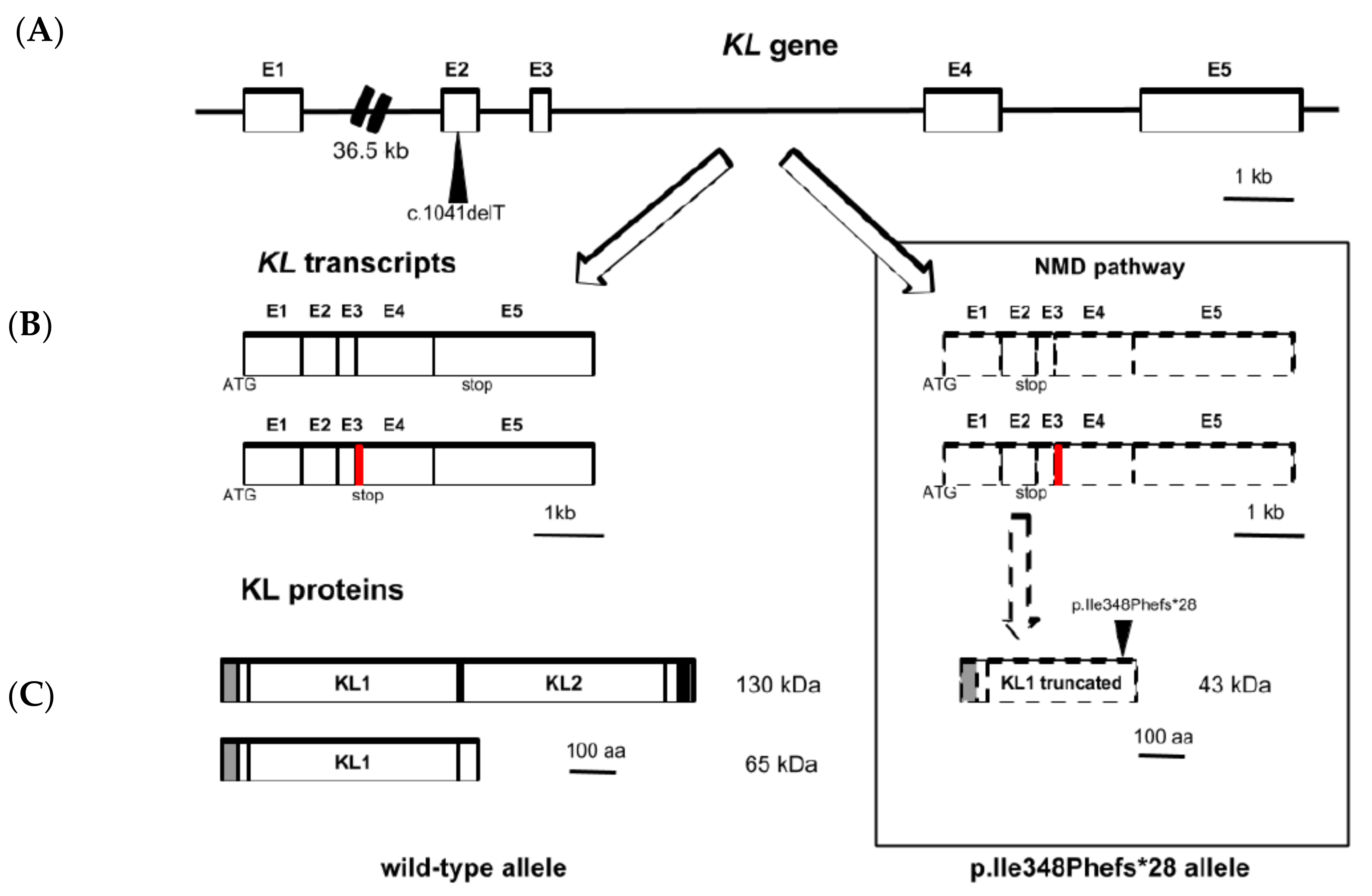
3.2. Sequencing and Variant Analysis of KL Gene
3.3. Soluble KL and FGF23 Serum Levels
3.4. FGF23 Signaling Pathway Activation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vervloet, M.G.; Sezer, S.; Massy, Z.A.; Johansson, L.; Cozzolino, M.; Fouque, D.; ERA–EDTA Working Group on Chronic Kidney Disease–Mineral and Bone Disorders and the European Renal Nutrition Working Group. The role of phosphate in kidney disease. Nat. Rev. Nephrol. 2017, 13, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Sherman, R.A. Hyperphosphatemia in Dialysis Patients: Beyond Nonadherence to Diet and Binders. Am. J. Kidney Dis. 2016, 67, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Shiizaki, K.; Kuro-o, M.; Moe, O.W. Fibroblast growth factor 23 and Klotho: physiology and pathophysiology of an endocrine network of mineral metabolism. Annu. Rev. Phisiol. 2013, 75, 503–533. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, K.; Amin, R.; Moe, O.W.; Hu, M.C.; Erben, R.G.; Östman Wernerson, A.; Lanske, B.; Olauson, H.; Larsson, T.E. The kidney is the principal organ mediating klotho effects. J. Am. Soc. Nephrol. 2006, 25, 2169–2175. [Google Scholar] [CrossRef]
- Kuro-o, M.; Matsumura, Y.; Aizawa, H.; Kawaguchi, H.; Suga, T.; Utsugi, T.; Ohyama, Y.; Kurabayashi, M.; Kaname, T.; Kume, E.; et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997, 390, 45–51. [Google Scholar] [CrossRef]
- Tsujikawa, H.; Kurotaki, Y.; Fujimori, T.; Fukuda, K.; Nabeshima, Y. Klotho, a gene related to a syndrome resembling human premature aging, functions in a negative regulatory circuit of vitamin D endocrine system. Mol. Endocrinol. 2003, 17, 2393–2403. [Google Scholar] [CrossRef]
- Stenvinkel, P.; Larsson, T.E. Chronic kidney disease: a clinical model of premature aging. Am. J. Kidney Dis. 2013, 62, 339–351. [Google Scholar] [CrossRef]
- Ichikawa, S.; Imel, E.A.; Kreiter, M.L.; Yu, X.; Mackenzie, D.S.; Sorenson, A.H.; Goetz, R.; Mohammadi, M.; White, K.E.; Econs, M.J. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J. Clin. Invest. 2007, 117, 2684–2691. [Google Scholar] [CrossRef]
- Brownstein, C.A.; Adler, F.; Nelson-Williams, C.; Iijima, J.; Li, P.; Imura, A.; Nabeshima, Y.; Reyes-Mugica, M.; Carpenter, T.O.; Lifton, R.P. A translocation causing increased alpha-Klotho level results in hypophosphatemic rickets and hyperparathyroidism. PNAS 2008, 105, 3455–3460. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods. 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Imura, A.; Urakawa, I.; Shimada, T.; Murakami, J.; Aono, Y.; Hasegawa, H.; Yamashita, T.; Nakatani, K.; Saito, Y.; et al. Establishment of sandwich ELISA for soluble alpha-Klotho measurement: Age-dependent change of soluble alpha-Klotho levels in healthy subjects. Biochem. Biophys. Res. Commun. 2010, 398, 513–518. [Google Scholar] [CrossRef]
- Askar, A.M. Hyperphosphatemia. The hidden killer in chronic kidney disease. Saudi. Med. J. 2015, 36, 13–19. [Google Scholar] [CrossRef]
- Araya, K.; Fukumoto, S.; Backenroth, R.; Takeuchi, Y.; Nakayama, K.; Ito, N.; Yoshii, N.; Yamazaki, Y.; Yamashita, T.; Silver, J.; et al. A novel mutation in fibroblast growth factor 23 gene as a cause of tumoral calcinosis. J. Clin. Endocrinol. Metab. 2005, 90, 5523–5527. [Google Scholar] [CrossRef]
- Benet-Pages, A.; Orlik, P.; Strom, T.M.; Lorenz-Depiereux, B. An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum. Mol. Genet. 2005, 14, 385–390. [Google Scholar] [CrossRef]
- Chefetz, I.; Heller, R.; Galli-Tsinopoulou, A.; Richard, G.; Wollnik, B.; Indelman, M.; Koerber, F.; Topaz, O.; Bergman, R.; Sprecher, E.; et al. A novel homozygous missense mutation in FGF23 causes Familial Tumoral Calcinosis associated with disseminated visceral calcification. Hum. Genet. 2005, 118, 261–266. [Google Scholar] [CrossRef]
- Larsson, T.; Yu, X.; Davis, S.I.; Draman, M.S.; Mooney, S.D.; Cullen, M.J.; White, K.E. A novel recessive mutation in fibroblast growth factor-23 causes familial tumoral calcinosis. J. Clin. Endocrinol. Metab. 2005, 90, 2424–2427. [Google Scholar] [CrossRef]
- Garringer, H.J.; Malekpour, M.; Esteghamat, F.; Mortazavi, S.M.; Davis, S.I.; Farrow, E.G.; Yu, X.; Arking, D.E.; Dietz, H.C.; White, K.E. Molecular genetic and biochemical analyses of FGF23 mutations in familial tumoral calcinosis. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E929–E937. [Google Scholar] [CrossRef]
- Masi, L.; Gozzini, A.; Franchi, A.; Campanacci, D.; Amedei, A.; Falchetti, A.; Franceschelli, F.; Marcucci, G.; Tanini, A.; Capanna, R.; et al. A novel recessive mutation of fibroblast growth factor-23 in tumoral calcinosis. J. Bone Joint Surg. Am. 2009, 91, 1190–1198. [Google Scholar] [CrossRef]
- Abbasi, F.; Ghafouri-Fard, S.; Javaheri, M.; Dideban, A.; Ebrahimi, A.; Ebrahim-Habibi, A. A new missense mutation in FGF23 gene in a male with hyperostosis-hyperphosphatemia syndrome (HHS). Gene 2014, 542, 269–271. [Google Scholar] [CrossRef]
- Chang, Y.F.; Imam, J.S.; Wilkinson, M.F. The nonsense-mediated decay RNA surveillance pathway. Annu. Rev. Biochem. 2007, 76, 51–74. [Google Scholar] [CrossRef]
- Hug, N.; Longman, D.; Cáceres, J.F. Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res. 2016, 44, 1483–1495. [Google Scholar] [CrossRef]
- Kurosu, H.; Ogawa, Y.; Miyoshi, M.; Yamamoto, M.; Nandi, A.; Rosenblatt, K.P.; Baum, M.G.; Schiavi, S.; Hu, M.C.; Moe, O.W.; et al. Regulation of fibroblast growth factor-23 signaling by klotho. J. Biol. Chem. 2006, 281, 6120–6123. [Google Scholar] [CrossRef]
- Shimada, T.; Hasegawa, H.; Yamazaki, Y.; Muto, T.; Hino, R.; Takeuchi, Y.; Fujita, T.; Nakahara, K.; Fukumoto, S.; Yamashita, T. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostatis. J. Bone Miner Res. 2004, 19, 429–435. [Google Scholar] [CrossRef]
- Urakawa, I.; Yamazaki, Y.; Shimada, T.; Iijima, K.; Hasegawa, H.; Okawa, K.; Fujita, T.; Fukumoto, S.; Yamashita, T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 2006, 444, 770–774. [Google Scholar] [CrossRef]
- Chen, G.; Liu, Y.; Goetz, R.; Fu, L.; Jayaraman, S.; Hu, M.C.; Moe, O.W.; Liang, G.; Li, X.; Mohammadi, M. α-Klotho is a non-enzymatic molecular scaffold for FGF23 hormone signalling. Nature 2018, 553, 461–466. [Google Scholar] [CrossRef]
- Turan, K.; Ata, P. Effects of intra- and extracellular factors on anti-aging klotho gene expression. Genet. Mol. Res. 2011, 10, 2009–2023. [Google Scholar] [CrossRef]
- Diener, S.; Schorpp, K.; Strom, T.M.; Hadian, K.; Lorenz-Depiereux, B. Development of A Cell-Based Assay to Identify Small Molecule Inhibitors of FGF23 Signaling. Assay Drug Dev. Technol. 2015, 13, 476–487. [Google Scholar] [CrossRef]
- Hu, M.C.; Kuro-o, M.; Moe, O.W. Renal and extrarenal actions of Klotho. Semin. Nephrol. 2013, 33, 118–129. [Google Scholar] [CrossRef]
- Matsumura, Y.; Aizawa, H.; Shiraki-Iida, T.; Nagai, R.; Kuro-o, M.; Nabeshima, Y. Identification of the human klotho gene and its two transcripts encoding membrane and secreted klotho protein. Biochem. Biophys. Res. Commun. 1998, 242, 626–630. [Google Scholar] [CrossRef]
- Shiraki-Iida, T.; Aizawa, H.; Matsumura, Y.; Sekine, S.; Iida, A.; Anazawa, H.; Nagai, R.; Kuro-o, M.; Nabeshima, Y. Structure of the mouse klotho gene and its two transcripts encoding membrane and secreted protein. FEBS Lett. 1998, 424, 6–10. [Google Scholar] [CrossRef]
- Neyra, J.A.; Hu, M.C. αKlotho and Chronic Kidney Disease. Vitam. Horm. 2016, 101, 257–310. [Google Scholar]
- Martín-Núñez, E.; Donate-Correa, J.; Muros-de-Fuentes, M.; Mora-Fernández, C.; Navarro-González, J.F. Implications of Klotho in vascular health and disease. World J. Cardiol. 2014, 6, 1262–1269. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Normal Ranges | Patient Values | |
|---|---|---|
| Calcium (mM) | 2.25–2.60 | 2.5 |
| Albumin (g/L) | 30–42 | 35.6 |
| Phosphorous (mM) | 0.85–1.5 | 2.7 |
| Potassium (mM) | 3.5–4.5 | 5.8 |
| Sodium (mM) | 136–146 | 135 |
| Bicarbonate (mM) | 22–29 | 21 |
| Chloride (mM) | 98–106 | 101 |
| Glucose (mM) | 3.9–5.8 | 4.8 |
| Urea (mM) | 2.5–7.5 | 27.6 |
| Creatinine (µM) | 62–106 | 620 |
| eGFR (mL/min) | >60 | <5 |
| PTH (pg/mL) | 10–46 | 533 |
| 25(OH)D3 (ng/mL) | 30–80 | 21.5 |
| 1,25(OH)2D3 (pg/mL) | 15–60 | 30 |
| Alkaline phosphatase (IU/L) | 30–160 | 25.1 |
| C-terminal Telopeptide (pg/mL) | 650–5300 | 2580 |
| C-terminal FGF23 (RU/mL) | 1–149 | 1375.9 |
| KL (pg/mL) | 239–1266 | 99.72 |
| Issue | Result | ||||
|---|---|---|---|---|---|
| Length of Protein | NMD | ||||
| AA Sequence Altered | Yes | ||||
| AA Changes | I348Ffs*28 | ||||
| Position of Altered AA | 348 (frameshift or PTC) | ||||
| Position (AA) of Stopcodon in Wild-Type/Mutated AA Sequence | 1013/375 | ||||
| Protein Features Affected | Start (AA) | End (AA) | Feature | Details | Result |
| 34 | 981 | TOPO_DOM | Extracellular | Lost | |
| 57 | 506 | REGION | Glycosyl hydrolase-1.1. | Lost | |
| 497 | 497 | MOD_RES | Phosphotyrosine | Lost | |
| 515 | 953 | REGION | Glycosyl hydrolase-1.2. | Lost | |
| 607 | 607 | CARBOHYD | N-linked (GlcNAc) | Lost | |
| 612 | 612 | CARBOHYD | N-linked (GlcNAc) | Lost | |
| 694 | 694 | CARBOHYD | N-linked (GlcNAc) | Lost | |
| 982 | 1002 | TRANSMEM | Helical | Lost | |
| 1003 | 1012 | TOPO_DOM | Cytoplasmic | Lost | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martín-Núñez, E.; Donate-Correa, J.; Kannengiesser, C.; De Brauwere, D.-P.; Leroy, C.; Oudin, C.; Friedlander, G.; Prieto-Morín, C.; Tagua, V.G.; Ureña-Torres, P.A.; et al. A Novel Heterozygous Deletion Variant in KLOTHO Gene Leading to Haploinsufficiency and Impairment of Fibroblast Growth Factor 23 Signaling Pathway. J. Clin. Med. 2019, 8, 500. https://doi.org/10.3390/jcm8040500
Martín-Núñez E, Donate-Correa J, Kannengiesser C, De Brauwere D-P, Leroy C, Oudin C, Friedlander G, Prieto-Morín C, Tagua VG, Ureña-Torres PA, et al. A Novel Heterozygous Deletion Variant in KLOTHO Gene Leading to Haploinsufficiency and Impairment of Fibroblast Growth Factor 23 Signaling Pathway. Journal of Clinical Medicine. 2019; 8(4):500. https://doi.org/10.3390/jcm8040500
Chicago/Turabian StyleMartín-Núñez, Ernesto, Javier Donate-Correa, Caroline Kannengiesser, David-Paul De Brauwere, Christine Leroy, Claire Oudin, Gérard Friedlander, Carol Prieto-Morín, Víctor G. Tagua, Pablo A. Ureña-Torres, and et al. 2019. "A Novel Heterozygous Deletion Variant in KLOTHO Gene Leading to Haploinsufficiency and Impairment of Fibroblast Growth Factor 23 Signaling Pathway" Journal of Clinical Medicine 8, no. 4: 500. https://doi.org/10.3390/jcm8040500
APA StyleMartín-Núñez, E., Donate-Correa, J., Kannengiesser, C., De Brauwere, D.-P., Leroy, C., Oudin, C., Friedlander, G., Prieto-Morín, C., Tagua, V. G., Ureña-Torres, P. A., & Navarro-González, J. F. (2019). A Novel Heterozygous Deletion Variant in KLOTHO Gene Leading to Haploinsufficiency and Impairment of Fibroblast Growth Factor 23 Signaling Pathway. Journal of Clinical Medicine, 8(4), 500. https://doi.org/10.3390/jcm8040500