Cross-Talk between Lipoproteins and Inflammation: The Role of Microvesicles



Abstract
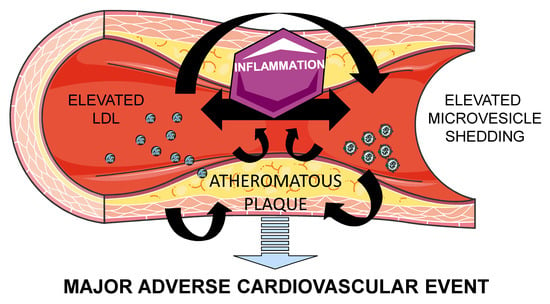
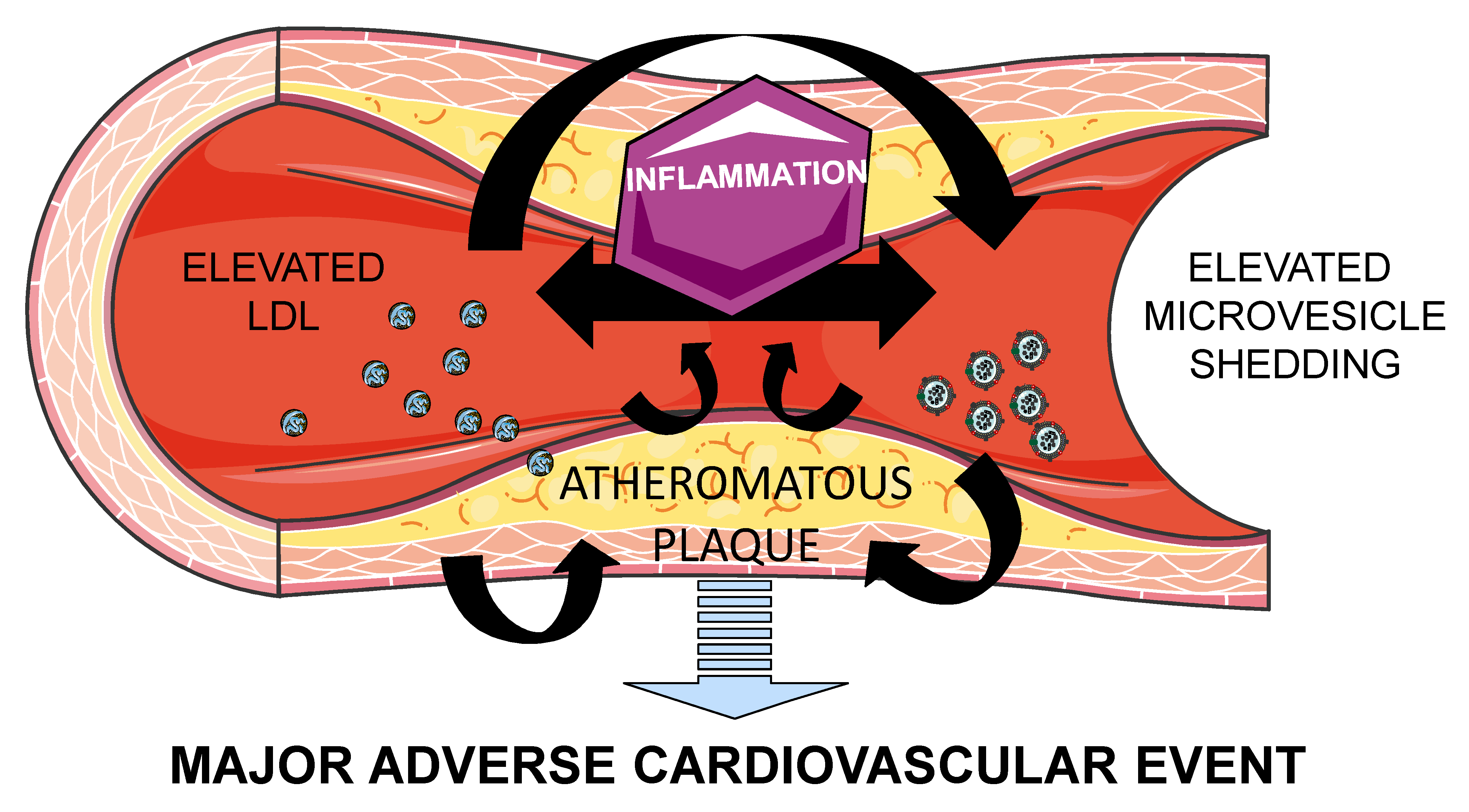{kind=link}
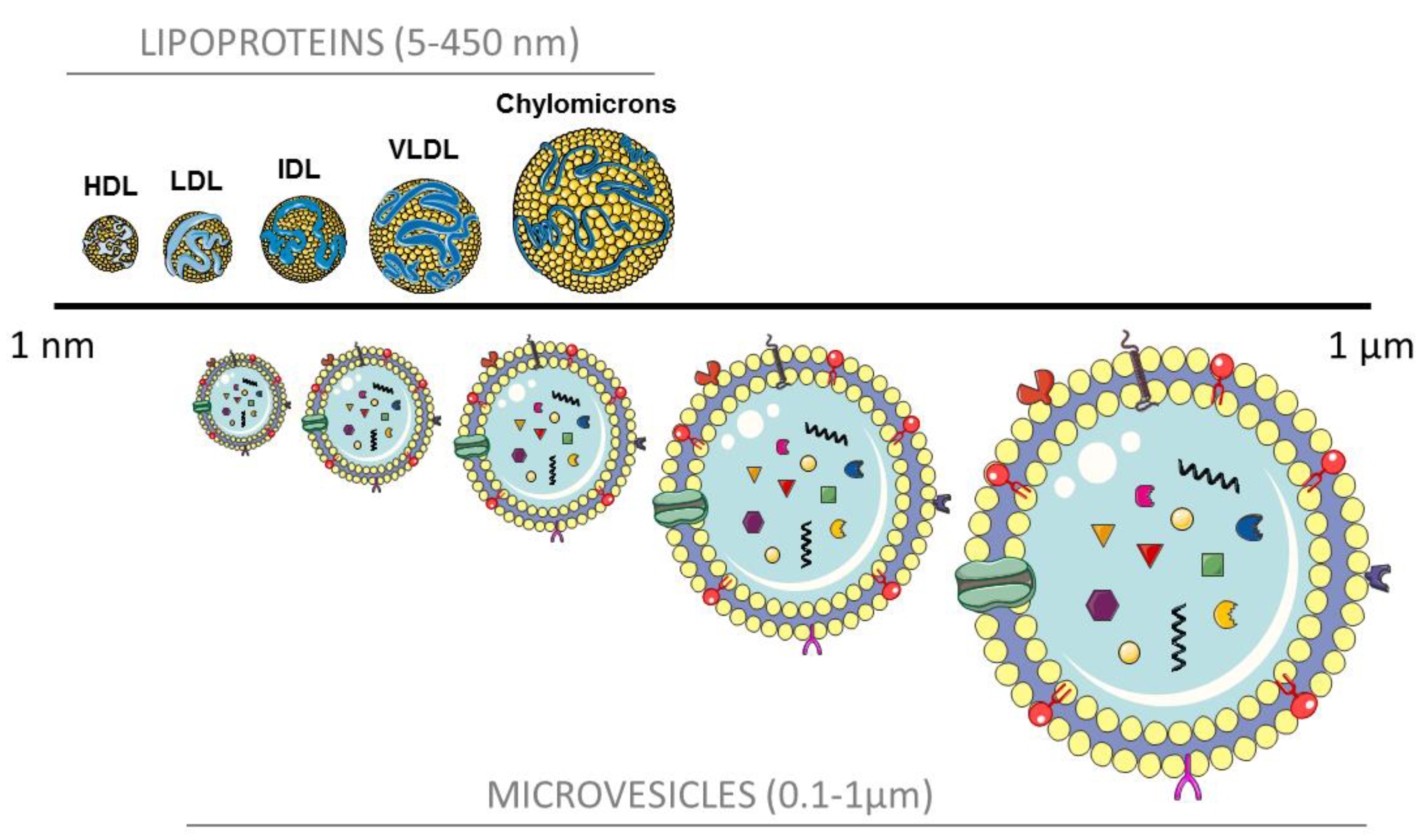{kind=link}
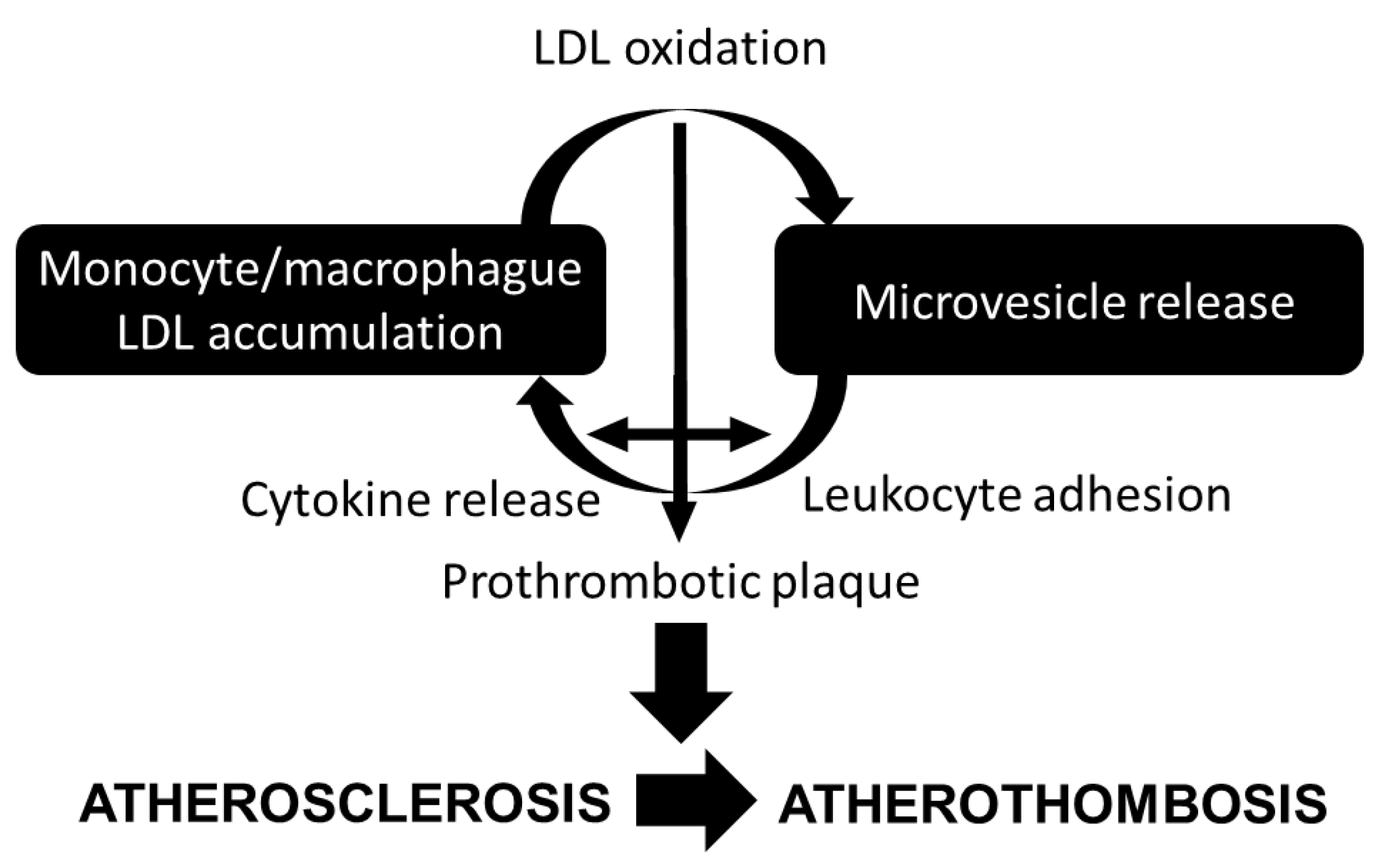{kind=link}
1. Introduction
2. Atherosclerosis: Lipids and Inflammation
3. Microvesicles
4. Role of Microvesicles in Atherosclerosis
Crosstalk between Lipoproteins and MV
5. Role of Microvesicles in Dyslipidemia
Pharmacological Treatment of Dyslipidemia and Circulating MV
6. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Badimon, L.; Vilahur, G. Thrombosis formation on atherosclerotic lesions and plaque rupture. J. Intern. Med. 2014, 276, 618–632. [Google Scholar] [CrossRef]
- Silverman, M.G.; Ference, B.A.; Im, K.; Wiviott, S.D.; Giugliano, R.P.; Grundy, S.M.; Braunwald, E.; Sabatine, M.S. Association Between Lowering LDL-C and Cardiovascular Risk Reduction Among Different Therapeutic Interventions: A Systematic Review and Meta-analysis. JAMA 2016, 316, 1289–1297. [Google Scholar] [CrossRef]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Prim. 2019, 5, 56. [Google Scholar] [CrossRef]
- Vilahur, G.; Ben-Aicha, S.; Badimon, L. New insights into the role of adipose tissue in thrombosis. Cardiovasc. Res. 2017, 113, 1046–1054. [Google Scholar] [CrossRef]
- Zerrad-Saadi, A.; Therond, P.; Chantepie, S.; Couturier, M.; Rye, K.-A.; Chapman, M.J.; Kontush, A. HDL3-Mediated Inactivation of LDL-Associated Phospholipid Hydroperoxides Is Determined by the Redox Status of Apolipoprotein A-I and HDL Particle Surface Lipid Rigidity. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 2169–2175. [Google Scholar] [CrossRef] [PubMed]
- Baker, P.W.; Rye, K.A.; Gamble, J.R.; Vadas, M.A.; Barter, P.J. Ability of reconstituted high density lipoproteins to inhibit cytokine-induced expression of vascular cell adhesion molecule-1 in human umbilical vein endothelial cells. J. Lipid Res. 1999, 40, 345–353. [Google Scholar] [PubMed]
- Jonas, K.; Kopeć, G. HDL Cholesterol as a Marker of Disease Severity and Prognosis in Patients with Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2019, 20, 3514. [Google Scholar] [CrossRef] [PubMed]
- Grün, J.L.; Manjarrez-Reyna, A.N.; Gómez-Arauz, A.Y.; Leon-Cabrera, S.; Rückert, F.; Fragoso, J.M.; Bueno-Hernández, N.; Islas-Andrade, S.; Meléndez-Mier, G.; Escobedo, G. High-Density Lipoprotein Reduction Differentially Modulates to Classical and Nonclassical Monocyte Subpopulations in Metabolic Syndrome Patients and in LPS-Stimulated Primary Human Monocytes In Vitro. J. Immunol. Res. 2018, 2018, 2737040. [Google Scholar] [CrossRef]
- Padró, T.; Cubedo, J.; Camino, S.; Béjar, M.T.; Ben-Aicha, S.; Mendieta, G.; Escolà-Gil, J.C.; Escate, R.; Gutiérrez, M.; Casani, L.; et al. Detrimental Effect of Hypercholesterolemia on High-Density Lipoprotein Particle Remodeling in Pigs. J. Am. Coll. Cardiol. 2017, 70, 165–178. [Google Scholar] [CrossRef]
- Ben-Aicha, S.; Escate, R.; Casaní, L.; Padró, T.; Peña, E.; Arderiu, G.; Mendieta, G.; Badimón, L.; Vilahur, G. High-density lipoprotein remodelled in hypercholesterolaemic blood induce epigenetically driven down-regulation of endothelial HIF-1α expression in a preclinical animal model. Cardiovasc. Res. 2019. [Google Scholar] [CrossRef]
- Marchio, P.; Guerra-Ojeda, S.; Vila, J.M.; Aldasoro, M.; Victor, V.M.; Mauricio, M.D. Targeting Early Atherosclerosis: A Focus on Oxidative Stress and Inflammation. Oxid. Med. Cell. Longev. 2019, 2019, 8563845. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, L.; Frumento, P.; Wallén, H.; de Faire, U.; Gigante, B. The predictive role of interleukin 6 trans-signalling in middle-aged men and women at low-intermediate risk of cardiovascular events. Eur. J. Prev. Cardiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Von Moltke, J.; Trinidad, N.J.; Moayeri, M.; Kintzer, A.F.; Wang, S.B.; Van Rooijen, N.; Brown, C.R.; Krantz, B.A.; Leppla, S.H.; Gronert, K.; et al. Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature 2012, 490, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Jukema, R.A.; Ahmed, T.A.N.; Tardif, J.-C. Does low-density lipoprotein cholesterol induce inflammation? If so, does it matter? Current insights and future perspectives for novel therapies. BMC Med. 2019, 17, 197. [Google Scholar] [CrossRef] [PubMed]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nũez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef]
- Varghese, J.F.; Patel, R.; Yadav, U.C.S. Sterol regulatory element binding protein (SREBP) -1 mediates oxidized low-density lipoprotein (oxLDL) induced macrophage foam cell formation through NLRP3 inflammasome activation. Cell. Signal. 2019, 53, 316–326. [Google Scholar] [CrossRef]
- Libby, P.; Ridker, P.M.; Maseri, A. Inflammation and atherosclerosis. Circulation 2002, 105, 1135–1143. [Google Scholar] [CrossRef]
- Obama, T.; Ohinata, H.; Takaki, T.; Iwamoto, S.; Sawada, N.; Aiuchi, T.; Kato, R.; Itabe, H. Cooperative Action of Oxidized Low-Density Lipoproteins and Neutrophils on Endothelial Inflammatory Responses Through Neutrophil Extracellular Trap Formation. Front. Immunol. 2019, 10, 1899. [Google Scholar] [CrossRef]
- Llorente-Cortés, V.; Royo, T.; Juan-Babot, O.; Badimon, L. Adipocyte differentiation-related protein is induced by LRP1-mediated aggregated LDL internalization in human vascular smooth muscle cells and macrophages. J. Lipid Res. 2007, 48, 2133–2140. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of Exosome Composition. Cell 2019, 177, 428–445.e18. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Théry, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Badimon, L.; Suades, R.; Arderiu, G.; Peña, E.; Chiva-Blanch, G.; Padró, T. Microvesicles in Atherosclerosis and Angiogenesis: From Bench to Bedside and Reverse. Front. Cardiovasc. Med. 2017, 4, 77. [Google Scholar] [CrossRef]
- Del Conde, I.; Shrimpton, C.N.; Thiagarajan, P.; López, J.A. Tissue-factor–bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood 2005, 106, 1604–1611. [Google Scholar] [CrossRef]
- Pfrieger, F.W.; Vitale, N. Cholesterol and the journey of extracellular vesicles. J. Lipid Res. 2018, 59, 2255–2261. [Google Scholar] [CrossRef]
- BIRO, E.; Akkerman, J.W.N.; Hoek, F.J.; Gorter, G.; Pronk, L.M.; Sturk, A.; Nieuwland, R. The phospholipid composition and cholesterol content of platelet-derived microparticles: A comparison with platelet membrane fractions. J. Thromb. Haemost. 2005, 3, 2754–2763. [Google Scholar] [CrossRef]
- Gudbergsson, J.M.; Jønsson, K.; Simonsen, J.B.; Johnsen, K.B. Systematic review of targeted extracellular vesicles for drug delivery—Considerations on methodological and biological heterogeneity. J. Control. Release 2019, 306, 108–120. [Google Scholar] [CrossRef]
- Williams, C.; Palviainen, M.; Reichardt, N.-C.; Siljander, P.R.-M.; Falcón-Pérez, J.M. Metabolomics Applied to the Study of Extracellular Vesicles. Metabolites 2019, 9, 276. [Google Scholar] [CrossRef]
- Turchinovich, A.; Drapkina, O.; Tonevitsky, A. Transcriptome of extracellular vesicles: State-of-the-art. Front. Immunol. 2019, 10, 202. [Google Scholar] [CrossRef]
- De Giusti, C.J.; Santalla, M.; Das, S. Exosomal non-coding RNAs (Exo-ncRNAs) in cardiovascular health. J. Mol. Cell. Cardiol. 2019, 137, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Wang, M. New insights into the immunomodulatory role of exosomes in cardiovascular disease. Rev. Cardiovasc. Med. 2019, 20, 153–160. [Google Scholar] [PubMed]
- Yang, W.; Zou, B.; Hou, Y.; Yan, W.; Chen, T.; Qu, S. Extracellular vesicles in vascular calcification. Clin. Chim. Acta 2019, 499, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Kita, S.; Maeda, N.; Shimomura, I. Interorgan communication by exosomes, adipose tissue, and adiponectin in metabolic syndrome. J. Clin. Investig. 2019, 129, 4041–4049. [Google Scholar] [CrossRef]
- Yu, H.; Wang, Z. Cardiomyocyte-Derived Exosomes: Biological Functions and Potential Therapeutic Implications. Front. Physiol. 2019, 10, 1049. [Google Scholar] [CrossRef]
- Jafarzadeh-Esfehani, R.; Soudyab, M.; Parizadeh, S.M.; Jaripoor, M.E.; Nejad, P.S.; Shariati, M.; Nabavi, A.S. Circulating exosomes and their role in stroke. Curr. Drug Targets 2019, 20. [Google Scholar] [CrossRef]
- Aghabozorgi, A.S.; Ahangari, N.; Eftekhaari, T.E.; Torbati, P.N.; Bahiraee, A.; Ebrahimi, R.; Pasdar, A. Circulating exosomal miRNAs in cardiovascular disease pathogenesis: New emerging hopes. J. Cell. Physiol. 2019, 234, 21796–21809. [Google Scholar] [CrossRef]
- Owens, A.P.; Mackman, N. Microparticles in Hemostasis and Thrombosis. Circ. Res. 2011, 108, 1284–1297. [Google Scholar] [CrossRef]
- Obermayer, G.; Afonyushkin, T.; Binder, C.J. Oxidized low-density lipoprotein in inflammation-driven thrombosis. J. Thromb. Haemost. 2018, 16, 418–428. [Google Scholar] [CrossRef]
- Habersberger, J.; Strang, F.; Scheichl, A.; Htun, N.; Bassler, N.; Merivirta, R.-M.; Diehl, P.; Krippner, G.; Meikle, P.; Eisenhardt, S.U.; et al. Circulating microparticles generate and transport monomeric C-reactive protein in patients with myocardial infarction. Cardiovasc. Res. 2012, 96, 64–72. [Google Scholar] [CrossRef]
- Pirro, M.; Bianconi, V.; Paciullo, F.; Mannarino, M.R.; Bagaglia, F.; Sahebkar, A. Lipoprotein(a) and inflammation: A dangerous duet leading to endothelial loss of integrity. Pharmacol. Res. 2017, 119, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Tsimerman, G.; Roguin, A.; Bachar, A.; Melamed, E.; Brenner, B.; Aharon, A. Involvement of microparticles in diabetic vascular complications. Thromb. Haemost. 2011, 106, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Bei, J.-J.; Liu, C.; Peng, S.; Liu, C.-H.; Zhao, W.-B.; Qu, X.-L.; Chen, Q.; Zhou, Z.; Yu, Z.-P.; Peter, K.; et al. Staphylococcal SSL5-induced platelet microparticles provoke proinflammatory responses via the CD40/TRAF6/NFKB signalling pathway in monocytes. Thromb. Haemost. 2016, 115, 632–645. [Google Scholar] [CrossRef] [PubMed]
- Suades, R.; Padró, T.; Vilahur, G.; Badimon, L. Circulating and platelet-derived microparticles in human blood enhance thrombosis on atherosclerotic plaques. Thromb. Haemost. 2012, 108, 1208–1219. [Google Scholar] [CrossRef] [PubMed]
- Hugel, B.; Martínez, M.C.; Kunzelmann, C.; Freyssinet, J.-M. Membrane Microparticles: Two Sides of the Coin. Physiology 2005, 20, 22–27. [Google Scholar] [CrossRef]
- Blum, A. The possible role of red blood cell microvesicles in atherosclerosis. Eur. J. Intern. Med. 2009, 20, 101–105. [Google Scholar] [CrossRef]
- Lin, H.-L.; Xu, X.-S.; Lu, H.-X.; Zhang, L.; Li, C.-J.; Tang, M.-X.; Sun, H.-W.; Liu, Y.; Zhang, Y. Pathological mechanisms and dose dependency of erythrocyte-induced vulnerability of atherosclerotic plaques. J. Mol. Cell. Cardiol. 2007, 43, 272–280. [Google Scholar] [CrossRef]
- Leroyer, A.S.; Isobe, H.; Lesèche, G.; Castier, Y.; Wassef, M.; Mallat, Z.; Binder, B.R.; Tedgui, A.; Boulanger, C.M. Cellular Origins and Thrombogenic Activity of Microparticles Isolated from Human Atherosclerotic Plaques. J. Am. Coll. Cardiol. 2007, 49, 772–777. [Google Scholar] [CrossRef]
- Mallat, Z.; Benamer, H.; Hugel, B.; Benessiano, J.; Steg, P.G.; Freyssinet, J.M.; Tedgui, A. Elevated levels of shed membrane microparticles with procoagulant potential in the peripheral circulating blood of patients with acute coronary syndromes. Circulation 2000, 101, 841–843. [Google Scholar] [CrossRef]
- Lee, H.; Zhang, D.; Zhu, Z.; Dela Cruz, C.S.; Jin, Y. Epithelial cell-derived microvesicles activate macrophages and promote inflammation via microvesicle-containing microRNAs. Sci. Rep. 2016, 6, 35250. [Google Scholar] [CrossRef]
- Shu, Z.; Tan, J.; Miao, Y.; Zhang, Q. The role of microvesicles containing microRNAs in vascular endothelial dysfunction. J. Cell. Mol. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Mahmood Hussain, M. A proposed model for the assembly of chylomicrons. Atherosclerosis 2000, 148, 1–15. [Google Scholar] [CrossRef]
- Wiklander, O.P.B.; Nordin, J.Z.; O’Loughlin, A.; Gustafsson, Y.; Corso, G.; Mäger, I.; Vader, P.; Lee, Y.; Sork, H.; Seow, Y.; et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J. Extracell. Vesicles 2015, 4, 26316. [Google Scholar] [CrossRef] [PubMed]
- Skålén, K.; Gustafsson, M.; Rydberg, E.K.; Hultén, L.M.; Wiklund, O.; Innerarity, T.L.; Borén, J. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature 2002, 417, 750–754. [Google Scholar] [CrossRef]
- Liu, M.-L.; Reilly, M.P.; Casasanto, P.; McKenzie, S.E.; Williams, K.J. Cholesterol enrichment of human monocyte/macrophages induces surface exposure of phosphatidylserine and the release of biologically-active tissue factor-positive microvesicles. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 430–435. [Google Scholar] [CrossRef]
- Cipollone, F.; Mezzetti, A.; Porreca, E.; Di Febbo, C.; Nutini, M.; Fazia, M.; Falco, A.; Cuccurullo, F.; Davì, G. Association Between Enhanced Soluble CD40L and Prothrombotic State in Hypercholesterolemia. Circulation 2002, 106, 399–402. [Google Scholar] [CrossRef]
- Williams, K.J.; Tabas, I. Lipoprotein retention—And clues for atheroma regression. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1536–1540. [Google Scholar] [CrossRef]
- Podrez, E.A.; Byzova, T.V.; Febbraio, M.; Salomon, R.G.; Ma, Y.; Valiyaveettil, M.; Poliakov, E.; Sun, M.; Finton, P.J.; Curtis, B.R.; et al. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat. Med. 2007, 13, 1086–1095. [Google Scholar] [CrossRef]
- Chen, K.; Febbraio, M.; Li, W.; Silverstein, R.L. A Specific CD36-Dependent Signaling Pathway Is Required for Platelet Activation by Oxidized Low-Density Lipoprotein. Circ. Res. 2008, 102, 1512–1519. [Google Scholar] [CrossRef]
- Wraith, K.S.; Magwenzi, S.; Aburima, A.; Wen, Y.; Leake, D.; Naseem, K.M. Oxidized low-density lipoproteins induce rapid platelet activation and shape change through tyrosine kinase and Rho kinase-signaling pathways. Blood 2013, 122, 580–589. [Google Scholar] [CrossRef]
- Wang, H.; Wang, Z.-H.; Kong, J.; Yang, M.-Y.; Jiang, G.-H.; Wang, X.-P.; Zhong, M.; Zhang, Y.; Deng, J.-T.; Zhang, W. Oxidized Low-Density Lipoprotein-Dependent Platelet-Derived Microvesicles Trigger Procoagulant Effects and Amplify Oxidative Stress. Mol. Med. 2012, 18, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.B.; Nielsen, M.H.; Handberg, A. In vitro Incubation of Platelets with oxLDL Does Not Induce Microvesicle Release When Measured by Sensitive Flow Cytometry. Front. Cardiovasc. Med. 2015, 2, 37. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Zhou, E.; Wang, X.; Tian, M.; Kong, J.; Li, J.; Ji, L.; Niu, C.; Shen, H.; Dong, S.; et al. Oxidized low-density lipoprotein-induced microparticles promote endothelial monocyte adhesion via intercellular adhesion molecule 1. Am. J. Physiol. Physiol. 2017, 313, C567–C574. [Google Scholar] [CrossRef] [PubMed]
- Rautou, P.-E.; Leroyer, A.S.; Ramkhelawon, B.; Devue, C.; Duflaut, D.; Vion, A.-C.; Nalbone, G.; Castier, Y.; Leseche, G.; Lehoux, S.; et al. Microparticles From Human Atherosclerotic Plaques Promote Endothelial ICAM-1–Dependent Monocyte Adhesion and Transendothelial Migration. Circ. Res. 2011, 108, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.L.; Scalia, R.; Mehta, J.L.; Williams, K.J. Cholesterol-induced membrane microvesicles as novel carriers of damage-associated molecular patterns: Mechanisms of formation, action, and detoxification. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2113–2121. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, N.; Nomura, S.; Kamihata, H.; Kimura, Y.; Iwasaka, T. Increased level of oxidized LDL-dependent monocytederived microparticles in acute coronary syndrome. Thromb. Haemost. 2004, 91, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Llorente-Cortés, V.; Otero-Viñas, M.; Camino-López, S.; Llampayas, O.; Badimon, L. Aggregated low-density lipoprotein uptake induces membrane tissue factor procoagulant activity and microparticle release in human vascular smooth muscle cells. Circulation 2004, 110, 452–459. [Google Scholar] [CrossRef]
- Huber, J.; Vales, A.; Mitulovic, G.; Blumer, M.; Schmid, R.; Witztum, J.L.; Binder, B.R.; Leitinger, N. Oxidized membrane vesicles and blebs from apoptotic cells contain biologically active oxidized phospholipids that induce monocyte-endothelial interactions. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 101–107. [Google Scholar] [CrossRef]
- Papac-Milicevic, N.; Busch, C.J.L.; Binder, C.J. Malondialdehyde Epitopes as Targets of Immunity and the Implications for Atherosclerosis. In Advances in Immunology; Academic Press Inc.: Cambridge, MA, USA, 2016; Volume 131, pp. 1–59. [Google Scholar]
- Bodega, G.; Alique, M.; Puebla, L.; Carracedo, J.; Ramírez, R.M. Microvesicles: ROS scavengers and ROS producers. J. Extracell. Vesicles 2019, 8, 1626654. [Google Scholar] [CrossRef]
- Todorova, D.; Simoncini, S.; Lacroix, R.; Sabatier, F.; Dignat-George, F. Extracellular vesicles in angiogenesis. Circ. Res. 2017, 120, 1658–1673. [Google Scholar] [CrossRef]
- Karpe, F. Postprandial lipoprotein metabolism and atherosclerosis. J. Intern. Med. 1999, 246, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Nappo, F.; Esposito, K.; Cioffi, M.; Giugliano, G.; Molinari, A.M.; Paolisso, G.; Marfella, R.; Giugliano, D. Postprandial endothelial activation in healthy subjects and in type 2 diabetic patients: Role of fat and carbohydrate meals. J. Am. Coll. Cardiol. 2002, 39, 1145–1150. [Google Scholar] [CrossRef]
- Ferreira, A.C.; Peter, A.A.; Mendez, A.J.; Jimenez, J.J.; Mauro, L.M.; Chirinos, J.A.; Ghany, R.; Virani, S.; Garcia, S.; Horstman, L.L.; et al. Postprandial Hypertriglyceridemia Increases Circulating Levels of Endothelial Cell Microparticles. Circulation 2004, 110, 3599–3603. [Google Scholar] [CrossRef] [PubMed]
- Tushuizen, M.E.; Nieuwland, R.; Scheffer, P.G.; Sturk, A.; Heine, R.J.; Diamant, M. Two consecutive high-fat meals affect endothelial-dependent vasodilation, oxidative stress and cellular microparticles in healthy men. J. Thromb. Haemost. 2006, 4, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Tamburrelli, C.; Gianfagna, F.; D’imperio, M.; De Curtis, A.; Rotilio, D.; Iacoviello, L.; De Gaetano, G.; Donati, M.B.; Cerletti, C. Postprandial cell inflammatory response to a standardised fatty meal in subjects at different degree of cardiovascular risk. Thromb. Haemost. 2012, 107, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Tushuizen, M.E.; Nieuwland, R.; Rustemeijer, C.; Hensgens, B.E.; Sturk, A.; Heine, R.J.; Diamant, M. Elevated endothelial microparticles following consecutive meals are associated with vascular endothelial dysfunction in type 2 diabetes. Diabetes Care 2007, 30, 728–730. [Google Scholar] [CrossRef][Green Version]
- Elevated Levels of Platelet Microparticles in Carotid Atherosclerosis and during the Postprandial State- ClinicalKey. Available online: https://www.clinicalkey.es/#!/content/playContent/1-s2.0-S0049384808005082?scrollTo=%23hl0000494 (accessed on 14 November 2019).
- Hjuler Nielsen, M.; Irvine, H.; Vedel, S.; Raungaard, B.; Beck-Nielsen, H.; Handberg, A. Elevated Atherosclerosis-Related Gene Expression, Monocyte Activation and Microparticle-Release Are Related to Increased Lipoprotein-Associated Oxidative Stress in Familial Hypercholesterolemia. PLoS ONE 2015, 10, e0121516. [Google Scholar] [CrossRef]
- Suades, R.; Padró, T.; Alonso, R.; Mata, P.; Badimon, L. High levels of TSP1+/CD142+ platelet-derived microparticles characterise young patients with high cardiovascular risk and subclinical atherosclerosis. Thromb. Haemost. 2015, 114, 1310–1321. [Google Scholar] [CrossRef]
- Pirro, M.; Schillaci, G.; Paltriccia, R.; Bagaglia, F.; Menecali, C.; Mannarino, M.R.; Capanni, M.; Velardi, A.; Mannarino, E. Increased ratio of CD31+/CD42− microparticles to endothelial progenitors as a novel marker of atherosclerosis in hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2530–2535. [Google Scholar] [CrossRef]
- Badimon, L.; Chiva-Blanch, G. Lipid Metabolism in Dyslipidemia and Familial Hypercholesterolemia. Mol. Nutr. Fats 2019, 307–322. [Google Scholar]
- Owens, A.P.; Passam, F.H.; Antoniak, S.; Marshall, S.M.; McDaniel, A.L.; Rudel, L.; Williams, J.C.; Hubbard, B.K.; Dutton, J.-A.; Wang, J.; et al. Monocyte tissue factor-dependent activation of coagulation in hypercholesterolemic mice and monkeys is inhibited by simvastatin. J. Clin. Investig. 2012, 122, 558–568. [Google Scholar] [CrossRef] [PubMed]
- Chiva-Blanch, G.; Padró, T.; Alonso, R.; Crespo, J.; Perez de Isla, L.; Mata, P.; Badimon, L. Liquid biopsy of extracellular microvesicles maps coronary calcification and atherosclerotic plaque in asymptomatic patients with familial hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 945–955. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.H.; Irvine, H.; Vedel, S.; Raungaard, B.; Beck-Nielsen, H.; Handberg, A. The Impact of Lipoprotein-Associated Oxidative Stress on Cell-Specific Microvesicle Release in Patients with Familial Hypercholesterolemia. Oxid. Med. Cell. Longev. 2016, 2016, 2492858. [Google Scholar] [CrossRef] [PubMed]
- Suades, R.; Padró, T.; Alonso, R.; López-Miranda, J.; Mata, P.; Badimon, L. Circulating CD45+/CD3+ lymphocyte-derived microparticles map lipid-rich atherosclerotic plaques in familial hypercholesterolaemia patients. Thromb. Haemost. 2013, 111, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Suades, R.; Padró, T.; Crespo, J.; Sionis, A.; Alonso, R.; Mata, P.; Badimon, L. Liquid Biopsy of Extracellular Microvesicles Predicts Future Major Ischemic Events in Genetically Characterized Familial Hypercholesterolemia Patients. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1172–1181. [Google Scholar] [CrossRef]
- Connolly, K.D.; Willis, G.R.; Datta, D.B.N.; Ellins, E.A.; Ladell, K.; Price, D.A.; Guschina, I.A.; Rees, D.A.; James, P.E. Lipoprotein-apheresis reduces circulating microparticles in individuals with familial hypercholesterolemia. J. Lipid Res. 2014, 55, 2064–2072. [Google Scholar] [CrossRef]
- Suades, R.; Padró, T.; Alonso, R.; Mata, P.; Badimon, L. Lipid-lowering therapy with statins reduces microparticle shedding from endothelium, platelets and inflammatory cells. Thromb. Haemost. 2013, 110, 366–377. [Google Scholar] [CrossRef]
- Nomura, S.; Inami, N.; Shouzu, A.; Omoto, S.; Kimura, Y.; Takahashi, N.; Tanaka, A.; Urase, F.; Maeda, Y.; Ohtani, H.; et al. The effects of pitavastatin, eicosapentaenoic acid and combined therapy on platelet-derived microparticles and adiponectin in hyperlipidemic, diabetic patients. Platelets 2009, 20, 16–22. [Google Scholar] [CrossRef]
- Nomura, S.; Shouzu, A.; Omoto, S.; Inami, N.; Ueba, T.; Urase, F.; Maeda, Y. Effects of eicosapentaenoic acid on endothelial cell-derived microparticles, angiopoietins and adiponectin in patients with type 2 diabetes. J. Atheroscler. Thromb. 2009, 16, 83–90. [Google Scholar] [CrossRef]
- Tramontano, A.F.; O’Leary, J.; Black, A.D.; Muniyappa, R.; Cutaia, M.V.; El-Sherif, N. Statin decreases endothelial microparticle release from human coronary artery endothelial cells: Implication for the Rho-kinase pathway. Biochem. Biophys. Res. Commun. 2004, 320, 34–38. [Google Scholar] [CrossRef]
- Sommeijer, D.W.; Joop, K.; Leyte, A.; Reitsma, P.H.; Cate, H.T. Pravastatin reduces fibrinogen receptor gpIIIa on platelet-derived microparticles in patients with type 2 diabetes. J. Thromb. Haemost. 2005, 3, 1168–1171. [Google Scholar] [CrossRef] [PubMed]
- Mobarrez, F.; He, S.; Bröijersen, A.; Wiklund, B.; Antovic, A.; Antovic, J.; Egberg, N.; Jörneskog, G.; Wallén, H. Atorvastatin reduces thrombin generation and expression of tissue factor, P-selectin and GPIIIa on platelet-derived microparticles in patients with peripheral arterial occlusive disease. Thromb. Haemost. 2011, 106, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Pawelczyk, M.; Chmielewski, H.; Kaczorowska, B.; Przybyła, M.; Baj, Z. The influence of statin therapy on platelet activity markers in hyperlipidemic patients after ischemic stroke. Arch. Med. Sci. 2015, 1, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Camargo, L.M.; França, C.N.; Izar, M.C.; Bianco, H.T.; Lins, L.S.; Barbosa, S.P.; Pinheiro, L.F.; Fonseca, F.A.H. Effects of simvastatin/ezetimibe on microparticles, endothelial progenitor cells and platelet aggregation in subjects with coronary heart disease under antiplatelet therapy. Braz. J. Med. Biol. Res. 2014, 47, 432–437. [Google Scholar] [CrossRef]
- Lins, L.C.A.; França, C.N.; Fonseca, F.A.H.; Barbosa, S.P.M.; Matos, L.N.; Aguirre, A.C.; Bianco, H.T.; do Amaral, J.B.; Izar, M.C. Effects of Ezetimibe on Endothelial Progenitor Cells and Microparticles in High-Risk Patients. Cell Biochem. Biophys. 2014, 70, 687–696. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2019. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiva-Blanch, G.; Badimon, L. Cross-Talk between Lipoproteins and Inflammation: The Role of Microvesicles. J. Clin. Med. 2019, 8, 2059. https://doi.org/10.3390/jcm8122059
Chiva-Blanch G, Badimon L. Cross-Talk between Lipoproteins and Inflammation: The Role of Microvesicles. Journal of Clinical Medicine. 2019; 8(12):2059. https://doi.org/10.3390/jcm8122059
Chicago/Turabian StyleChiva-Blanch, Gemma, and Lina Badimon. 2019. "Cross-Talk between Lipoproteins and Inflammation: The Role of Microvesicles" Journal of Clinical Medicine 8, no. 12: 2059. https://doi.org/10.3390/jcm8122059
APA StyleChiva-Blanch, G., & Badimon, L. (2019). Cross-Talk between Lipoproteins and Inflammation: The Role of Microvesicles. Journal of Clinical Medicine, 8(12), 2059. https://doi.org/10.3390/jcm8122059