4. Discussion
A total of 528 patients were included in our study, 70.11% of patients were males, as had been the case in another similar study, however oral cavity tumours were more prevalent in our study, with 72.4% versus 62%. The tumour stage was predominantly (64.2%) advanced, (stage III and IV), similar to previous studies [
15].
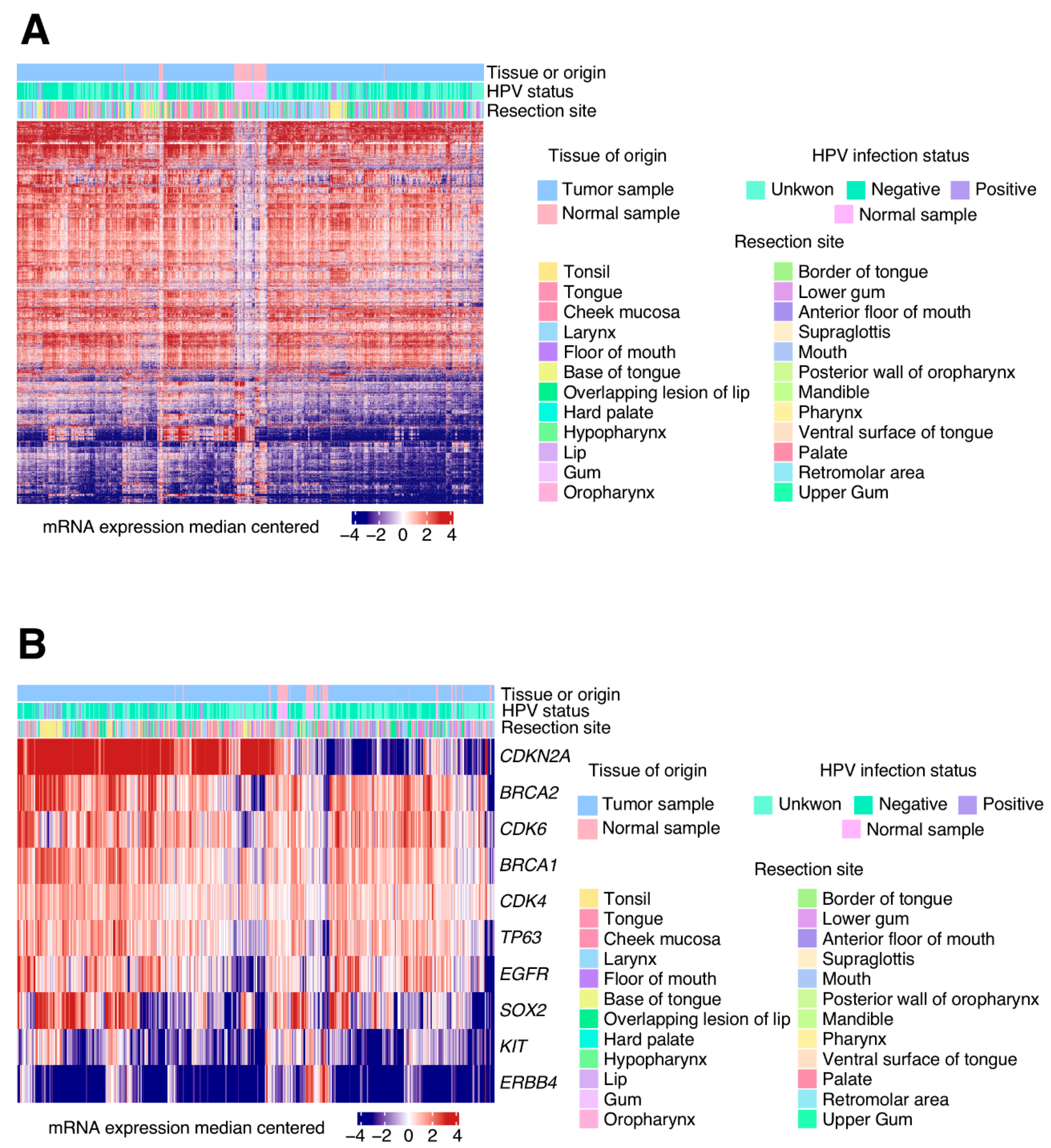
Transcriptional alterations analysis identified 3491 DEGs in HNSCC tumour samples when compared to healthy oral tissue. The oncogenes and tumour suppressor genes from the COSMIC cancer genes list were selected from the total DEGs, reducing these DEGs to 148. According to previous results, the more relevant DEGs were CDKN2A, BRCA2, CDK6, BRCA1, CDK4, TP63, EGFR, SOX2, KIT and ERBB4 [
16,
17].
The upregulated D-cyclin-dependent kinases, CDK4 and CDK6, were pro-tumorigenic proteins and core controllers of G1/S cell cycle phase transition [
18]. Their tyrosin kinase domain phosphorylated the retinoblastoma protein (RB), impairing it from forming a complex with the E2F transcription factor. Once released, E2F induces the expression of the several genes that are necessary for the cell’s progression into the DNA synthesis phase. There are different regulators of CDK activity that could act as tumour suppressors, even by dually inhibiting cell cycle progression and enhancing pro-apoptotic factors. In this sense, HPV (+) cells show
P16INK4A upregulation, a tumour suppressor gene transcribed from the
CDKN2A locus [
19]. This polypeptide inhibits CDK4/6 kinase activity and arrests the cell cycle at G1/S checkpoint. At the same time, P16
INK4A can also stabilize TP53, a potent tumour suppressor and pro-apoptotic protein, by sequestering the murine double murine 2 (MDM2) ubiquitin into the nucleus. The high levels of p16 in HPV+ tumours unlikely result in significant stabilisation of p53 or inhibition of the cell cycle, as RB and p53 are degraded because of viral genes E7 and E6 activity. On the other hand, HPV (−) cancers centre around genes that control cell motility, invasion, EMT and angiogenesis [
20].
The upregulation of the epidermal growth factor receptor (EGFR) into tumour cells can intensify its signalling and promote both abnormal rates of cell proliferation and cell survival [
21]. The EGFR gene belongs to the ERBB family of kinase receptor proteins alongside three other members: ERBB2, ERBB3 and ERBB4. Intriguingly, contrary to what was expected and published for other cancers [
22,
23], the ERBB4 gene is found deeply downregulated (log
2(FC) = −3.67) in the HNSCC tumour samples. Other pathways, in which members are upregulated, are the PIK3CA/AKT/mTOR and WNT/B-catenin signalling axes, some of the Hippo pathway transductors and the DNA repair machinery members. It has been proven that these mechanisms were also implicated in previous HNSCC studies, not only by DEGs but also by other epigenetic biomarkers [
24].
We also found recurrent repression of KIT receptor and other elements of its signalling route. This is not the first time this pro-tumorigenic pathway has been found to be downregulated in cancer. In thyroid carcinoma, lower levels of KIT positively correlate with more malignant phenotypes [
25].
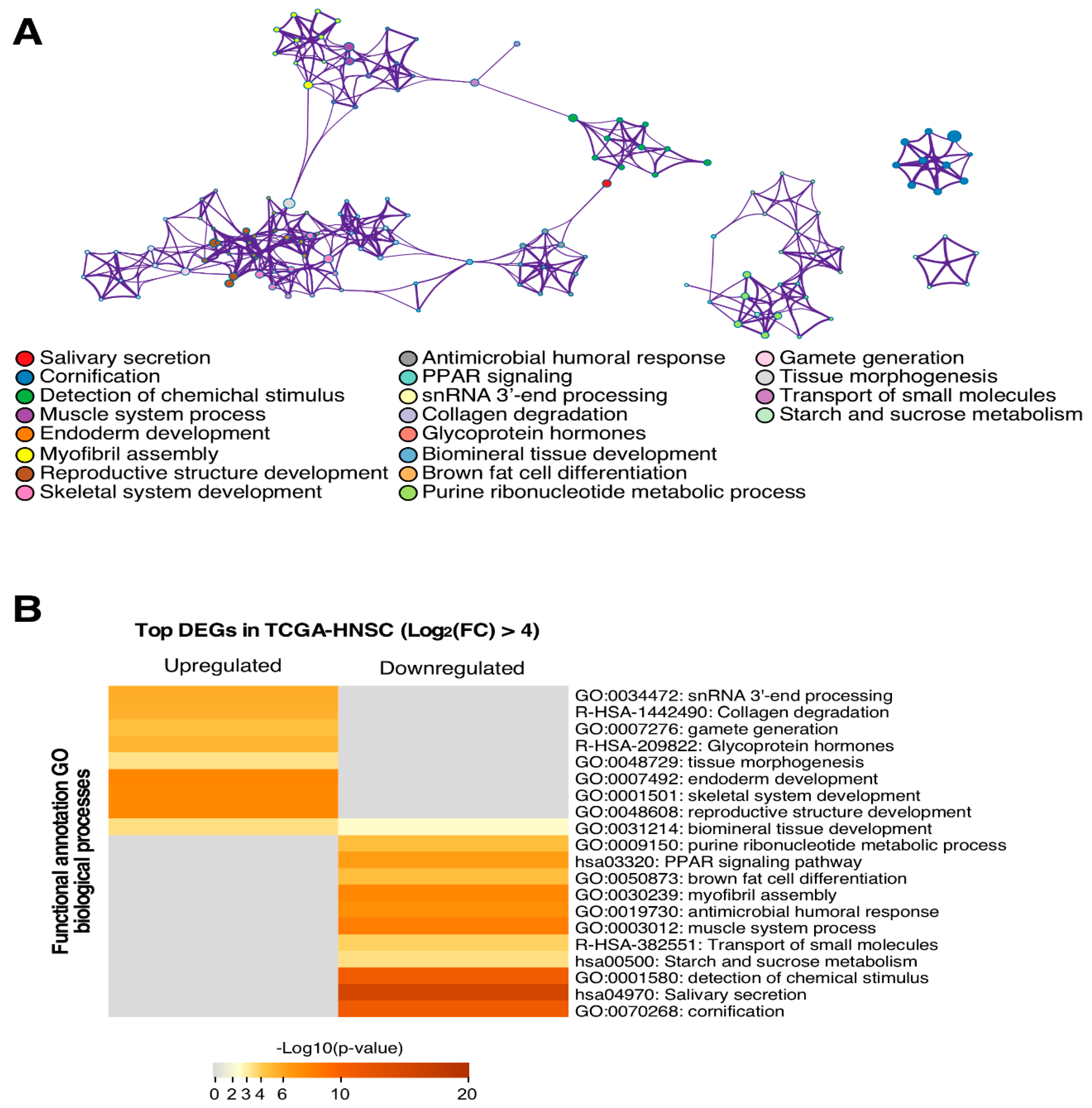
Functional analysis reveals that the more relevant signalling pathways in upregulated genes were those that were involved in tissue development, such as endoderm development, tissue morphogenesis, or skeletal system development. On the other hand, those downregulated genes were more implicated in the cell differentiation molecular pathway, salivary secretion, cornification or starch and sucrose metabolism. Altered molecular mechanism identification is useful for designing new treatment strategies such as immunotherapy, since these targeted therapies are effective against determined molecules [
26].
In order to help to develop new therapies and programs for HNSCC prevention, as there is a lot of information in our analysis, we wanted to make our results more useful by performing a studies profile in HNSCC attending on localization. We could observe that, especially in oropharynx, DEGs’ number were significantly different (1209 genes exclusively different express in oropharynx) comparing results with the other localization, as none of the other group showed more than 224 DEGs. Although cautiously, these differences in DEGs results, the majority of the HPV (+) tumours are concentrated in the oropharynx group, where samples from the tonsil are included. Differential expression profiles developed by other authors, as well as by our group some years before, in HNSCC, revealed heterogeneity in results that reflects the nature of cancer [
27,
28].
Data obtained from survival analysis reveal different oncogenes overexpressed and whose higher expression correlates with worse survival. Other genes classified as tumour suppressors, whose lower expression correlates with worse survival but which, in the general cohort, are found with greater expression than normal samples. Concretely, EXT2 and IGF2BP2, are two known tumour suppressor genes whose highest expression correlates with worse survival. Taken altogether, there are genes classified as oncogenes or tumour suppressor genes in the COSMIC and that in our cohort show survival impacts that support this hypothesis, but there are some others that deviate from this theory, which behaviours show contrary to those expected genes that are classified as oncogenes, overexpressed and whose higher expression correlates with worse survival. CDKN2A, which is used in the diagnosis of HNSCC to check malignancy, has different behaviour in our results, as its overexpression is usually associated with better survival. It may be due to the fact that this correlation can be made if we are in context of HPV infection.
Analyses that report differential expression genes and somatic copy number alterations, show that is complicated to establish a cause-effect edge. Some genes are overexpressed and amplified, such as, for example, FOXL2, but there are other deleted genes that are over expressed, such as MUC4. This can be explained because, although there is a percentage of samples where the gene is not present, in the other, they have a tendency to increase its expression in comparison with normal tissue. The same can occur with amplified or repressed genes. Although a gene is amplified, there are other known mechanisms that act in a superior level that copy number alterations, like epigenetic changes or cotranscriptional repression.
Today’s primary challenge is to differentiate somatic mutations with oncogenic potential from all those which crop up later in a tumour’s lifetime without contributing to the tumour development. In this sense, the concept of positive selection has opened a new avenue for the discovery of novel tumorigenic roles in cancer. Mutations that improve the fitness of the tumour cells would be under positive selection pressure, meaning that they would recur at elevated rates across patients. This is the principle that is widely used to identify these signals and differentiate them from alterations with real oncogenic power [
29].
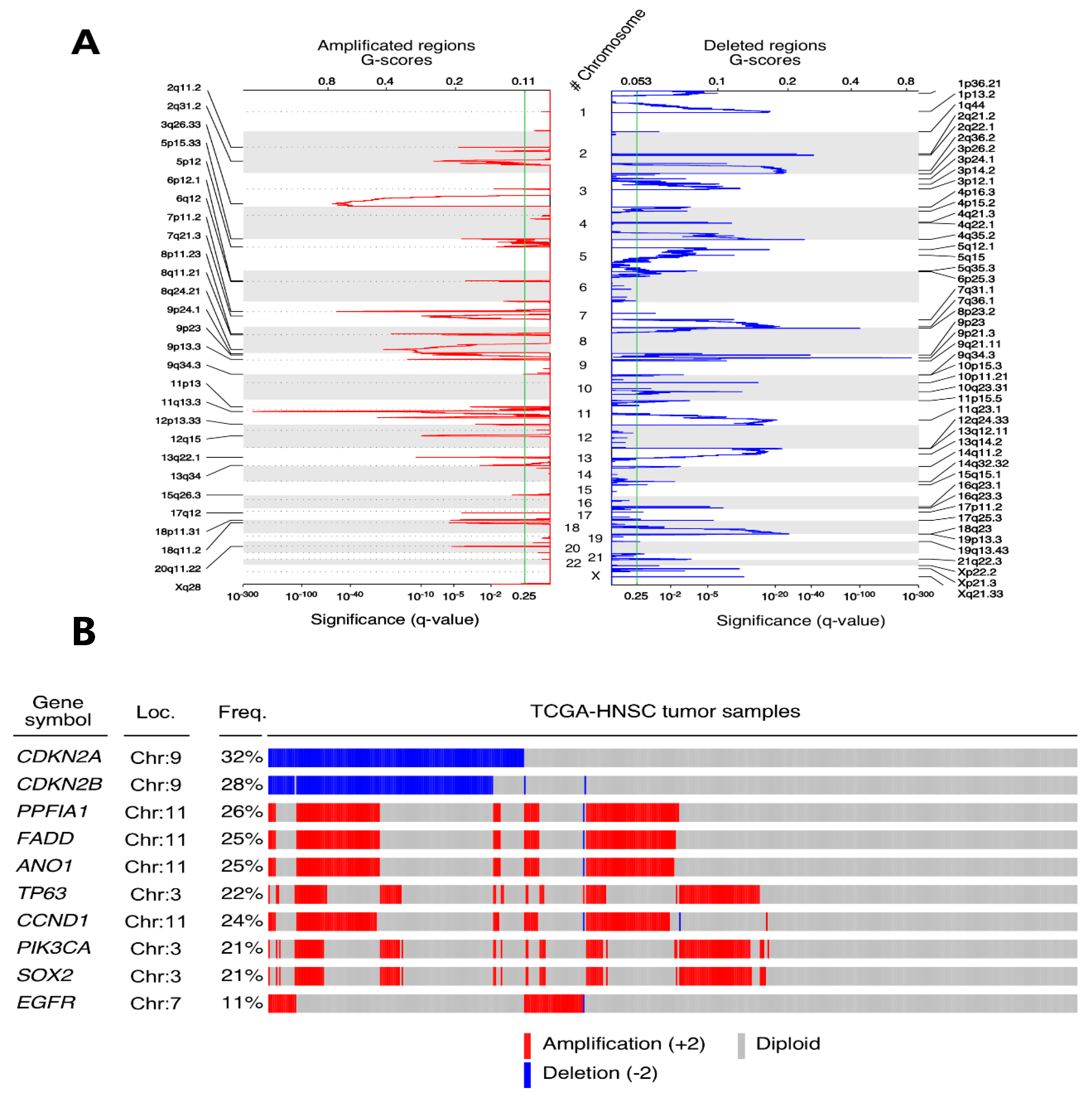
SCNAs are very frequent genomic aberrations in cancer. Despite the fact that they directly affect the genome architecture, the main force through which they trigger their oncogenic potential is transcriptional deregulation. In fact, the amplifications of many oncogenes and the deletions of tumour suppressors were related to the cancer’s aggressiveness, endangering the patients’ survival.
In this study, we used the GISTIC2.0 algorithm results to identify recurrent SCNAs in the TCGA-HNSCC tumour samples. Our results demonstrated the co-amplification of segments 11q13 and 11q22, both embedding caspase cascade inhibitors, as has been previously described [
8]. Modifications of 27 arm-level with gains in 3q, 5p and 8q, and losses in 3p and 8p chromosomal arms, have already been described in lung squamous cell carcinomas [
30]. Taking into account the five most SCN-altered genes in HNSCC, deletion was observed in CDKN2A, CDKN2B, and amplification in PPFIA1, FADD and
ANO1. These results concur with recent studies [
31] and also with other known cancer driver amplifications, such as SOX2, PIK3CA, EGFR and CCDN1. SOX2 is a well-known oncogene that has already been shown SCNA in HNSCC and lung squamous cell carcinoma [
32]. PIK3CA amplifications have been associated with an increased risk of local recurrence [
33]. EGFR and CCDN1 amplification have been associated with clinical stage, tumour differentiation, and lymph node metastasis in HNSCC [
31].
The TCGA mutation-calling pipeline offers four different methods to elucidate every single variation inside the tumour’s whole exome sequencing (WES) results. They are used in parallel and independently to better compare final results. In this sense, VarScan has been designed to be more permissive in the introduction of false positives, which was demonstrated to give a better performance [
34] when dealing with “normal allele alteration” problems. On the other hand, MuTect2 is the algorithm with the most stringent false-positive filter [
34]. These methods and others have been challenged in many reviews, showing that MuTect2 has the best performance dealing with both low- and high-frequency somatic mutation discovery [
35,
36]. In this study, we decided to use the combination of results from both the VarScan and MuTect2 pipelines.
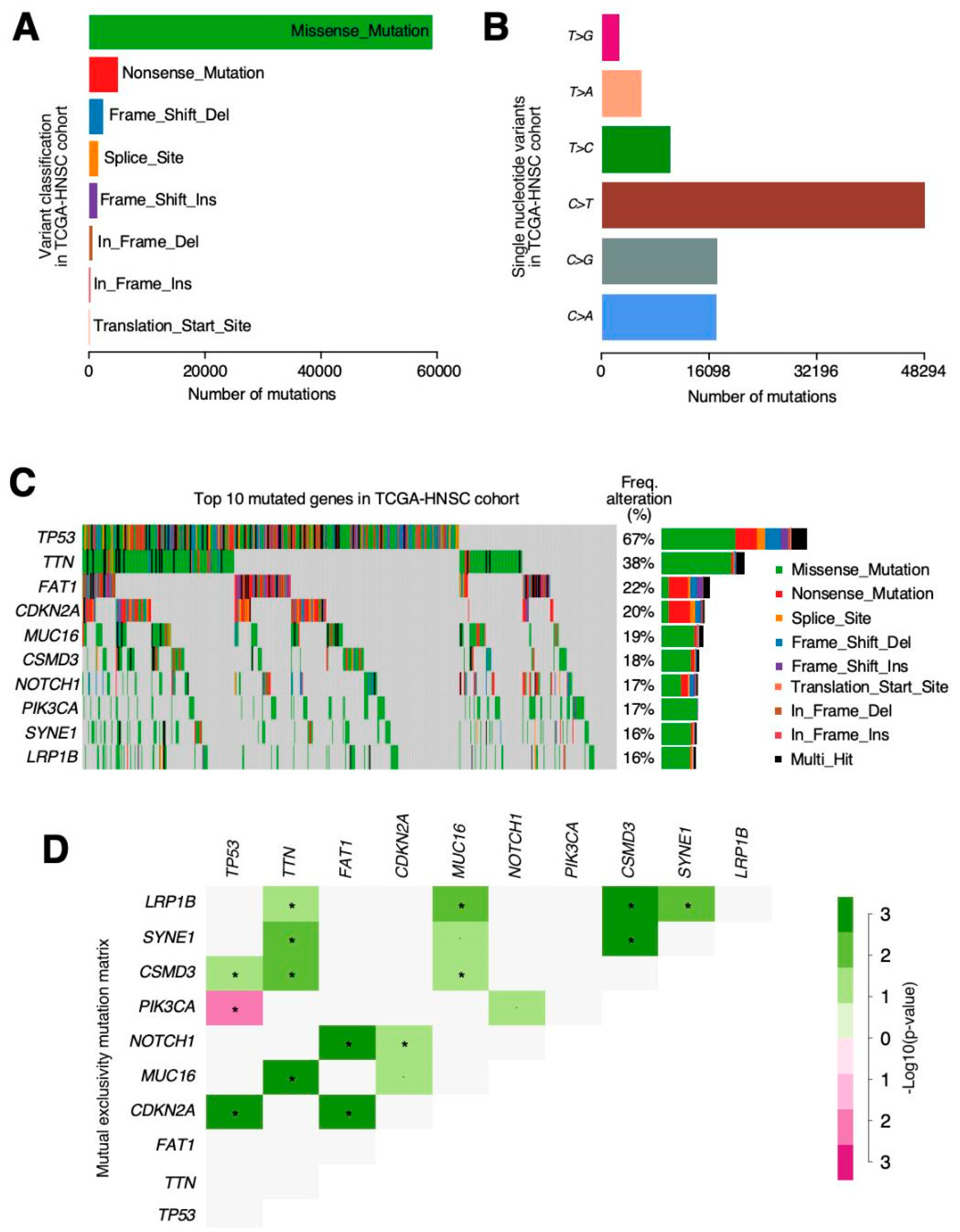
The most relevant mutations in our cohort were
TP53 (72%) followed by
TTN (39%),
FAT1 (23%) and
MUC16 (19%). TP53 has also been observed as the most prevalent mutation in previous studies [
37,
38] and also CDKN2A, EGFR and FAT1 were observed as being the most relevant [
38]. Among all mutations, we highlighted the mutual exclusivity pattern found between the
TP53 and
PIK3CA mutations. PIK3CA mutations have been associated with tobacco and alcohol consumption in previous studies [
39]. Maybe, HPV status can explain this mutually exclusive mutations based on HPV status, as p53 mutations are very rare in HPV (+) cancers, while PIK3CA mutations are common, at least in a genetically well-defined subgroup of HPV (+) cancers [
40].
Although these results do not concur with Babur et al. [
41], the mutual exclusivity pattern is necessary in order to delineate the functional relations and involvement in common pathways of cancer-causing alterations. The identification of these patterns permits clinicians and scientists, not only to establish new tumour classifications or new preventive therapies, but also to design potential treatment targets and identify tumour vulnerabilities. Therefore, further analysis of mutual exclusivity patterns in HNSCC is still necessary [
42].
cBioportal is a web application that simplifies data acquisition and processing from many high-quality publications, including the TCGA-HNSCC cohort. According to the cBioPortal analysis, the top five alteration genes were CDKN2A, CDKN2B, FGF3, FGF4, and FGF19 whereas in our results, those were CDKN2A, CDKN2B, PPFIA1, FADD, and ANO1. Theses discrepancies can be explained because there are two ways of using cBioportal: first, we can consult a dataset’s highlighting alterations in the “summary mode” or, second, a “by gene” query is available if we have a pre-selected genes list of our interest. cBioportal summary mode on TCGA-HNSCC “provisional” dataset show FGF3, FGF4 and FGF19 as copy number altered genes in 25% of the patients and no mention about PPFIA1, FADD or ANO1 genes. Nevertheless, the same platform when using the “by gene” query and the same dataset reports copy number alterations in these genes as 26% in the case of PPFIA1 and 25% in the cases of ANO1 and FADD.
We also have found differences in mutation data, while the top five genes in cBioPortal were TP53, FAT1, CDKN2A, NOTCH1, and PIK3CA, whereas in our results, they were TP53, TTN, FAT1, and MUC16. We did both summary and “by gene” queries in cBioportal and results were very similar as in copy number alteration matter. In this case, a reviewer search returned TP53, FAT1, CDKN2A, NOTCH1 and PIK3CA as the top mutated genes in the TCGA-HNSCC “provisional” dataset. There were no mentions about TTN or MUC16 mutation frequency, as we have reported. Once again, if we do a personalized query including all these genes, we face the same issue where TTN and MUC16 show larger mutation proportion (42% and 20%, respectively) than NOTCH1 or PIK3CA, both mutated in 18% of the samples. These discrepancies in cBioportal reports are intriguing and may be much better explained from its creator team and should not suppose any issue for the acceptance of our results as accurate and veracious.
Identification of those genes that are drivers in the tumorigenesis process is difficult as, even in the same tumour samples, different somatic alterations can be identified [
43]. After analysing missense mutations and truncated mutations in oncogenes and tumour suppressors (results not shown), we have found that generally, in oncogenes, missense mutations are superior to truncated mutations but not in tumour suppressors, except from those more famous in HNSCC as CDKN2, FAT1 or PTEN. This means that missense mutations also can produce downregulation in a protein activity, so they are not always associated with gain of function. Some tumour suppressors analysed in this study were not described as tumour suppressors in HNSCC and it is possible that missense mutations that we have found were really random.
Although we were unable to assess the HPV infection with SCNA or somatic mutations, as only a small proportion were HPV (+), previous studies that have associated these alterations with HPV infections have shown enrichment mutations in HPV-negative in TP53, CDKN2A and PIK3CA, and also copy number gains in EGFR and CCND1, similar to our results [
44]. Despite the already mentioned limited HPV (+) proportion, we performed differentially expression gene analysis in HPV (−) and HPV (+), obtaining similar results as other authors before us [
45].
Clustering genes in similar signalling pathways or even targets similar to other cancers can help to determine the treatment with targeted drugs that are already available or under development. HNSCC classical classification is not always able to be associated with a determined prognostic. Genetic expression analysis, somatic mutations and SCNA can help us to determine patterns that will enable the tumoral behaviour to be determined or the possible targeted therapies to be identified.

 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}