An Intriguing Involvement of Mitochondria in Cystic Fibrosis




{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Cystic Fibrosis: News on the Disease
2.1. CFTR Protein
2.1.1. Domain Structure of CFTR
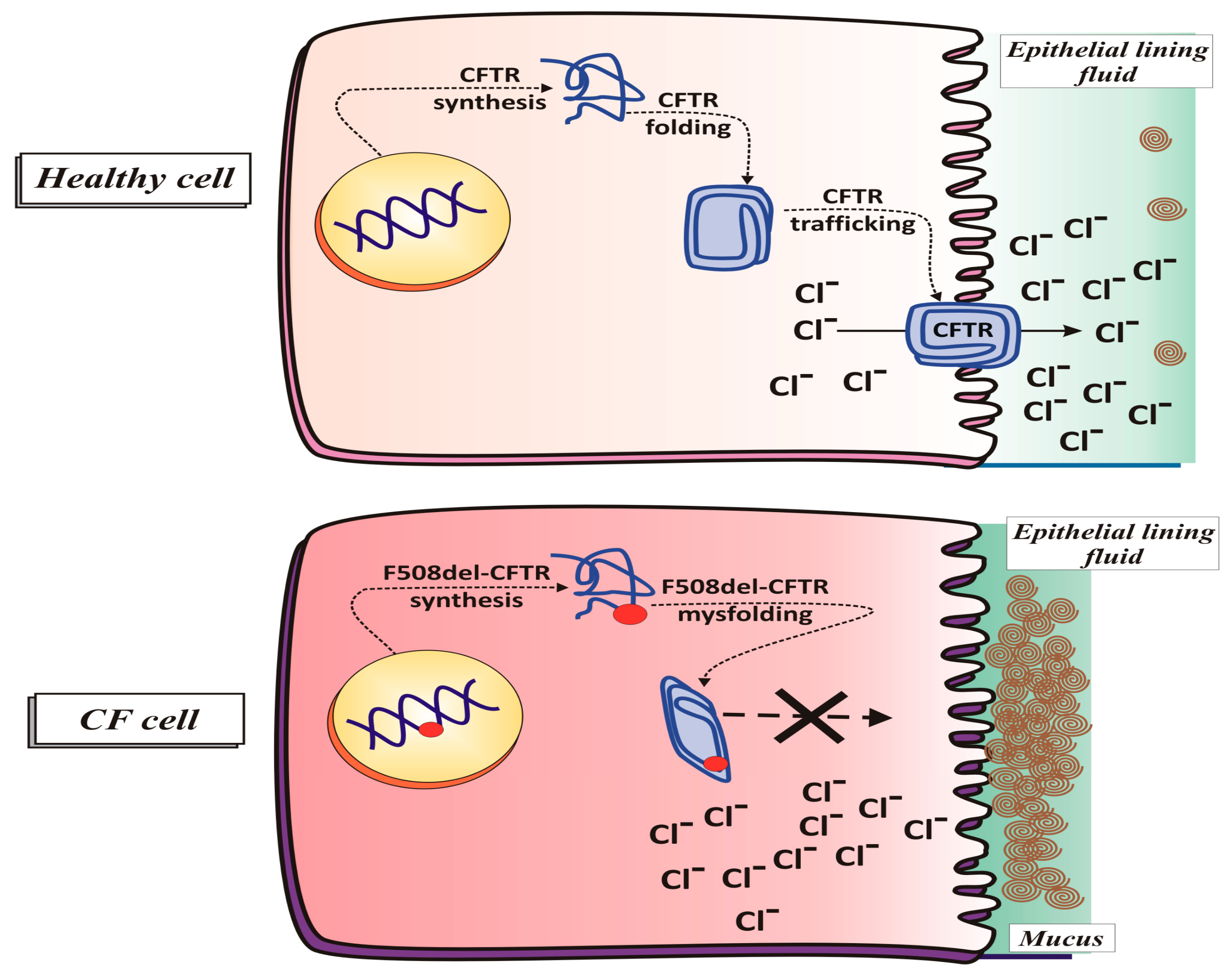
2.1.2. CFTR Synthesis and Trafficking
2.1.3. CFTR Function in Physiological Conditions
2.1.4. CFTR Function in Pathological Conditions (Cystic Fibrosis)
2.1.5. Classes of Mutations
F508del CFTR
2.2. Clinical Trials in Cystic Fibrosis
2.2.1. Treatments of CF Symptoms
2.2.2. Latest Breakthrough Therapies
3. Lung: Information and Facts
3.1. Lung: A Metabolically Active Organ
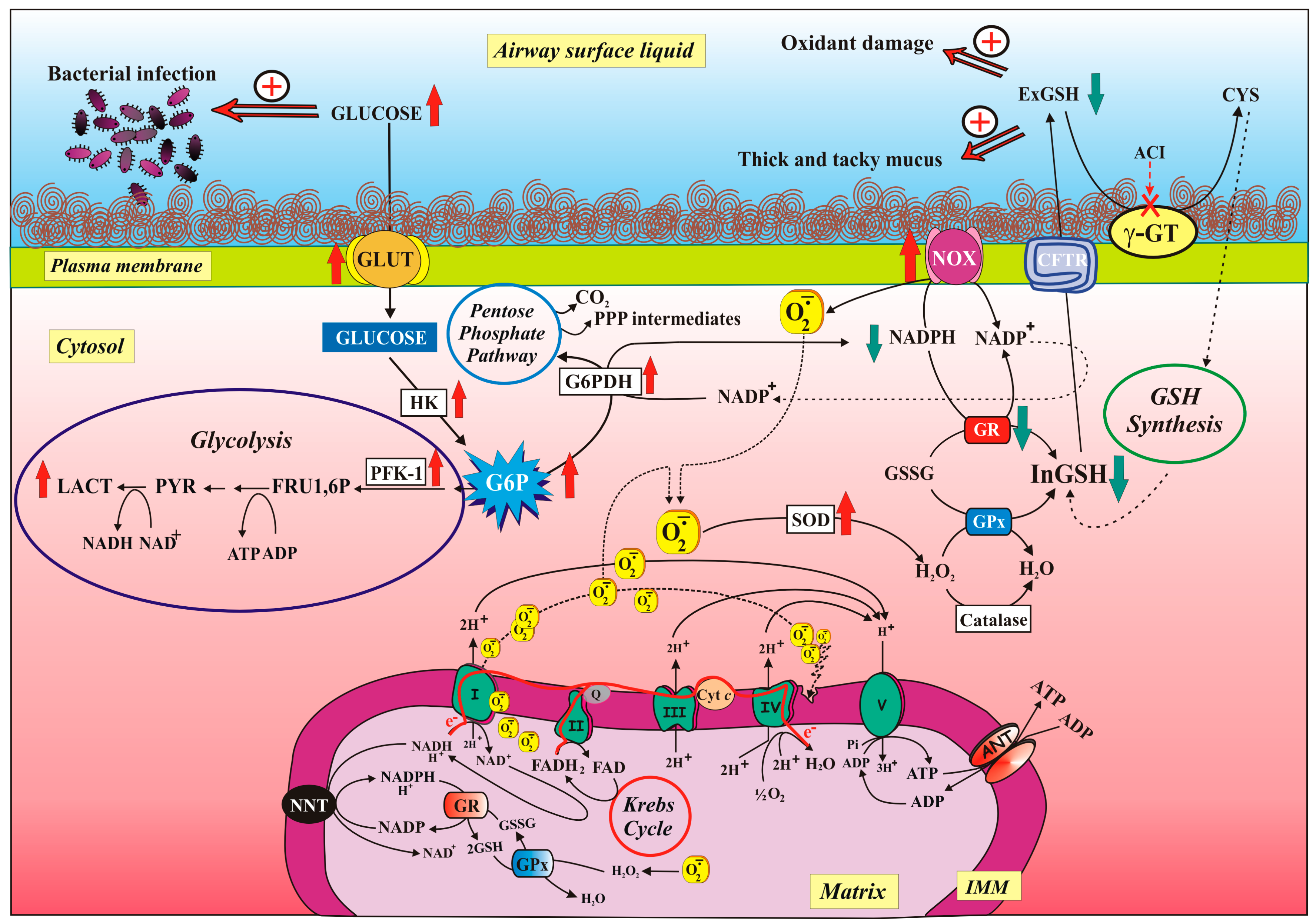
3.2. Lung Redox Homeostasis
3.3. Airways Surface Liquid: Characteristics and Functions
3.4. Glucose Movement Across the Airway Epithelium
4. Mitochondria
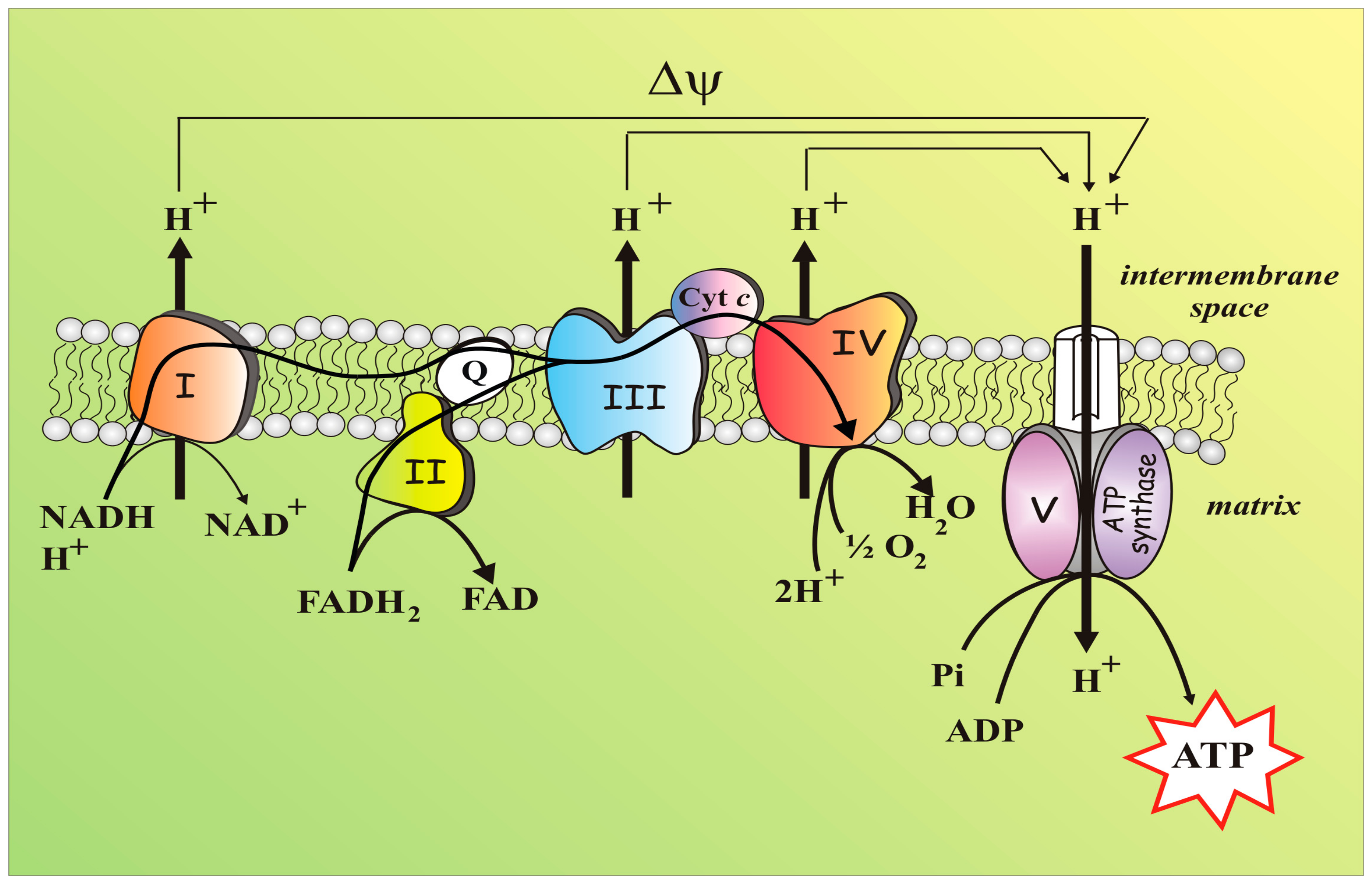
4.1. Mitochondria: A Short Brief and Essential Presentation
4.2. Mitochondria, An Essential Part of the Redox Balance
4.3. GSH as Tool to Combat Ox Stress (Infections)
5. Mitochondria in CF: What Is Known?
5.1. What Was Already Known about Mitochondria in CF?
5.2. The Latest Findings on Mitochondria in CF
5.2.1. Characterization of Mitochondrial Function in Cells with Impaired CFTR Function
- -
- -
- Studying how the balance between the production and neutralization of ROS is maintained in the presence of antioxidant enzymes, measuring the activity of superoxide dismutase (SOD) and catalase;
- -
- Measuring both the GSH-dependent enzyme, i.e., GPx and GR activities, and the GSH levels, either inside or outside the cell;
- -
- Analysing the redox states of the NAD and NADP pyridine nucleotide pools, which play critical roles in defining the activity of energy producing pathways and in both driving oxidative stress and maintaining antioxidant defences, respectively;
- -
- Identify the involvement of CFTR—if any—as part of the GSH cycle.
5.2.2. Defective CFTR and NOX/GR Activity Imbalance Contribute to ROS Overproduction
5.2.3. Modulation of Glucose-Related Metabolic Pathways Helps both Reduce Glucose Level in ASL and Fight Oxidative Stress
6. Conclusion Remarks
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| 6AN | 6-aminonicotinamide |
| ABC | ATP Binding Cassette |
| ACI | Acivicin |
| ANT | Adenine nucleotide translocase |
| AOX | Antioxidant system |
| ASL | Airway surface liquid |
| ATP | Adenosine 5′-triphosphate |
| CF | Cystic Fibrosis |
| CFTR | Cystic Fibrosis Transmembrane Conductance Regulator |
| CITR | Citrate |
| COPII | Protein Complex II |
| COX | Mitochondrial Complex IV |
| CYS | Cysteine |
| DPI | Diphenyliodonium |
| ΔΨ | Mitochondrial membrane potential |
| ENaC | Epithelial sodium channel |
| ER | Endoplasmic reticulum |
| ETC | Electron transport chain |
| exGSH | Extracellular GSH |
| GI | Glycolytic index |
| GLU | Glucose |
| GLUT | Glucose transporter |
| G6P | Glucose-6-phpsphate |
| G6PDH | Glucose-6-phosphate dehydrogenase |
| GPx | Glutathione peroxidase |
| GR | Glutathione reductase |
| GSH | Reduced glutathione |
| GSSG | Glutathione disulphide |
| CYS | Cysteine |
| γ-GT | γ-glutamyltransferase |
| γ-GCS | γ-glutamylcysteine synthase |
| HK | Hexokinase |
| H2O2 | Hydrogen peroxide |
| ICDH | Isocitrate dehydrogenase |
| IMS | Intermembrane space |
| inGSH | Intracellular GSH |
| L-LAC | L-lactate |
| MDH | Malate dehydrogenase |
| ME | Malic enzyme |
| mtCx-I | Mitochondrial Complex I |
| mGSH | mitochondrial GSH |
| Mirs | microRNAs |
| NAC | N-acetylcysteine |
| NOX | NAD(P)H oxidases |
| O2 | Molecular oxygen |
| O2−• | Superoxide anion radical |
| OLIGO | Oligomycin |
| OXPHOS | Oxidative phosphorylation |
| PFK | Phosphofructokinase |
| PKA | Protein kinase A |
| PPP | Pentose phosphate pathway |
| ROS | Reactive oxygen species |
| ROT | Rotenone |
| SOD | Superoxide dismutase |
| TCA | Tricarboxylic acid cycle |
| TMA | 4,6,4′-trimethylangelicin |
References
- McBride, H.M. Open questions: Seeking a holistic approach for mitochondrial research. BMC Biol. 2015, 13, 8. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pagliarini, D.J.; Rutter, J. Hallmarks of a new era in mitochondrial biochemistry. Genes Dev. 2013, 27, 2615–2627. [Google Scholar] [CrossRef] [PubMed]
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef]
- Riordan, J.R. CFTR and prospects for therapy. Annu. Rev. Biochem. 2008, 77, 701–726. [Google Scholar] [CrossRef] [PubMed]
- Farrel, P.M. The prevalence of cystic fibrosis in the European Union. J. Cyst. Fibros. 2008, 7, 450–453. [Google Scholar] [CrossRef]
- McCormick, J.; Mehta, G.; Olesen, H.V.; Viviani, L.; Macek, M., Jr.; Mehta, A.; European Registry Working Group. Comparative demographics of the European cystic fibrosis population: A cross-sectional database analysis. Lancet 2010, 375, 1007–1013. [Google Scholar] [CrossRef]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.-L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef]
- Rowe, S.M.; Miller, S.; Sorscher, E.J. Cystic fibrosis. N. Engl. J. Med. 2005, 352, 1992–2001. [Google Scholar] [CrossRef]
- Chan, H.C.; Ruan, Y.C.; He, Q.; Chen, M.H.; Chen, H.; Xu, W.M.; Chen, W.Y.; Xie, C.; Zhang, X.H.; Zhou, Z. The cystic fibrosis transmembrane conductance regulator in reproductive health and disease. J. Physiol. 2009, 587, 2187–2195. [Google Scholar] [CrossRef]
- Frizzell, R.A.; Hanrahan, J.W. Physiology of epithelian chloride and fluid secretion. Cold Spring Harb. Perspect. Med. 2012, 2, a009563. [Google Scholar] [CrossRef]
- Quinton, P.M. Physiological basis of cystic fibrosis: A historical perspective. Physiol. Rev. 1999, 79, S3–S22. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, B.P.; Freedman, S.D. Cystic fibrosis. Lancet 2009, 373, 1891–1904. [Google Scholar] [CrossRef]
- Yoshimura, K.; Nakamura, H.; Trapnell, B.C.; Chu, C.S.; Dalemans, W.; Pavirani, A.; Lecocq, J.P.; Crystal, R.G. Expression of the cystic fibrosis transmembrane conductance regulator gene in cells of non-epithelial origin. Nucleic Acids Res. 1991, 19, 5417–5423. [Google Scholar] [CrossRef] [PubMed]
- Levesque, P.C.; Hart, P.J.; Hume, J.R.; Kenyon, J.L.; Horowitz, B. Expression of cystic fibrosis transmembrane regulator Cl channels in heart. Circ. Res. 1992, 71, 1002–1007. [Google Scholar] [CrossRef]
- Horowitz, B.; Tsung, S.S.; Hart, P.; Levesque, P.C.; Hume, J.R. Alternative splicing of CFTRCl channels inheart. Am. J. Physiol. 1993, 264, H2214–H2220. [Google Scholar]
- Tizzano, E.F.; Chitayat, D.; Buchwald, M. Cell-specific localization of CFTR mRNA shows developmentally regulated expression in human fetal tissues. Hum. Mol. Genet. 1993, 2, 219–224. [Google Scholar] [CrossRef]
- Mulberg, A.E.; Wiedner, E.B.; Bao, X.; Marshall, J.; Jefferson, D.M.; Altschuler, S.M. Cystic fibrosis transmembrane conductance regulator protein expression in brain. Neuroreport 1994, 5, 1684–1688. [Google Scholar] [CrossRef]
- Kulka, M.; Gilchrist, M.; Duszyk, M.; Befus, A.D. Expression and functional characterization of CFTR in mast cells. J. Leukoc. Biol. 2002, 71, 54–64. [Google Scholar]
- Lange, T.; Jungmann, P.; Haberle, J.; Falk, S.; Duebbers, A.; Bruns, R.; Ebner, A.; Hinterdorfer, P.; Oberleithner, H.; Schillers, H. Reduced number of CFTR molecules in erythrocyte plasma membrane of cystic fibrosis patients. Mol. Membr. Biol. 2006, 23, 317–323. [Google Scholar] [CrossRef]
- Swahn, H.; Harris, A. Cell-selective regulation of CFTR gene expression: Relevance to gene editing therapeutics. Genes 2019, 10, 235. [Google Scholar] [CrossRef]
- Schwiebert, E.M.; Benos, D.J.; Egan, M.E.; Stutts, M.J.; Guggino, W.B. CFTR is a conductance regulator as well as a chloride channel. Physiol. Rev. 1999, 79, S145–S166. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, D.N.; Welsh, M.J. Structure and function of the CFTR chloride channel. Physiol. Rev. 1999, 79, S23–S45. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.H.; Rich, D.P.; Marshall, J.; Gregory, R.J.; Welsh, M.J.; Smith, A.E. Phosphorylation of the R domain by cAMP-dependent protein kinase regulates the CFTR chloride channel. Cell 1991, 66, 1027–1036. [Google Scholar] [CrossRef]
- Berger, A.L.; Ikuma, M.; Welsh, M.J. Normal gating of CFTR requires ATP binding to both nucleotide-binding domains and hydrolysis at the second nucleotide-binding domain. Proc. Natl. Acad. Sci. USA 2005, 102, 455–460. [Google Scholar] [CrossRef]
- Vergani, P.; Lockless, S.W.; Nairn, A.C.; Gadsby, D.C. CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature 2005, 433, 876–880. [Google Scholar] [CrossRef]
- Csanady, L.; Vergani, P.; Gadsby, D.C. Strict coupling between CFTR’s catalytic cycle and gating of its Cl− ion pore revealed by distributions of open channel burst durations. Proc. Natl. Acad. Sci. USA 2010, 107, 1241–1246. [Google Scholar] [CrossRef]
- Naren, A.P.; Cobb, B.; Li, C.; Roy, K.; Nelson, D.; Heda, G.D.; Liao, J.; Kirk, K.L.; Sorscher, E.J.; Hanrahan, J.; et al. A macromolecular complex of beta 2 adrenergic receptor, CFTR, and ezrin/radixin/moesin-binding phosphoprotein 50 is regulated by PKA. Proc. Natl. Acad. Sci. USA 2003, 100, 342–346. [Google Scholar] [CrossRef]
- Li, C.; Naren, A.P. Macromolecular complexes of cystic fibrosis transmembrane conductance regulator and its interacting partners. Pharmacol. Ther. 2005, 108, 208–223. [Google Scholar] [CrossRef]
- Li, C.; Naren, A.P. Analysis of CFTR interactome in the macromolecular complexes. Methods Mol. Biol. 2011, 741, 255–270. [Google Scholar]
- Zhang, W.; Penmatsa, H.; Ren, A.; Punchihewa, C.; Lemoff, A.; Yan, B.; Fujii, N.; Naren, A.P. Functional regulation of cystic fibrosis transmembrane conductance regulator-containing macromolecular complexes: A small-molecule inhibitor approach. Biochem. J. 2011, 435, 451–462. [Google Scholar] [CrossRef]
- Guerra, L.; Fanelli, T.; Favia, M.; Riccardi, S.M.; Busco, G.; Cardone, R.A.; Carrabino, S.; Weinman, E.J.; Reshkin, S.J.; Conese, M.; et al. Na+/H+ exchanger regulatory factor isoform 1 overexpression modulates cystic fibrosis transmembrane conductance regulator (CFTR) expression and activity in human airway 16HBE14o- cells and rescues DeltaF508 CFTR functional expression in cystic fibrosis cells. J. Biol. Chem. 2005, 280, 40925–40933. [Google Scholar] [PubMed]
- Favia, M.; Guerra, L.; Fanelli, T.; Cardone, R.A.; Monterisi, S.; Di Sole, F.; Castellani, S.; Chen, M.; Seidler, U.; Reshkin, S.J.; et al. Na+/H+ exchanger regulatory factor 1 overexpression-dependent increase of cytoskeleton organization is fundamental in the rescue of F508del cystic fibrosis transmembrane conductance regulator in human airway CFBE41o- cells. Mol. Biol. Cell 2010, 21, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Rogan, M.P.; Stoltz, D.A.; Hornick, D.B. Cystic fibrosis transmembrane conductance regulator intracellular processing, trafficking, and opportunities for mutation-specific treatment. Chest 2011, 139, 1480–1490. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Janich, S.; Cohn, J.A.; Wilson, J.M. The common variant of cystic fibrosis transmembrane conductance regulator is recognized by hsp70 and degraded in a pre-Golgi nonlysosomal compartment. Proc. Natl. Acad. Sci. USA 1993, 90, 9480–9484. [Google Scholar] [CrossRef] [PubMed]
- Cheung, J.C.; Deber, C.M. Misfolding of the cystic fibrosis transmembrane conductance regulator and disease. Biochemistry 2008, 47, 1465–1473. [Google Scholar] [CrossRef] [PubMed]
- Pind, S.; Riordan, J.R.; Williams, D.B. Participation of the endoplasmic reticulum chaperone calnexin (p88, IP90) in the biogenesis of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 1994, 269, 12784–12788. [Google Scholar] [PubMed]
- Turnbull, E.L.; Rosser, M.F.; Cyr, D.M. The role of the UPS in cystic fibrosis. BMC Biochem. 2007, 8, S11. [Google Scholar] [CrossRef]
- Cheng, J.; Wang, H.; Guggino, W.B. Modulation of mature cystic fibrosis transmembrane regulator protein by the PDZ domain protein CAL. J. Biol. Chem. 2004, 279, 1892–1898. [Google Scholar] [CrossRef]
- Saint-Criq, V.; Gray, M.A. Role of CFTR in epithelial physiology. Cell. Mol. Life Sci. 2017, 74, 93–115. [Google Scholar] [CrossRef]
- Kogan, I.; Ramjeesingh, M.; Li, C.; Kidd, J.F.; Wang, Y.; Leslie, E.M.; Cole, S.P.; Bear, C.E. CFTR directly mediates nucleotide-regulated glutathione flux. EMBO J. 2003, 22, 1981–1989. [Google Scholar] [CrossRef]
- Boucher, R.C. Status of gene therapy for cystic fibrosis lung disease. J. Clin. Investig. 1999, 103, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Fahy, J.V.; Dickey, B.F. Airway mucus function and dysfunction. N. Engl. J. Med. 2010, 363, 2233–2247. [Google Scholar] [CrossRef] [PubMed]
- Hull, J. Cystic fibrosis transmembrane conductance regulator dysfunction and its treatment. J. R. Soc. Med. 2012, 105 (Suppl. 2), S2–S8, review. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Shrestha, C.L.; Kopp, B.T. Cystic fibrosis transmembrane conductance regulator (CFTR) modulators have differential effects on cystic fibrosis macrophage function. Sci. Rep. 2018, 8, 17066. [Google Scholar] [CrossRef]
- Ratner, D.; Mueller, C. Immune responses in cystic fibrosis: Are they intrinsically defective? Am. J. Respir. Cell Mol. Biol. 2012, 46, 715–722. [Google Scholar] [CrossRef]
- Tazi, M.F.; Dakhlallah, D.A.; Caution, K.; Gerber, M.M.; Chang, S.W.; Khalil, H.; Kopp, B.T.; Ahmed, A.E.; Krause, K.; Davis, I.; et al. Elevated Mirc1/Mir17-92 cluster expression negatively regulates autophagy and CFTR (cystic fibrosis transmembrane conductance regulator) function in CF macrophages. Autophagy 2016, 12, 2026–2037. [Google Scholar] [CrossRef]
- Painter, R.G.; Valentine, V.G.; Lanson, N.A., Jr.; Leidal, K.; Zhang, Q.; Lombard, G.; Thompson, C.; Viswanathan, A.; Nauseef, W.M.; Wang, G.; et al. CFTR expression in human neutrophils and the phagolysosomal chlorination defect in cystic fibrosis. Biochemistry 2006, 45, 10260–10269. [Google Scholar] [CrossRef]
- Bonfield, T.; Chmiel, J.F. Impaired innate immune cells in cystic fibrosis: Is it really a surprise? J. Cyst. Fibr. 2017, 16, 433–435. [Google Scholar] [CrossRef]
- Lukacs, G.L.; Verkman, A.S. CFTR: Folding, misfolding and correcting the ΔF508 conformational defect. Trends Mol. Med. 2012, 18, 81–91. [Google Scholar] [CrossRef]
- Kälin, N.; Claaß, A.; Sommer, M.; Puchelle, E.; Tümmler, B. ΔF508 CFTR protein expression in tissues from patients with cystic fibrosis. J. Clin. Investig. 1999, 103, 1379–1389. [Google Scholar] [CrossRef]
- Bronsveld, I.; Mekus, F.; Bijman, J.; Ballmann, M.; de Jonge, H.R.; Laabs, U.; Halley, D.J.; Ellemunter, H.; Mastella, G.; Thomas, S.; et al. Chloride conductance and genetic background modulate the cystic fibrosis phenotype of ΔF508 homozygous twins and siblings. J. Clin. Investig. 2001, 108, 1705–1715. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gentzsch, M.; Choudhury, A.; Chang, X.B.; Pagano, R.E.; Riordan, J.R. Misassembled mutant DeltaF508 CFTR in the distal secretory pathway alters cellular lipid trafficking. J. Cell Sci. 2007, 120, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Swiatecka-Urban, A.; Brown, A.; Moreau-Marquis, S.; Renuka, J.; Coutermarsh, B.; Barnaby, R.; Karlson, K.H.; Flotte, T.R.; Fukuda, M.; Langford, G.M.; et al. The short apical membrane half-life of rescued ΔF508-cystic fibrosis transmembrane conductance regulator (CFTR) results from accelerated endocytosis of ΔF508-CFTR in polarized human airway epithelial cells. J. Biol. Chem. 2005, 280, 36762–36772. [Google Scholar] [CrossRef] [PubMed]
- Castellani, C.; Assael, B.M. Cystic fibrosis: A clinical view. Cell. Mol. Life Sci. 2017, 74, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Burgel, P.-R.; Bellis, G.; Olesen, H.; Viviani, L.; Zolin, A.; Blasi, F.; Elborn, J.S. Future trends in cystic fibrosis demography in 34 European countries. Eur. Respir. J. 2015, 46, 133–141. [Google Scholar]
- MacConnachie, A.M. Dornase-alfa (DNase, Pulmozyme) for cystic fibrosis. Intensive Crit. Care Nurs. 1998, 14, 101–102. [Google Scholar] [CrossRef]
- Reeves, E.P.; Molloy, K.; Pohl, K.; McElvaney, N.G. Hypertonic saline in treatment of pulmonary disease in cystic fibrosis. Sci. World J. 2012, 2012, 465230. [Google Scholar] [CrossRef] [PubMed]
- Suri, R. The use of human deoxyribonuclease (rhDNase) in the management of cystic fibrosis. BioDrugs 2005, 19, 135–144. [Google Scholar] [CrossRef]
- Pisi, G.; Chetta, A. Airway clearance therapy in cystic fibrosis patients. Acta Biomed. 2009, 80, 102–106. [Google Scholar]
- Adler, F.R.; Aurora, P.; Barker, D.H.; Barr, M.L.; Blackwell, L.S.; Bosma, O.H.; Brown, S.; Cox, D.R.; Jensen, J.L.; Kurland, G.; et al. Lung transplantation for cystic fibrosis. Proc. Am. Thorac. Soc. 2009, 6, 619–633. [Google Scholar] [CrossRef]
- Favia, M.; Mancini, M.T.; Bezzerri, V.; Guerra, L.; Laselva, O.; Abbattiscianni, A.C.; Debellis, L.; Reshkin, S.J.; Gambari, R.; Cabrini, G.; et al. Trimethylangelicin promotes the functional rescue of mutant F508del CFTR protein in cystic fibrosis airway cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L48–L61. [Google Scholar] [CrossRef] [PubMed]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.; Burton, B.; Stack, J.H.; Straley, K.S.; Decker, C.J.; Miller, M.; McCartney, J.; Olson, E.R.; et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. USA 2011, 108, 18843–18848. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.Q.; Xiong, G.; Wilber, M.; Cohen, N.A.; Kreindler, J.L. A role for two-pore K+ channels in modulating Na+ absorption and Cl− secretion in normal human bronchial epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L4–L12. [Google Scholar] [CrossRef] [PubMed]
- Schiffhauer, E.S.; Vij, N.; Kovbasnjuk, O.; Kang, P.W.; Walker, D.; Lee, S.; Zeitlin, P.L. Dual activation of CFTR and CLCN2 by lubiprostone in murine nasal epithelia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L324–L331. [Google Scholar] [CrossRef] [PubMed]
- Griesenbach, U.; Davies, J.C.; Alton, E. Cystic fibrosis gene therapy: A mutation-independent treatment. Curr. Opin. Pulm. Med. 2016, 22, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Summer, R. Cellular metabolism in lung health and disease. Annu. Rev. Physiol. 2019, 81, 403–428. [Google Scholar] [CrossRef]
- O’Neil, J.J.; Tierney, D.F. Rat lung metabolism: Glucose utilization by isolated perfused lungs and tissue slices. Am. J. Physiol. 1974, 226, 867–873. [Google Scholar] [CrossRef]
- Tierney, D.F. Intermediary metabolism of the lung. Fed. Proc. 1974, 33, 2232–2237. [Google Scholar]
- Mustafa, M.G.; Cross, C.E. Effects of short-term ozone exposure on lung mitochondrial oxidative and energy metabolism. Arch. Biochem. Biophys. 1974, 162, 585–594. [Google Scholar] [CrossRef]
- Hussien, R.; Brooks, G.A. Mitochondrial and plasma membrane lactate transporter and lactate dehydrogenase isoform expression in breast cancer cell lines. Physiol. Genomics 2011, 43, 255–264. [Google Scholar] [CrossRef]
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K.; et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell 2008, 134, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate metabolism in human lung tumors. Cell 2017, 171, 358–371.e9. [Google Scholar] [CrossRef] [PubMed]
- Hüttemann, M.; Lee, I.; Gao, X.; Pecina, P.; Pecinova, A.; Liu, J.; Aras, S.; Sommer, N.; Sanderson, T.H.; Tost, M.; et al. Cytochrome c oxidase subunit 4 isoform 2-knockout mice show reduced enzyme activity, airway hyporeactivity, and lung pathology. FASEB J. 2012, 26, 3916–3930. [Google Scholar] [CrossRef] [PubMed]
- Squadrito, G.L.; Cueto, R.; Dellinger, B.; Pryor, W.A. Quinoid redox cycling as a mechanism for sustained free radical generation by inhaled airborne particulate matter. Free Radic. Biol. Med. 2001, 31, 1132–1138. [Google Scholar] [CrossRef]
- Dellinger, B.; Pryor, W.A.; Cueto, R.; Squadrito, G.L.; Hegde, V.; Deutsch, W.A. Role of free radicals in the toxicity of airborne fine particulate matter. Chem. Res. Toxicol. 2001, 14, 1371–1377. [Google Scholar] [CrossRef]
- Aravamudan, B.; Thompson, M.A.; Pabelick, C.M.; Prakash, Y.S. Mitochondria in lung diseases. Expert Rev. Respir. Med. 2013, 7, 631–646. [Google Scholar] [CrossRef]
- Segal, B.H.; Grimm, M.J.; Khan, A.N.; Han, W.; Blackwell, T.S. Regulation of innate immunity by NADPH oxidase. Free Radic. Biol. Med. 2012, 53, 72–80. [Google Scholar] [CrossRef]
- van der Vliet, A. Nox enzymes in allergic airway inflammation. Biochim. Biophys. Acta 2011, 1810, 1035–1044. [Google Scholar] [CrossRef]
- Brown, R.K.; Kelly, F.J. Evidence of increased oxidative damage in patients with cystic fibrosis. Pediatr. Res. 1994, 36, 1–7. [Google Scholar] [CrossRef]
- Yagi, K. Lipid peroxides and human diseases. Chem. Phys. Lipids 1987, 45, 337–351. [Google Scholar] [CrossRef]
- Galli, F.; Battistoni, A.; Gambari, R.; Pompella, A.; Bragonzi, A.; Pilolli, F.; Iuliano, L.; Piroddi, M.; Dechecchi, M.C.; Cabrini, G.; et al. Oxidative stress and antioxidant therapy in cystic fibrosis. Biochim. Biophys. Acta 2012, 1822, 690–713. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, V.D.; Vliagoftis, H. Airway epithelium interactions with aeroallergens: Role of secreted cytokines and chemokines in innate immunity. Front. Immunol. 2015, 6, 147. [Google Scholar] [CrossRef] [PubMed]
- Collawn, J.F.; Lazrak, A.; Bebok, Z.; Matalon, S. The CFTR and ENaC debate: How important is ENaC in CF lung disease? Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L1141–L1146. [Google Scholar] [CrossRef] [PubMed]
- Kunzelmann, K.; Kathöfer, S.; Greger, R. Na+ and Cl− conductances in airway epithelial cells: Increased Na+ conductance in cystic fibrosis. Pflugers. Arch 1995, 431, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Boucher, R.C. Relationship of airway epithelial ion transport to chronic bronchitis. Proc. Am. Thorac. Soc. 2004, 1, 66–70. [Google Scholar] [CrossRef]
- Zhao, R.; Liang, X.; Zhao, M.; Liu, S.L.; Huang, Y.; Idell, S.; Li, X.; Ji, H.L. Correlation of apical fluid-regulating channel proteins with lung function in human COPD lungs. PLoS ONE 2014, 9, e109725. [Google Scholar] [CrossRef]
- Matthay, M.A.; Folkesson, H.G.; Clerici, C. Lung epithelial fluid transport and the resolution of pulmonary edema. Physiol. Rev. 2002, 82, 569–600. [Google Scholar] [CrossRef]
- Zeitlin, P.L. Cystic fibrosis and estrogens: A perfect storm. J. Clin. Investig. 2008, 118, 3841–3844. [Google Scholar] [CrossRef]
- Rhoades, R.A. Net uptake of glucose, glycerol, and fatty acids by the isolated perfused rat lung. Am. J. Physiol. 1974, 226, 144–149. [Google Scholar] [CrossRef]
- Bearham, J.; Garnett, J.P.; Schroeder, V.; Biggart, M.G.; Baines, D.L. Effective glucose metabolism maintains low intracellular glucose in airway epithelial cells after exposure to hyperglycaemia. Am. J. Physiol. Cell Physiol. 2019. [Google Scholar] [CrossRef]
- Kalsi, K.K.; Baker, E.H.; Fraser, O.; Chung, Y.L.; Mace, O.J.; Tarelli, E.; Philips, B.J.; Baines, D.L. Glucose homeostasis across human airway epithelial cell monolayers: Role of diffusion, transport and metabolism. Pflugers Arch. 2009, 457, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Garnett, J.P.; Nguyen, T.T.; Moffatt, J.D.; Pelham, E.R.; Kalsi, K.K.; Baker, E.H.; Baines, D.L. Proinflammatory mediators disrupt glucose homeostasis in airway surface liquid. J. Immunol. 2012, 189, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Philips, B.J.; Redman, J.; Brennan, A.; Wood, D.; Holliman, R.; Baines, D.; Baker, E.H. Glucose in bronchial aspirates increases the risk of respiratory MRSA in intubated patients. Thorax 2005, 60, 761–764. [Google Scholar] [CrossRef] [PubMed]
- Baker, E.H.; Clark, N.; Brennan, A.L.; Fisher, D.A.; Gyi, K.M.; Hodson, M.E.; Philips, B.J.; Baines, D.L.; Wood, D.M. Hyperglycemia and cystic fibrosis alter respiratory fluid glucose concentrations estimated by breath condensate analysis. J. Appl. Physiol. 1985, 102, 1969–1975. [Google Scholar] [CrossRef]
- Wood, D.M.; Brennan, A.L.; Philips, B.J.; Baker, E.H. Effect of hyperglycaemia on glucose concentration of human nasal secretions. Clin. Sci. (Lond.) 2004, 106, 527–533. [Google Scholar] [CrossRef]
- Bilodeau, C.; Bardou, O.; Maillé, É.; Berthiaume, Y.; Brochiero, E. Deleterious impact of hyperglycemia on cystic fibrosis airway ion transport and epithelial repair. J. Cyst. Fibros. 2016, 15, 43–51. [Google Scholar] [CrossRef]
- Meo, S.A. Significance of spirometry in diabetic patients. Int. J. Diabetes Mellit. 2009, 2, 47–50. [Google Scholar] [CrossRef]
- Duchen, M.R. Mitochondria in health and disease: Perspectives on a new mitochondrial biology. Mol. Asp. Med. 2004, 25, 365–451. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Murphy, M.P. Mitochondria—A neglected drug target. Curr. Opin. Investig. Drugs 2009, 10, 1022–1024. [Google Scholar]
- Scarpulla, R.C. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 2008, 88, 611–638. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G.; Fergusson, S.J. Bioenergetics; Academic Press: Cambridge, MA, USA, 2013. [Google Scholar]
- Diaz, F.; Kotarsky, H.; Fellman, V.; Moraes, C.T. Mitochondrial disorders caused by mutations in respiratory chain assembly factors. Semin. Fetal Neonatal Med. 2011, 16, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Efremov, R.G.; Sazanov, L.A. Respiratory complex I: ‘Steam engine’ of the cell? Curr. Opin. Struct. Biol. 2011, 21, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J.; Jin, X.; Willmore, W.G. Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox Biol. 2013, 2, 123–139. [Google Scholar] [CrossRef]
- Mitchell, P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 1961, 191, 144–148. [Google Scholar] [CrossRef]
- Neupert, W.; Herrmann, J.M. Translocation of proteins into mitochondria. Annu. Rev. Biochem. 2007, 76, 723–749. [Google Scholar] [CrossRef]
- Mailloux, R.J.; Harper, M.E. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic. Biol. Med. 2011, 51, 1106–1115. [Google Scholar] [CrossRef]
- Mailloux, R.J. Mitochondrial antioxidants and the maintenance of cellular hydrogen peroxide levels. Oxid. Med. Cell. Longev. 2018, 2018, 7857251. [Google Scholar] [CrossRef]
- Georgieva, E.; Ivanova, D.; Zhelev, Z.; Bakalova, R.; Gulubova, M.; Aoki, I. Mitochondrial dysfunction and redox imbalance as a diagnostic marker of “free radical diseases”. Anticancer Res. 2017, 37, 5373–5381. [Google Scholar]
- Apostolova, N.; Victor, V.M. Molecular strategies for targeting antioxidants to mitochondria: Therapeutic implications. Antioxid. Redox Signal. 2015, 22, 686–729. [Google Scholar] [CrossRef]
- Hirrlinger, J.; Dringen, R. The cytosolic redox state of astrocytes: Maintenance, regulation and functional implications for metabolite trafficking. Brain Res. Rev. 2010, 63, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C. Mechanisms of inflammatory neurodegeneration: iNOS and NADPH oxidase. Biochem. Soc. Trans. 2007, 35, 1119–1121. [Google Scholar] [CrossRef] [PubMed]
- Sorce, S.; Krause, K.H. NOX enzymes in the central nervous system: From signaling to disease. Antioxid. Redox Signal. 2009, 11, 2481–2504. [Google Scholar] [CrossRef] [PubMed]
- Handy, D.E.; Loscalzo, J. Redox regulation of mitochondrial function. Antioxid. Redox Signal. 2012, 16, 1323–1367. [Google Scholar] [CrossRef] [PubMed]
- de Bari, L.; Favia, M.; Bobba, A.; Lassandro, R.; Guerra, L.; Atlante, A. Aberrant GSH reductase and NOX activities concur with defective CFTR to pro-oxidative imbalance in cystic fibrosis airways. J. Bioenerg. Biomembr. 2018, 50, 117–129. [Google Scholar] [CrossRef]
- Magni, G.; Orsomando, G.; Raffelli, N.; Ruggieri, S. Enzymology of mammalian NAD metabolism in health and disease. Front. Biosci. 2008, 13, 6135–6154. [Google Scholar] [CrossRef]
- Legan, S.K.; Rebrin, I.; Mockett, R.J.; Radyuk, S.N.; Klichko, V.I.; Sohal, R.S.; Orr, W.C. Overexpression of glucose-6-phosphate dehydrogenase extends the life span of Drosophila melanogaster. J. Biol. Chem. 2008, 283, 32492–32499. [Google Scholar] [CrossRef]
- Zhao, Y.; Seefeldt, T.; Chen, W.; Wang, X.; Matthees, D.; Hu, Y.; Guan, X. Effects of glutathione reductase inhibition on cellular thiol redox state and related systems. Arch. Biochem. Biophys. 2009, 485, 56–62. [Google Scholar] [CrossRef]
- Ribas, V.; García-Ruiz, C.; Fernández-Checa, J.C. Glutathione and mitochondria. Front. Pharmacol. 2014, 5, 151. [Google Scholar] [CrossRef]
- Velsor, L.W.; Kariya, C.; Kachadourian, R.; Day, B.J. Mitochondrial oxidative stress inthe lungs of cystic fibrosis transmembrane conductance regulator protein mutant mice. Am. J. Respir. Cell Mol. Biol. 2006, 35, 579–586. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cantin, A.M.; North, S.L.; Hubbard, R.C.; Crystal, R.G. Normal alveolar epithelial lining fluid contains high levels of glutathione. J. Appl. Physiol. 1985, 63, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Aldini, G.; Altomare, A.; Baron, G.; Vistoli, G.; Carini, M.; Borsani, L.; Sergio, F. N-Acetylcysteine as an antioxidant and disulphide breaking agent: The reasons why. Free Radic. Res. 2018, 52, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Favia, M.; Atlante, A. Mitochondria and cystic fibrosis transmembrane conductance regulator dialogue: Some news. J. Rare Dis. Res. Treat. 2016, 1, 23–29. [Google Scholar]
- Stutts, M.J.; Knowles, M.R.; Gatzy, J.T.; Boucher, R.C. Oxygen consumption and ouabain binding sites in cystic fibrosis nasal epithelium. Pediatr. Res. 1986, 20, 1316–1320. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F.; Freeman, B.A.; Levitt, J.G.; Crapo, J.D. The effect of hyperoxia on superoxide production by lung submitochondrial particles. Arch. Biochem. Biophys. 1982, 217, 401–410. [Google Scholar] [CrossRef]
- Awasthi, A.; Prasad, B.; Kumar, J. Altered mitochondrial function and cystic fibrosis. Hered. Genet. S7 2015. [Google Scholar] [CrossRef]
- Shapiro, B.L.; Feigal, R.J.; Lam, L.F. Mitochondrial NADH dehydrogenase in cystic fibrosis. Proc. Nat. Acad. Sci. USA 1979, 76, 2979–2983. [Google Scholar] [CrossRef] [PubMed]
- Battino, M.; Rugolo, M.; Romeo, G.; Lenaz, G. Kinetic alterations of cytochrome-c oxidase in cystic fibrosis. FEBS Lett. 1986, 199, 155–158. [Google Scholar] [CrossRef]
- Valdivieso, A.G.; Santa-Coloma, T.A. CFTR activity and mitochondrial function. Redox Biol. 2013, 1, 190–202. [Google Scholar] [CrossRef]
- Picci, L.; Brentagni, L.; Mastella, G.; Scarso, E.; Pizzochero, P.; Mattiazzo, P.; Chiandetti, L.; Anglani, F.; Zacchello, F. 2D-electrophoresis of mitochondrial proteins from cystic fibrosis patients. Adv. Exp. Med. Biol. 1991, 290, 379–381. [Google Scholar] [PubMed]
- de Meer, K.; Jeneson, J.A.; Gulmans, V.A.; van der Laag, J.; Berger, R. Efficiency of oxidative work performance of skeletal muscle in patients with cystic fibrosis. Thorax 1995, 50, 980–983. [Google Scholar] [CrossRef] [PubMed]
- Chomyn, A. Mitochondrial genetic control of assembly and function of complex I in mammalian cells. J. Bioenerg. Biomembr. 2001, 33, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Hajek, P.; Chomyn, A.; Chan, E.; Seo, B.B.; Matsuno-Yagi, A.; Yagi, T.; Attardi, G. Lack of complex I activity in human cells carrying a mutation in MtDNA-encoded ND4 subunit is corrected by the Saccharomyces cerevisiae NADH-quinone oxidoreductase (NDI1) gene. J. Biol. Chem. 2001, 276, 38808–38813. [Google Scholar] [CrossRef] [PubMed]
- Atlante, A.; Favia, M.; Bobba, A.; Guerra, L.; Casavola, V.; Reshkin, S.J. Characterization of mitochondrial function in cells with impaired cystic fibrosis transmembrane conductance regulator (CFTR) function. J. Bioenerg. Biomembr. 2016, 48, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Cleeter, M.W.; Cooper, J.M.; Schapira, A.H. Irreversible inhibition of mitochondrial complex I by 1-methyl-4-phenylpyridinium: Evidence for free radical involvement. J. Neurochem. 1992, 58, 786–789. [Google Scholar] [CrossRef] [PubMed]
- Esposito, L.A.; Melov, S.; Panov, A.; Cottrell, B.A.; Wallace, D.C. Mitochondrial disease in mouse results in increased oxidative stress. Proc. Nat. Acad. Sci. USA 1999, 96, 4820–4825. [Google Scholar] [CrossRef]
- Liang, F.Q.; Godley, B.F. Oxidative stress-induced mitochondrial DNA damage in human retinal pigment epithelial cells: A possible mechanism for RPE aging and age-related macular degeneration. Exp. Eye Res. 2003, 76, 397–403. [Google Scholar] [CrossRef]
- Linsdell, P.; Hanrahan, J.W. Glutathione permeability of CFTR. Am. J. Physiol. 1998, 275, C323–C326. [Google Scholar] [CrossRef]
- Gao, L.; Kim, K.J.; Yankaskas, J.R.; Forman, H.J. Abnormal glutathione transport in cystic fibrosis airway epithelia. Am. J. Physiol. Lung Cell. Mol. Physiol. 1999, 277, L113–L118. [Google Scholar] [CrossRef]
- Kelly-Aubert, M.; Trudel, S.; Fritsch, J.; Nguyen-Khoa, T.; Baudouin-Legros, M.; Moriceau, S.; Jeanson, L.; Djouadi, F.; Matar, C.; Conti, M.; et al. GSH monoethyl ester rescues mitochondrial defects in cystic fibrosis models. Hum. Mol. Genet. 2011, 20, 2745–2759. [Google Scholar] [CrossRef] [PubMed]
- Passarelli, C.; Tozzi, G.; Pastore, A.; Bertini, E.; Piemonte, F. GSSG-mediated complex I defect in isolated cardiac mitochondria. Int. J. Mol. Med. 2010, 26, 95–99. [Google Scholar] [PubMed]
- Cantin, A.M. Potential for antioxidant therapy of cystic fibrosis. Curr. Opin. Pulm. Med. 2004, 10, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.F.; Nilsson, M.; Sims, N.R. Glutathione monoethylester prevents mitochondrial glutathione depletion during focal cerebral ischemia. Neurochem. Int. 2004, 44, 153–159. [Google Scholar] [CrossRef]
- Anderson, M.E.; Powrie, F.; Puri, R.N.; Meister, A. Glutathione monoethyl ester: Preparation, uptake by tissues, and conversion to glutathione. Arch. Biochem. Biophys. 1985, 239, 538–548. [Google Scholar] [CrossRef]
- Kang, J.; Pervaiz, S. Mitochondria: Redox metabolism and dysfunction. Biochem. Res. Int. 2012, 2012, 896751. [Google Scholar] [CrossRef]
- Dunn, J.D.; Alvarez, L.A.J.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485. [Google Scholar] [CrossRef]
- Willems, P.H.; Rossignol, R.; Dieteren, C.E.; Murphy, M.P.; Koopman, W.J. Redox homeostasis and mitochondrial dynamics. Cell Metab. 2015, 22, 207–218. [Google Scholar] [CrossRef]
- Hudson, V.M. New insights into the pathogenesis of cystic fibrosis: Pivotal role of glutathione system dysfunction and implications for therapy. Treat. Respir. Med. 2004, 3, 353–363. [Google Scholar] [CrossRef]
- van der Vliet, A. NADPH oxidases in lung biology and pathology: Host defense enzymes, and more. Free Radic. Biol. Med. 2008, 44, 938–955. [Google Scholar] [CrossRef]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Blacker, T.S.; Duchen, M.R. Investigating mitochondrial redox state using NADH and NADPH autofluorescence. Free Radic. Biol. Med. 2016, 100, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Wetmore, D.R.; Joseloff, E.; Pilewski, J.; Lee, D.P.; Lawton, K.A.; Mitchell, M.W.; Milburn, M.V.; Ryals, J.A.; Guo, L. Metabolomic profiling reveals biochemical pathways and biomarkers associated with pathogenesis in cystic fibrosis cells. J. Biol. Chem. 2010, 285, 30516–30522. [Google Scholar] [CrossRef] [PubMed]
- Hudson, V.M. Rethinking cystic fibrosis pathology: The critical role of abnormal reduced glutathione (GSH) transport caused by CFTR mutation. Free Radic. Biol. Med. 2001, 30, 1440–1461. [Google Scholar] [CrossRef]
- Włodek, P.; Sokołowska, M.; Smoleński, O.; Włodek, L. The γ-glutamyltransferase activity and non-protein sulfhydryl compounds levels in rat kidney of different age groups. Acta Biochim. Pol. 2002, 49, 501–507. [Google Scholar] [PubMed]
- Corti, A.; Franzini, M.; Paolicchi, A.; Pompella, A. Gamma-glutamyltransferase of cancer cells at the crossroads of tumor progression, drug resistance and drug targeting. Anticancer Res. 2010, 30, 1169–1181. [Google Scholar]
- Ma, T.; Thiagarajah, J.R.; Yang, H.; Sonawane, N.D.; Folli, C.; Galietta, L.J.V.; Verkman, A.S. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J. Clin. Investig. 2002, 110, 1651–1658. [Google Scholar] [CrossRef]
- Pezzulo, A.A.; Gutiérrez, J.; Duschner, K.S.; McConnell, K.S.; Taft, P.J.; Ernst, S.E.; Yahr, T.L.; Rahmouni, K.; Klesney-Tait, J.; Stoltz, D.A.; et al. Glucose depletion in the airway surface liquid is essential for sterility of the airways. PLoS ONE 2011, 6, e16166. [Google Scholar] [CrossRef]
- Tabary, O.; Corvol, H.; Boncoeur, E.; Chadelat, K.; Fitting, C.; Cavaillon, J.M.; Clément, A.; Jacquot, J. Adherence of airway neutrophils and infiammatory response are increased in CF airway epithelial cell neutrophil interactions. Am. J. Phys. Lung Cell. Mol. Phys. 2006, 290, L588–L596. [Google Scholar]
- Favia, M.; de Bari, L.; Lassandro, R.; Atlante, A. Modulation of glucose-related metabolic pathways controls glucose level in airway surface liquid and fight oxidative stress in cystic fibrosis cells. J. Bioenerg. Biomembr. 2019, 51, 203–218. [Google Scholar] [CrossRef]
- Bardon, A.; Ceder, O.; Kollberg, H. Increased activity of four glycolytic enzymes in cultured fibroblasts from cystic fibrosis patients. Res. Commun. Chem. Pathol. Pharmacol. 1986, 51, 405–408. [Google Scholar] [PubMed]
- Atlante, A.; de Bari, L.; Bobba, A.; Amadoro, G. A disease with a sweet tooth: Exploring the Warburg effect in Alzheimer’s disease. Biogerontology 2017, 18, 301–319. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Favia, M.; de Bari, L.; Bobba, A.; Atlante, A. An Intriguing Involvement of Mitochondria in Cystic Fibrosis. J. Clin. Med. 2019, 8, 1890. https://doi.org/10.3390/jcm8111890
Favia M, de Bari L, Bobba A, Atlante A. An Intriguing Involvement of Mitochondria in Cystic Fibrosis. Journal of Clinical Medicine. 2019; 8(11):1890. https://doi.org/10.3390/jcm8111890
Chicago/Turabian StyleFavia, Maria, Lidia de Bari, Antonella Bobba, and Anna Atlante. 2019. "An Intriguing Involvement of Mitochondria in Cystic Fibrosis" Journal of Clinical Medicine 8, no. 11: 1890. https://doi.org/10.3390/jcm8111890
APA StyleFavia, M., de Bari, L., Bobba, A., & Atlante, A. (2019). An Intriguing Involvement of Mitochondria in Cystic Fibrosis. Journal of Clinical Medicine, 8(11), 1890. https://doi.org/10.3390/jcm8111890