Chronic Stress, Inflammation, and Colon Cancer: A CRH System-Driven Molecular Crosstalk



{kind=link}
{kind=link}
Abstract
:1. Introduction
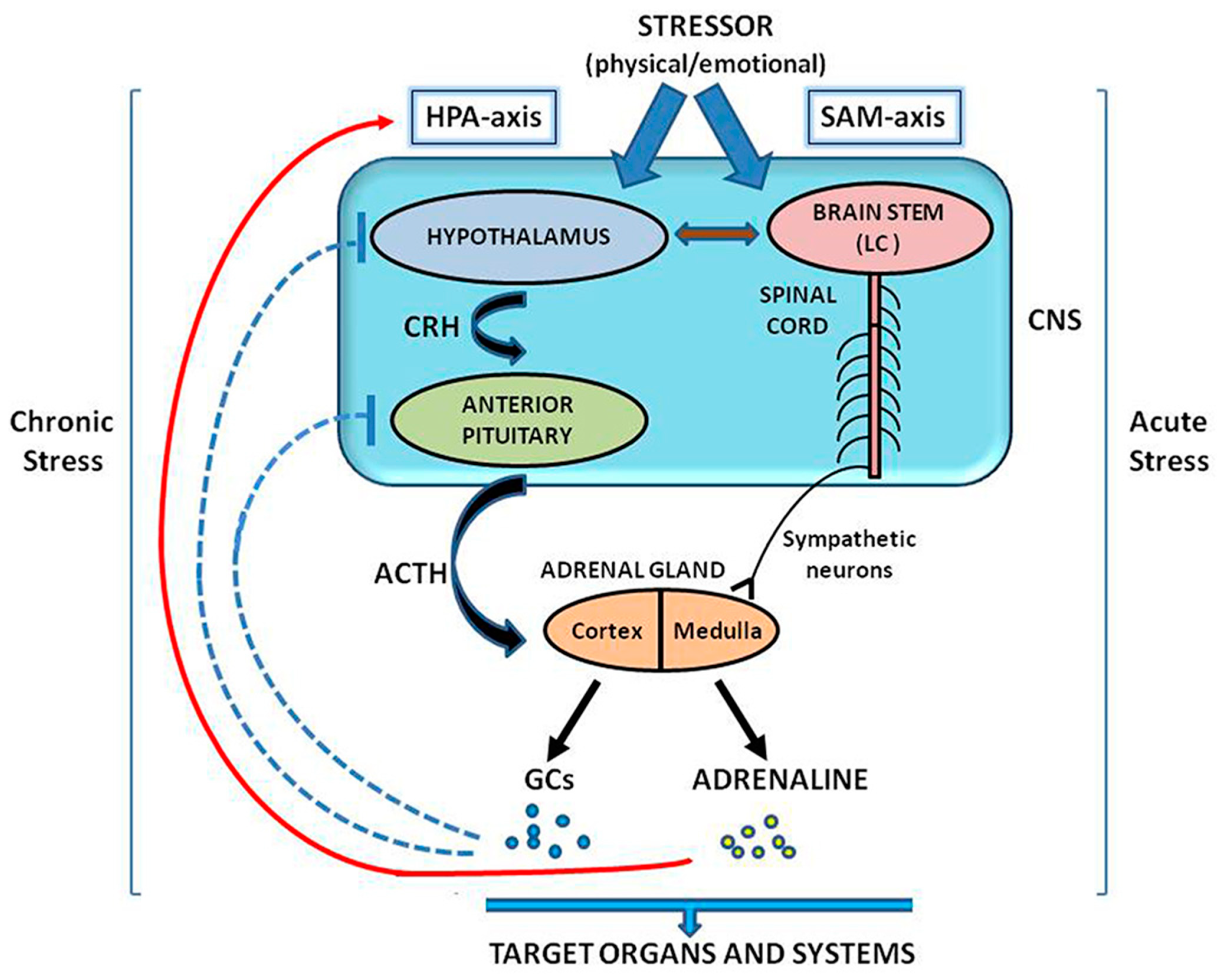
2. The CRH System in the Regulation of Stress Response
3. Intestine: A Stress Target
3.1. Stress and Intestinal Inflammation. Impact of the CRH–Immune System Crosstalk
3.2. Inflammatory Signals and CRC Initiation and Progression: The Role of the Immune Microenvironment
3.3. Distribution of Peripheral CRH Family Members in Normal and Inflamed Intestine
4. Peripheral CRH-Driven Mediators of Intestinal Inflammation
4.1. Immunity
4.1.1. Adaptive Immunity
B and T cells
4.1.2. Innate Immunity
Dendritic Cells
Macrophages
Intestinal Epithelium
Mast Cells
4.2. Fibroblasts and Endothelial Cells
4.3. Enteric Neurons
4.4. Gut Microbiota
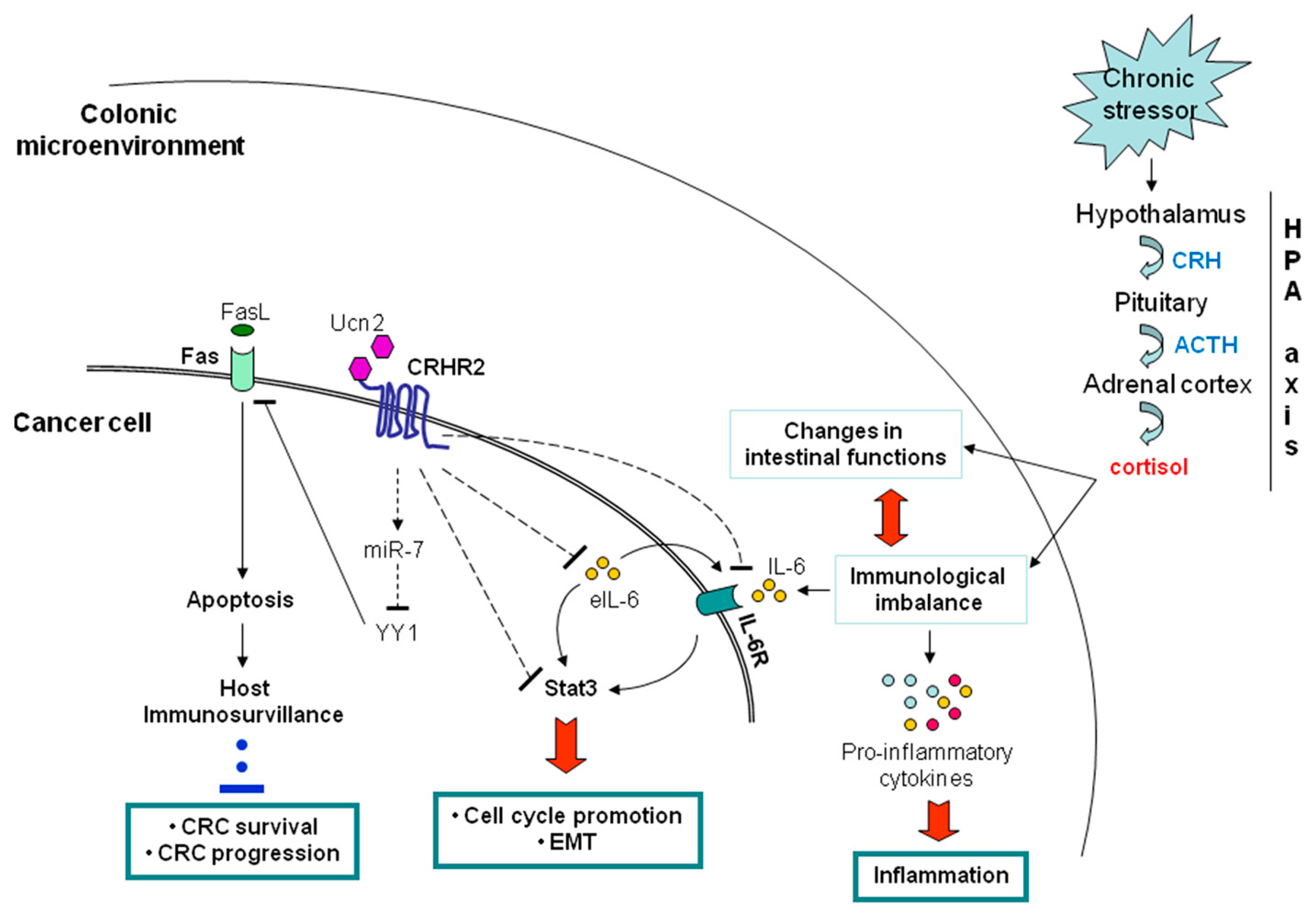
5. Inflammation and CRC Crosstalk via the CRH System
5.1. Peripheral CRH System and CRC
5.1.1. Negative Contribution of the Peripheral CRHR2/Ucn2 Signaling in CRC Progression and Metastatic Potential
5.1.2. Contribution of CRHR2/Ucn2 Signaling in CRC Immune Surveillance
6. Conclusions––What Is Next?
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Zänkert, S.; Bellingrath, S.; Wüst, S.; Kudielka, B.M. HPA axis responses to psychological challenge linking stress and disease: What do we know on sources of intra- and interindividual variability? Psychoneuroendocrinology 2019, 105, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Afrisham, R.; Paknejad, M.; Soliemanifar, O.; Sadegh-Nejadi, S.; Meshkani, R.; Ashtary-Larky, D. The Influence of Psychological Stress on the Initiation and Progression of Diabetes and Cancer. Int. J. Endocrinol. Metab. 2019, 17, e67400. [Google Scholar] [CrossRef] [Green Version]
- Sharif, K.; Watad, A.; Coplan, L.; Lichtbroun, B.; Krosser, A.; Lichtbroun, M.; Bragazzi, N.L.; Amital, H.; Afek, A.; Shoenfeld, Y. The role of stress in the mosaic of autoimmunity: An overlooked association. Autoimmun. Rev. 2018, 17, 967–983. [Google Scholar] [CrossRef]
- Myers, B.; McKlveen, J.M.; Herman, J.P. Neural Regulation of the Stress Response: The Many Faces of Feedback. Cell. Mol. Neurobiol. 2012, 32, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.M.; Vale, W.W. The role of the hypothalamic–pituitary–adrenal axis in neuroendocrine responses to stress. Dialogues Clin. Neurosci. 2006, 8, 383–395. [Google Scholar] [PubMed]
- Godoy, L.D.; Rossignoli, M.T.; Delfino-Pereira, P.; Garcia-Cairasco, N.; de Lima Umeoka, E.H. A Comprehensive Overview on Stress Neurobiology: Basic Concepts and Clinical Implications. Front. Behav. Neurosci. 2018, 12, 127. [Google Scholar] [CrossRef] [Green Version]
- Herman, J.P.; McKlveen, J.M.; Ghosal, S.; Kopp, B.; Wulsin, A.; Makinson, R.; Scheimann, J.; Myers, B. Regulation of the Hypothalamic-Pituitary-Adrenocortical Stress Response. Compr. Physiol. 2016, 6, 603–621. [Google Scholar]
- Bale TL, V.W. CRF and CRF receptors: Role in stress responsivity and other behaviors. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 525–557. [Google Scholar] [CrossRef]
- Buckinx, R.; Adriaensen, D.; Van Nassauw, L.; Timmermans, J.-P. Corticotrophin-releasing factor, related peptides, and receptors in the normal and inflamed gastrointestinal tract. Front. Neurosci. 2011, 5, 54. [Google Scholar] [CrossRef] [PubMed]
- FS, D. Enhancing versus Suppressive Effects of Stress on Immune Function: Implications for Immunoprotection versus Immunopathology. Allergy Asthma Clin. Immunol. 2008, 15, 2–11. [Google Scholar]
- Stengel, A.; Taché, Y. Neuroendocrine control of the gut during stress: Corticotropin-releasing factor signaling pathways in the spotlight. Annu. Rev. Physiol. 2009, 71, 219–239. [Google Scholar] [CrossRef] [PubMed]
- Taché, Y.; Kiank, C.; Stengel, A. A role for corticotropin-releasing factor in functional gastrointestinal disorders. Curr. Gastroenterol. Rep. 2009, 11, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Binder, E.B.; Nemeroff, C.B. The CRF system, stress, depression and anxiety-insights from human genetic studies. Mol. Psychiatry 2010, 15, 574–588. [Google Scholar] [CrossRef] [PubMed]
- Taché, Y.; Bonaz, B. Corticotropin-releasing factor receptors and stress-related alterations of gut motor function. J. Clin. Investig. 2007, 117, 33–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, S.M. Stress and the Gastrointestinal Tract IV. Modulation of intestinal inflammation by stress: Basic mechanisms and clinical relevance. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 280, G315–G318. [Google Scholar] [CrossRef] [PubMed]
- Larauche, M.; Kiank, C.; Tache, Y. Corticotropin releasing factor signaling in colon and ileum: Regulation by stress and pathophysiological implications. J. Physiol. Pharmacol. 2009, 60 (Suppl. 7), 33–46. [Google Scholar]
- Chatoo, M.; Li, Y.; Ma, Z.; Coote, J.; Du, J.; Chen, X. Involvement of Corticotropin-Releasing Factor and Receptors in Immune Cells in Irritable Bowel Syndrome. Front. Endocrinol. 2018, 9, 21. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Keller, D.S.; Windsor, A.; Cohen, R.; Chand, M. Colorectal cancer in inflammatory bowel disease: Review of the evidence. Tech. Coloproctol. 2019, 23, 3–13. [Google Scholar] [CrossRef]
- Zisman, T.L.; Rubin, D.T. Colorectal cancer and dysplasia in inflammatory bowel disease. World J. Gastroenterol. 2008, 14, 2662–2669. [Google Scholar] [CrossRef]
- Mattar, M.C.; Lough, D.; Pishvaian, M.J.; Charabaty, A. Current management of inflammatory bowel disease and colorectal cancer. Gastrointest. Cancer Res. 2011, 4, 53–61. [Google Scholar] [PubMed]
- Vale, W.; Spiess, J.; Rivier, C.; Rivier, J. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science 1981, 213, 1394–1397. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, G. Regulation of the hypothalamic–pituitary–adrenal axis by neuropeptides. Horm. Mol. Biol. Clin. Investig. 2011, 7, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Keller-Wood, M.E.; Dallman, M.F. Corticosteroid inhibition of ACTH secretion. Endocr. Rev. 1984, 5, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Gaffey, A.E.; Bergeman, C.S.; Clark, L.A.; Wirth, M.M. Aging and the HPA axis: Stress and resilience in older adults. Neurosci. Biobehav. Rev. 2016, 68, 928–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segerstrom, S.C.; Miller, G.E. Psychological stress and the human immune system: A meta-analytic study of 30 years of inquiry. Psychol. Bull. 2004, 130, 601–630. [Google Scholar] [CrossRef]
- Chrousos, G.P. Stress and disorders of the stress system. Nat. Rev. Endocrinol. 2009, 5, 374–381. [Google Scholar] [CrossRef]
- Dragoş, D.; Tănăsescu, M.D. The effect of stress on the defense systems. J. Med. Life 2010, 3, 10–18. [Google Scholar]
- Chrousos, G.P.; Gold, P.W. The concepts of stress and stress system disorders. Overview of physical and behavioral homeostasis. JAMA 1992, 267, 1244–1252. [Google Scholar] [CrossRef]
- Jones, B.E.; Yang, T.Z. The efferent projections from the reticular formation and the locus coeruleus studied by anterograde and retrograde axonal transport in the rat. J. Comp. Neurol. 1985, 242, 56–92. [Google Scholar] [CrossRef]
- Lewis, D.I.; Coote, J.H. Excitation and inhibition of rat sympathetic preganglionic neurones by catecholamines. Brain Res. 1990, 530, 229–234. [Google Scholar] [CrossRef]
- Reiche, E.M.V.; Nunes, S.O.V.; Morimoto, H.K. Stress, depression, the immune system, and cancer. Lancet Oncol. 2004, 5, 617–625. [Google Scholar] [CrossRef]
- Tsigos, C.; Kyrou, I.; Kassi, E.; Chrousos, G.P. Stress, Endocrine Physiology and Pathophysiology; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., Dungan, K., Grossman, A., Hershman, J.M., Kaltsas, G., Koch, C., Kopp, P., et al., Eds.; Endotext; MDText, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Isaac, L. Clonidine in the central nervous system: Site and mechanism of hypotensive action. J. Cardiovasc. Pharmacol. 1980, 2 (Suppl. 1), S5–S19. [Google Scholar] [CrossRef]
- Wank, S.A. Cholecystokinin receptors. Am. J. Physiol. 1995, 269, G628–G646. [Google Scholar] [CrossRef] [PubMed]
- McCorry, L.K. Physiology of the autonomic nervous system. Am. J. Pharm. Educ. 2007, 71, 78. [Google Scholar] [CrossRef] [PubMed]
- Ducarouge, B.; Muriel Jacquier, S. Stress neuromediators are key regulators of the intestinal barrier: Link to inflammation and cancer. Trends Cell Mol. Biol. 2011, 6, 59–88. [Google Scholar]
- Carabotti, M.; Scirocco, A.; Maselli, M.A.; Severi, C. The gut–brain axis: Interactions between enteric microbiota, central and enteric nervous systems. Ann. Gastroenterol. 2015, 28, 203–209. [Google Scholar] [PubMed]
- Gross, K.J.; Pothoulakis, C. Role of neuropeptides in inflammatory bowel disease. Inflamm. Bowel Dis. 2007, 13, 918–932. [Google Scholar] [CrossRef]
- Ader, R.; Cohen, N.; Felten, D. Psychoneuroimmunology: Interactions between the nervous system and the immune system. Lancet 1995, 345, 99–103. [Google Scholar] [CrossRef]
- Blalock, J.E. The syntax of immune-neuroendocrine communication. Immunol. Today 1994, 15, 504–511. [Google Scholar] [CrossRef]
- Anstead, M.I.; Hunt, T.A.; Carlson, S.L.; Burki, N.K. Variability of peripheral blood lymphocyte beta-2-adrenergic receptor density in humans. Am. J. Respir. Crit. Care Med. 1998, 157, 990–992. [Google Scholar] [CrossRef] [PubMed]
- Landmann, R. Beta-adrenergic receptors in human leukocyte subpopulations. Eur. J. Clin. Investig. 1992, 22 (Suppl. 1), 30–36. [Google Scholar]
- Maisel, A.S.; Fowler, P.; Rearden, A.; Motulsky, H.J.; Michel, M.C. A new method for isolation of human lymphocyte subsets reveals differential regulation of beta-adrenergic receptors by terbutaline treatment. Clin. Pharmacol. Ther. 1989, 46, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Tsagarakis, S.; Grossman, A. Corticotropin-releasing hormone: Interactions with the immune system. Neuroimmunomodulation 1994, 1, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Fridmanis, D.; Roga, A.; Klovins, J. ACTH Receptor (MC2R) Specificity: What Do We Know About Underlying Molecular Mechanisms? Front. Endocrinol. 2017, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Weik, U.; Herforth, A.; Kolb-Bachofen, V.; Deinzer, R. Acute stress induces proinflammatory signaling at chronic inflammation sites. Psychosom. Med. 2008, 70, 906–912. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Song, C.; Lin, A.; De Jongh, R.; Van Gastel, A.; Kenis, G.; Bosmans, E.; De Meester, I.; Benoy, I.; Neels, H.; et al. The effects of psychological stress on humans: Increased production of pro-inflammatory cytokines and a Th1-like response in stress-induced anxiety. Cytokine 1998, 10, 313–318. [Google Scholar] [CrossRef]
- Brydon, L.; Edwards, S.; Mohamed-Ali, V.; Steptoe, A. Socioeconomic status and stress-induced increases in interleukin-6. Brain Behav. Immun. 2004, 18, 281–290. [Google Scholar] [CrossRef]
- Won, E.; Kim, Y.-K. Stress, the Autonomic Nervous System, and the Immune-kynurenine Pathway in the Etiology of Depression. Curr. Neuropharmacol. 2016, 14, 665–673. [Google Scholar] [CrossRef] [Green Version]
- Stephens, M.A.C.; Wand, G. Stress and the HPA axis: Role of glucocorticoids in alcohol dependence. Alcohol Res. 2012, 34, 468–483. [Google Scholar]
- Bhatia, V.; Tandon, R.K. Stress and the gastrointestinal tract. J. Gastroenterol. Hepatol. 2005, 20, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 2009, 9, 799–809. [Google Scholar] [CrossRef]
- Groschwitz, K.R.; Hogan, S.P. Intestinal barrier function: Molecular regulation and disease pathogenesis. J. Allergy Clin. Immunol. 2009, 124, 3–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukudo, S.; Nomura, T.; Hongo, M. Impact of corticotropin-releasing hormone on gastrointestinal motility and adrenocorticotropic hormone in normal controls and patients with irritable bowel syndrome. Gut 1998, 42, 845–849. [Google Scholar] [CrossRef]
- Grivennikov, S.I. Inflammation and colorectal cancer: Colitis-associated neoplasia. Semin. Immunopathol. 2013, 35, 229–244. [Google Scholar] [CrossRef] [PubMed]
- Paschos, K.A.; Kolios, G.; Chatzaki, E. The corticotropin-releasing factor system in inflammatory bowel disease: Prospects for new therapeutic approaches. Drug Discov. Today 2009, 14, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Mearin, F.; Lacy, B.E.; Chang, L.; Chey, W.D.; Lembo, A.J.; Simren, M.; Spiller, R. Bowel Disorders. Gastroenterology 2016, 150, 1393–1407. [Google Scholar]
- Matricon, J.; Barnich, N.; Ardid, D. Immunopathogenesis of inflammatory bowel disease. Self. Nonself. 2010, 1, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Demaude, J.; Salvador-Cartier, C.; Fioramonti, J.; Ferrier, L.; Bueno, L. Phenotypic changes in colonocytes following acute stress or activation of mast cells in mice: Implications for delayed epithelial barrier dysfunction. Gut 2006, 55, 655–661. [Google Scholar] [CrossRef]
- Radek, K.A. Antimicrobial anxiety: The impact of stress on antimicrobial immunity. J. Leukoc. Biol. 2010, 88, 263–277. [Google Scholar] [CrossRef]
- Costantini, T.W.; Bansal, V.; Peterson, C.Y.; Loomis, W.H.; Putnam, J.G.; Rankin, F.; Wolf, P.; Eliceiri, B.P.; Baird, A.; Coimbra, R. Efferent vagal nerve stimulation attenuates gut barrier injury after burn: Modulation of intestinal occludin expression. J. Trauma 2010, 68, 1349–1354. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.; Saunders, P.R.; Hanssen, N.P.; Yang, P.C.; Yates, D.; Groot, J.A.; Perdue, M.H. Corticotropin-releasing hormone mimics stress-induced colonic epithelial pathophysiology in the rat. Am. J. Physiol. 1999, 277, G391–G399. [Google Scholar] [CrossRef] [PubMed]
- Saunders, P.R.; Hanssen, N.P.; Perdue, M.H. Cholinergic nerves mediate stress-induced intestinal transport abnormalities in Wistar-Kyoto rats. Am. J. Physiol. 1997, 273, G486–G490. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.C.-H.; Wang, J.-T.; Wei, S.-C.; Ni, Y.-H. Host-microbial interactions and regulation of intestinal epithelial barrier function: From physiology to pathology. World J. Gastrointest. Pathophysiol. 2012, 3, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Pastorelli, L.; De Salvo, C.; Mercado, J.R.; Vecchi, M.; Pizarro, T.T. Central role of the gut epithelial barrier in the pathogenesis of chronic intestinal inflammation: Lessons learned from animal models and human genetics. Front. Immunol. 2013, 4, 280. [Google Scholar] [CrossRef] [PubMed]
- Margolis, K.G.; Gershon, M.D. Neuropeptides and inflammatory bowel disease. Curr. Opin. Gastroenterol. 2009, 25, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Mashaghi, A.; Marmalidou, A.; Tehrani, M.; Grace, P.M.; Pothoulakis, C.; Dana, R. Neuropeptide substance P and the immune response. Cell. Mol. Life Sci. 2016, 73, 4249–4264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Meira, L.B.; Bugni, J.M.; Green, S.L.; Lee, C.-W.; Pang, B.; Borenshtein, D.; Rickman, B.H.; Rogers, A.B.; Moroski-Erkul, C.A.; McFaline, J.L.; et al. DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J. Clin. Investig. 2008, 118, 2516–2525. [Google Scholar] [CrossRef] [Green Version]
- Markman, J.L.; Shiao, S.L. Impact of the immune system and immunotherapy in colorectal cancer. J. Gastrointest. Oncol. 2015, 6, 208–223. [Google Scholar]
- Zeng, Z.S.; Huang, Y.; Cohen, A.M.; Guillem, J.G. Prediction of colorectal cancer relapse and survival via tissue RNA levels of matrix metalloproteinase-9. J. Clin. Oncol. 1996, 14, 3133–3140. [Google Scholar] [CrossRef] [PubMed]
- Cianci, R.; Franza, L.; Schinzari, G.; Rossi, E.; Ianiro, G.; Tortora, G.; Gasbarrini, A.; Gambassi, G.; Cammarota, G. The Interplay between Immunity and Microbiota at Intestinal Immunological Niche: The Case of Cancer. Int. J. Mol. Sci. 2019, 20, 501. [Google Scholar] [CrossRef]
- Zou, S.; Fang, L.; Lee, M.-H. Dysbiosis of gut microbiota in promoting the development of colorectal cancer. Gastroenterol. Rep. 2018, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P. Molecular mechanistic pathway of colorectal carcinogenesis associated with intestinal microbiota. Anaerobe 2018, 49, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-C.; Lin, W.-C.; Kong, M.-S.; Shi, H.N.; Walker, W.A.; Lin, C.-Y.; Huang, C.-T.; Lin, Y.-C.; Jung, S.-M.; Lin, T.-Y. Oral inoculation of probiotics Lactobacillus acidophilus NCFM suppresses tumour growth both in segmental orthotopic colon cancer and extra-intestinal tissue. Br. J. Nutr. 2012, 107, 1623–1634. [Google Scholar] [CrossRef] [PubMed]
- Gagnière, J.; Raisch, J.; Veziant, J.; Barnich, N.; Bonnet, R.; Buc, E.; Bringer, M.-A.; Pezet, D.; Bonnet, M. Gut microbiota imbalance and colorectal cancer. World J. Gastroenterol. 2016, 22, 501–518. [Google Scholar] [CrossRef] [PubMed]
- Rothemich, A.; Arthur, J.C. The Azoxymethane/Il10−/− Model of Colitis-Associated Cancer (CAC). Methods Mol. Biol. 2019, 1960, 215–225. [Google Scholar]
- Cani, P.D.; Plovier, H.; Van Hul, M.; Geurts, L.; Delzenne, N.M.; Druart, C.; Everard, A. Endocannabinoids--at the crossroads between the gut microbiota and host metabolism. Nat. Rev. Endocrinol. 2016, 12, 133–143. [Google Scholar] [CrossRef]
- Tsilimigras, M.C.B.; Fodor, A.; Jobin, C. Carcinogenesis and therapeutics: The microbiota perspective. Nat. Microbiol. 2017, 2, 17008. [Google Scholar] [CrossRef]
- Boleij, A.; Hechenbleikner, E.M.; Goodwin, A.C.; Badani, R.; Stein, E.M.; Lazarev, M.G.; Ellis, B.; Carroll, K.C.; Albesiano, E.; Wick, E.C.; et al. The Bacteroides fragilis toxin gene is prevalent in the colon mucosa of colorectal cancer patients. Clin. Infect. Dis. 2015, 60, 208–215. [Google Scholar] [CrossRef]
- Geis, A.L.; Fan, H.; Wu, X.; Wu, S.; Huso, D.L.; Wolfe, J.L.; Sears, C.L.; Pardoll, D.M.; Housseau, F. Regulatory T-cell Response to Enterotoxigenic Bacteroides fragilis Colonization Triggers IL17-Dependent Colon Carcinogenesis. Cancer Discov. 2015, 5, 1098–1109. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.-Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Thiele Orberg, E.; Fan, H.; Tam, A.J.; Dejea, C.M.; Destefano Shields, C.E.; Wu, S.; Chung, L.; Finard, B.B.; Wu, X.; Fathi, P.; et al. The myeloid immune signature of enterotoxigenic Bacteroides fragilis-induced murine colon tumorigenesis. Mucosal Immunol. 2017, 10, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Harusato, A.; Viennois, E.; Etienne-Mesmin, L.; Matsuyama, S.; Abo, H.; Osuka, S.; Lukacs, N.W.; Naito, Y.; Itoh, Y.; Li, J.-D.; et al. Early-Life Microbiota Exposure Restricts Myeloid-Derived Suppressor Cell-Driven Colonic Tumorigenesis. Cancer Immunol. Res. 2019, 7, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Evrard, B.; Coudeyras, S.; Dosgilbert, A.; Charbonnel, N.; Alamé, J.; Tridon, A.; Forestier, C. Dose-dependent immunomodulation of human dendritic cells by the probiotic Lactobacillus rhamnosus Lcr35. PLoS ONE 2011, 6, e18735. [Google Scholar] [CrossRef] [PubMed]
- Kuugbee, E.D.; Shang, X.; Gamallat, Y.; Bamba, D.; Awadasseid, A.; Suliman, M.A.; Zang, S.; Ma, Y.; Chiwala, G.; Xin, Y.; et al. Structural Change in Microbiota by a Probiotic Cocktail Enhances the Gut Barrier and Reduces Cancer via TLR2 Signaling in a Rat Model of Colon Cancer. Dig. Dis. Sci. 2016, 61, 2908–2920. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Li, J.; Liu, Y.; Yue, W.; Luo, X. Toll-like receptor 2 monoclonal antibody or/and Toll-like receptor 4 monoclonal antibody increase counts of Lactobacilli and Bifidobacteria in dextran sulfate sodium-induced colitis in mice. J. Gastroenterol. Hepatol. 2012, 27, 110–119. [Google Scholar] [CrossRef]
- Gamallat, Y.; Meyiah, A.; Kuugbee, E.D.; Hago, A.M.; Chiwala, G.; Awadasseid, A.; Bamba, D.; Zhang, X.; Shang, X.; Luo, F.; et al. Lactobacillus rhamnosus induced epithelial cell apoptosis, ameliorates inflammation and prevents colon cancer development in an animal model. Biomed. Pharmacother. 2016, 83, 536–541. [Google Scholar] [CrossRef]
- Guo, Y.; Bao, C.; Ma, D.; Cao, Y.; Li, Y.; Xie, Z.; Li, S. Network-Based Combinatorial CRISPR-Cas9 Screens Identify Synergistic Modules in Human Cells. ACS Synth. Biol. 2019, 8, 482–490. [Google Scholar] [CrossRef]
- Taché, Y.; Perdue, M.H. Role of peripheral CRF signalling pathways in stress-related alterations of gut motility and mucosal function. Neurogastroenterol. Motil. 2004, 16 (Suppl. 1), 137–142. [Google Scholar] [CrossRef]
- Muramatsu, Y.; Fukushima, K.; Iino, K.; Totsune, K.; Takahashi, K.; Suzuki, T.; Hirasawa, G.; Takeyama, J.; Ito, M.; Nose, M.; et al. Urocortin and corticotropin-releasing factor receptor expression in the human colonic mucosa. Peptides 2000, 21, 1799–1809. [Google Scholar] [CrossRef]
- Chen, A.; Blount, A.; Vaughan, J.; Brar, B.; Vale, W. Urocortin II gene is highly expressed in mouse skin and skeletal muscle tissues: Localization, basal expression in corticotropin-releasing factor receptor (CRFR) 1- and CRFR2-null mice, and regulation by glucocorticoids. Endocrinology 2004, 145, 2445–2457. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Hoy, J.J.; Idumalla, P.S.; Clifton, M.S.; Pecoraro, N.C.; Bhargava, A. Urocortin 2 expression in the rat gastrointestinal tract under basal conditions and in chemical colitis. Peptides 2007, 28, 1453–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sand, E.; Themner-Persson, A.; Ekblad, E. Corticotropin releasing factor-distribution in rat intestine and role in neuroprotection. Regul. Pept. 2011, 166, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Gao, N.; Hu, H.-Z.; Wang, X.; Wang, G.-D.; Fang, X.; Gao, X.; Xia, Y.; Wood, J.D. Distribution and chemical coding of corticotropin-releasing factor-immunoreactive neurons in the guinea pig enteric nervous system. J. Comp. Neurol. 2006, 494, 63–74. [Google Scholar] [CrossRef]
- Kimura, T.; Amano, T.; Uehara, H.; Ariga, H.; Ishida, T.; Torii, A.; Tajiri, H.; Matsueda, K.; Yamato, S. Urocortin I is present in the enteric nervous system and exerts an excitatory effect via cholinergic and serotonergic pathways in the rat colon. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G903–G910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Ren, W.; Qu, M.-H.; Bishop, G.A.; Wang, G.-D.; Wang, X.-Y.; Xia, Y.; Wood, J.D. Differential actions of urocortins on neurons of the myenteric division of the enteric nervous system in guinea pig distal colon. Br. J. Pharmacol. 2010, 159, 222–236. [Google Scholar] [CrossRef]
- Li, B.; Lee, C.; Filler, T.; Hock, A.; Wu, R.Y.; Li, Q.; Chen, S.; Koike, Y.; Ip, W.; Chi, L.; et al. Inhibition of corticotropin-releasing hormone receptor 1 and activation of receptor 2 protect against colonic injury and promote epithelium repair. Sci. Rep. 2017, 7, 46616. [Google Scholar] [CrossRef]
- Kiank, C.; Taché, Y.; Larauche, M. Stress-related modulation of inflammation in experimental models of bowel disease and post-infectious irritable bowel syndrome: Role of corticotropin-releasing factor receptors. Brain Behav. Immun. 2010, 24, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Wlk, M.; Wang, C.C.; Venihaki, M.; Liu, J.; Zhao, D.; Anton, P.M.; Mykoniatis, A.; Pan, A.; Zacks, J.; Karalis, K.; et al. Corticotropin-releasing hormone antagonists possess anti-inflammatory effects in the mouse ileum. Gastroenterology 2002, 123, 505–515. [Google Scholar] [CrossRef]
- Moss, A.C.; Anton, P.; Savidge, T.; Newman, P.; Cheifetz, A.S.; Gay, J.; Paraschos, S.; Winter, M.W.; Moyer, M.P.; Karalis, K.; et al. Urocortin II mediates pro-inflammatory effects in human colonocytes via corticotropin-releasing hormone receptor 2alpha. Gut 2007, 56, 1210–1217. [Google Scholar] [CrossRef] [PubMed]
- van Tol, E.A.; Petrusz, P.; Lund, P.K.; Yamauchi, M.; Sartor, R.B. Local production of corticotropin releasing hormone is increased in experimental intestinal inflammation in rats. Gut 1996, 39, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.-Q.; Wu, S.V.; Wang, L.; Taché, Y. Corticotropin releasing factor in the rat colon: Expression, localization and upregulation by endotoxin. Peptides 2010, 31, 322–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatzaki, E.; Anton, P.A.; Million, M.; Lambropoulou, M.; Constantinidis, T.; Kolios, G.; Taché, Y.; Grigoriadis, D.E. Corticotropin-releasing factor receptor subtype 2 in human colonic mucosa: Down-regulation in ulcerative colitis. World J. Gastroenterol. 2013, 19, 1416–1423. [Google Scholar] [CrossRef] [PubMed]
- Saruta, M.; Takahashi, K.; Suzuki, T.; Torii, A.; Kawakami, M.; Sasano, H. Urocortin 1 in colonic mucosa in patients with ulcerative colitis. J. Clin. Endocrinol. Metab. 2004, 89, 5352–5361. [Google Scholar] [CrossRef] [PubMed]
- Fukudo, S. Role of corticotropin-releasing hormone in irritable bowel syndrome and intestinal inflammation. J. Gastroenterol. 2007, 42 (Suppl. 1), 48–51. [Google Scholar] [CrossRef]
- Im, E.; Rhee, S.H.; Park, Y.S.; Fiocchi, C.; Taché, Y.; Pothoulakis, C. Corticotropin-releasing hormone family of peptides regulates intestinal angiogenesis. Gastroenterology 2010, 138, 2457–2467. [Google Scholar] [CrossRef] [PubMed]
- Kokkotou, E.; Torres, D.; Moss, A.C.; O’Brien, M.; Grigoriadis, D.E.; Karalis, K.; Pothoulakis, C. Corticotropin-releasing hormone receptor 2-deficient mice have reduced intestinal inflammatory responses. J. Immunol. 2006, 177, 3355–3361. [Google Scholar] [CrossRef]
- Reubi, J.C.; Waser, B.; Vale, W.; Rivier, J. Expression of CRF1 and CRF2 receptors in human cancers. J. Clin. Endocrinol. Metab. 2003, 88, 3312–3320. [Google Scholar] [CrossRef]
- Dermitzaki, E.; Tsatsanis, C.; Minas, V.; Chatzaki, E.; Charalampopoulos, I.; Venihaki, M.; Androulidaki, A.; Lambropoulou, M.; Spiess, J.; Michalodimitrakis, E.; et al. Corticotropin-releasing factor (CRF) and the urocortins differentially regulate catecholamine secretion in human and rat adrenals, in a CRF receptor type-specific manner. Endocrinology 2007, 148, 1524–1538. [Google Scholar] [CrossRef]
- Hughes, P.A.; Zola, H.; Penttila, I.A.; Blackshaw, L.A.; Andrews, J.M.; Krumbiegel, D. Immune activation in irritable bowel syndrome: Can neuroimmune interactions explain symptoms? Am. J. Gastroenterol. 2013, 108, 1066–1074. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Li, M.; Lu, B.; Wang, X.; Chen, C.; Zhang, M. Corticotropin-releasing factor augments LPS-induced immune/inflammatory responses in JAWSII cells. Immunol. Res. 2016, 64, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Stengel, A.; Taché, Y. Corticotropin-releasing factor signaling and visceral response to stress. Exp. Biol. Med. 2010, 235, 1168–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, D.Q.; Targan, S.R. Immunopathogenesis of inflammatory bowel disease. World J. Gastroenterol. 2008, 14, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, S.P.; Jenkins, D.; Spiller, R.C. Distinctive clinical, psychological, and histological features of postinfective irritable bowel syndrome. Am. J. Gastroenterol. 2003, 98, 1578–1583. [Google Scholar] [CrossRef]
- Spiller, R.C.; Jenkins, D.; Thornley, J.P.; Hebden, J.M.; Wright, T.; Skinner, M.; Neal, K.R. Increased rectal mucosal enteroendocrine cells, T lymphocytes, and increased gut permeability following acute Campylobacter enteritis and in post-dysenteric irritable bowel syndrome. Gut 2000, 47, 804–811. [Google Scholar] [CrossRef] [PubMed]
- Vermillion, D.L.; Ernst, P.B.; Collins, S.M. T-lymphocyte modulation of intestinal muscle function in the Trichinella-infected rat. Gastroenterology 1991, 101, 31–38. [Google Scholar] [CrossRef]
- Ohman, L.; Lindmark, A.-C.; Isaksson, S.; Posserud, I.; Strid, H.; Sjövall, H.; Simrén, M. B-cell activation in patients with irritable bowel syndrome (IBS). Neurogastroenterol. Motil. 2009, 21, 644–650. [Google Scholar] [CrossRef]
- Forshammar, J.; Isaksson, S.; Strid, H.; Stotzer, P.-O.; Sjövall, H.; Simrén, M.; Ohman, L. A pilot study of colonic B cell pattern in irritable bowel syndrome. Scand. J. Gastroenterol. 2008, 43, 1461–1466. [Google Scholar] [CrossRef]
- Baker, C.; Richards, L.J.; Dayan, C.M.; Jessop, D.S. Corticotropin-releasing hormone immunoreactivity in human T and B cells and macrophages: Colocalization with arginine vasopressin. J. Neuroendocrinol. 2003, 15, 1070–1074. [Google Scholar] [CrossRef]
- Yuan, P.-Q.; Wu, S.V.; Elliott, J.; Anton, P.A.; Chatzaki, E.; Million, M.; Taché, Y. Expression of corticotropin releasing factor receptor type 1 (CRF1) in the human gastrointestinal tract and upregulation in the colonic mucosa in patients with ulcerative colitis. Peptides 2012, 38, 62–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arseneau, K.O.; Tamagawa, H.; Pizarro, T.T.; Cominelli, F. Innate and adaptive immune responses related to IBD pathogenesis. Curr. Gastroenterol. Rep. 2007, 9, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Frdrichie, M.; Pohin, M.; Powrie, F. Cytokine Networks in the Pathophysiology of Inflammatory Bowel Disease. Immunity 2019, 50, 992–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, V.K.; Leu, S.J. Enhancing effect of corticotropin-releasing neurohormone on the production of interleukin-1 and interleukin-2. Neurosci. Lett. 1990, 120, 151–154. [Google Scholar] [CrossRef]
- Ekman, R.; Servenius, B.; Castro, M.G.; Lowry, P.J.; Cederlund, A.S.; Bergman, O.; Sjögren, H.O. Biosynthesis of corticotropin-releasing hormone in human T-lymphocytes. J. Neuroimmunol. 1993, 44, 7–13. [Google Scholar] [CrossRef]
- Kravchenco, I.V.; Furalev, V.A. Secretion of immunoreactive corticotropin releasing factor and adrenocorticotropic hormone by T- and B-lymphocytes in response to cellular stress factors. Biochem. Biophys. Res. Commun. 1994, 204, 828–834. [Google Scholar] [CrossRef] [PubMed]
- Gravanis, A.; Margioris, A.N. The corticotropin-releasing factor (CRF) family of neuropeptides in inflammation: Potential therapeutic applications. Curr. Med. Chem. 2005, 12, 1503–1512. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Kempuraj, D.; Papadopoulou, N.; Kourelis, T.; Donelan, J.; Manola, A.; Theoharides, T.C. Urocortin induces interleukin-6 release from rat cardiomyocytes through p38 MAP kinase, ERK and NF-kappaB activation. J. Mol. Endocrinol. 2009, 42, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Gay, J.; Kokkotou, E.; O’Brien, M.; Pothoulakis, C.; Karalis, K.P. Corticotropin-releasing hormone deficiency is associated with reduced local inflammation in a mouse model of experimental colitis. Endocrinology 2008, 149, 3403–3409. [Google Scholar] [CrossRef]
- Anton, P.M.; Gay, J.; Mykoniatis, A.; Pan, A.; O’Brien, M.; Brown, D.; Karalis, K.; Pothoulakis, C. Corticotropin-releasing hormone (CRH) requirement in Clostridium difficile toxin A-mediated intestinal inflammation. Proc. Natl. Acad. Sci. USA 2004, 101, 8503–8508. [Google Scholar] [CrossRef]
- Gonzalez-Rey, E.; Chorny, A.; Varela, N.; Robledo, G.; Delgado, M. Urocortin and adrenomedullin prevent lethal endotoxemia by down-regulating the inflammatory response. Am. J. Pathol. 2006, 168, 1921–1930. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rey, E.; Fernandez-Martin, A.; Chorny, A.; Delgado, M. Therapeutic effect of urocortin and adrenomedullin in a murine model of Crohn’s disease. Gut 2006, 55, 824–832. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Lu, Z.; Xiaoteng, W.; Yue, H.; Bin, L.; Lina, M.; Zhe, C. Corticotropin-releasing Factor Changes the Phenotype and Function of Dendritic Cells in Mouse Mesenteric Lymph Nodes. J. Neurogastroenterol. Motil. 2015, 21, 571–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hojo, M.; Ohkusa, T.; Tomeoku, H.; Koido, S.; Asaoka, D.; Nagahara, A.; Watanabe, S. Corticotropin-releasing factor secretion from dendritic cells stimulated by commensal bacteria. World J. Gastroenterol. 2011, 17, 4017–4022. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Ohkusa, T.; Kan, S.; Takakura, K.; Saito, K.; Komita, H.; Ito, Z.; Kobayashi, H.; Takami, S.; Uchiyama, K.; et al. Production of corticotropin-releasing factor and urocortin from human monocyte-derived dendritic cells is stimulated by commensal bacteria in intestine. World J. Gastroenterol. 2014, 20, 14420–14429. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Kwon, Y.S.; Park, C.O.; Oh, S.H.; Lee, J.H.; Wu, W.H.; Chang, N.S.; Lee, M.-G.; Lee, K.H. Corticotropin-releasing factor decreases IL-18 in the monocyte-derived dendritic cell. Exp. Dermatol. 2009, 18, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Z.; Zhang, L.; Wang, X.; Tao, L.; Lv, B. PDIA3 gene induces visceral hypersensitivity in rats with irritable bowel syndrome through the dendritic cell-mediated activation of T cells. PeerJ 2016, 4, e2644. [Google Scholar] [CrossRef] [Green Version]
- Long, Y.; Wang, W.; Wang, H.; Hao, L.; Qian, W.; Hou, X. Characteristics of intestinal lamina propria dendritic cells in a mouse model of postinfectious irritable bowel syndrome. J. Gastroenterol. Hepatol. 2012, 27, 935–944. [Google Scholar] [CrossRef]
- Tsatsanis, C.; Androulidaki, A.; Alissafi, T.; Charalampopoulos, I.; Dermitzaki, E.; Roger, T.; Gravanis, A.; Margioris, A.N. Corticotropin-releasing factor and the urocortins induce the expression of TLR4 in macrophages via activation of the transcription factors PU.1 and AP-1. J. Immunol. 2006, 176, 1869–1877. [Google Scholar] [CrossRef]
- Smith, E.M.; Gregg, M.; Hashemi, F.; Schott, L.; Hughes, T.K. Corticotropin Releasing Factor (CRF) activation of NF-kappaB-directed transcription in leukocytes. Cell. Mol. Neurobiol. 2006, 26, 1021–1036. [Google Scholar] [CrossRef]
- Vanner, S.; Greenwood-Van Meerveld, B.; Mawe, G.; Shea-Donohue, T.; Verdu, E.F.; Wood, J.; Grundy, D. Fundamentals of Neurogastroenterology: Basic Science. Gastroenterology 2016, 150, 1280–1291. [Google Scholar] [CrossRef] [Green Version]
- Muller, P.A.; Koscsó, B.; Rajani, G.M.; Stevanovic, K.; Berres, M.-L.; Hashimoto, D.; Mortha, A.; Leboeuf, M.; Li, X.-M.; Mucida, D.; et al. Crosstalk between muscularis macrophages and enteric neurons regulates gastrointestinal motility. Cell 2014, 158, 300–313. [Google Scholar] [CrossRef] [PubMed]
- Agelaki, S.; Tsatsanis, C.; Gravanis, A.; Margioris, A.N. Corticotropin-releasing hormone augments proinflammatory cytokine production from macrophages in vitro and in lipopolysaccharide-induced endotoxin shock in mice. Infect. Immun. 2002, 70, 6068–6074. [Google Scholar] [CrossRef] [PubMed]
- Black, P.H. Stress and the inflammatory response: A review of neurogenic inflammation. Brain Behav. Immun. 2002, 16, 622–653. [Google Scholar] [CrossRef]
- Tsatsanis, C.; Androulidaki, A.; Dermitzaki, E.; Gravanis, A.; Margioris, A.N. Corticotropin releasing factor receptor 1 (CRF1) and CRF2 agonists exert an anti-inflammatory effect during the early phase of inflammation suppressing LPS-induced TNF-alpha release from macrophages via induction of COX-2 and PGE2. J. Cell. Physiol. 2007, 210, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Koshida, H.; Kotake, Y. Corticotropin-releasing hormone enhances the superoxide anion production of rabbit peritoneal macrophages stimulated with N-formyl-methionyl-leucyl-phenylalanine. Life Sci. 1994, 54, 539–543. [Google Scholar] [CrossRef]
- Tsatsanis, C.; Androulidaki, A.; Dermitzaki, E.; Charalampopoulos, I.; Spiess, J.; Gravanis, A.; Margioris, A.N. Urocortin 1 and Urocortin 2 induce macrophage apoptosis via CRFR2. FEBS Lett. 2005, 579, 4259–4264. [Google Scholar] [CrossRef]
- Chaniotou, Z.; Giannogonas, P.; Theoharis, S.; Teli, T.; Gay, J.; Savidge, T.; Koutmani, Y.; Brugni, J.; Kokkotou, E.; Pothoulakis, C.; et al. Corticotropin-releasing factor regulates TLR4 expression in the colon and protects mice from colitis. Gastroenterology 2010, 139, 2083–2092. [Google Scholar] [CrossRef]
- Patsos, G.; Corfield, A. Management of the human mucosal defensive barrier: Evidence for glycan legislation. Biol. Chem. 2009, 390, 581–590. [Google Scholar] [CrossRef]
- Huttner, K.M.; Bevins, C.L. Antimicrobial peptides as mediators of epithelial host defense. Pediatr. Res. 1999, 45, 785–794. [Google Scholar] [CrossRef]
- Kawahito, Y.; Sano, H.; Mukai, S.; Asai, K.; Kimura, S.; Yamamura, Y.; Kato, H.; Chrousos, G.P.; Wilder, R.L.; Kondo, M. Corticotropin releasing hormone in colonic mucosa in patients with ulcerative colitis. Gut 1995, 37, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Chatzaki, E.; Crowe, P.D.; Wang, L.; Million, M.; Taché, Y.; Grigoriadis, D.E. CRF receptor type 1 and 2 expression and anatomical distribution in the rat colon. J. Neurochem. 2004, 90, 309–316. [Google Scholar] [CrossRef] [PubMed]
- la Fleur, S.E.; Wick, E.C.; Idumalla, P.S.; Grady, E.F.; Bhargava, A. Role of peripheral corticotropin-releasing factor and urocortin II in intestinal inflammation and motility in terminal ileum. Proc. Natl. Acad. Sci. USA 2005, 102, 7647–7652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castagliuolo, I.; Lamont, J.T.; Qiu, B.; Fleming, S.M.; Bhaskar, K.R.; Nikulasson, S.T.; Kornetsky, C.; Pothoulakis, C. Acute stress causes mucin release from rat colon: Role of corticotropin releasing factor and mast cells. Am. J. Physiol. 1996, 271, G884–G892. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, C.J.; Qiu, B.; Lam, S.K. Reduction of colonic mucus by repeated short-term stress enhances experimental colitis in rats. J. Physiol. Paris 2001, 95, 81–87. [Google Scholar] [CrossRef]
- Söderholm, J.D.; Yang, P.-C.; Ceponis, P.; Vohra, A.; Riddell, R.; Sherman, P.M.; Perdue, M.H. Chronic stress induces mast cell-dependent bacterial adherence and initiates mucosal inflammation in rat intestine. Gastroenterology 2002, 123, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H. Intestinal permeability regulation by tight junction: Implication on inflammatory bowel diseases. Intest. Res. 2015, 13, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Yue, H.; Bin, L.; Chaoying, C.; Meng, Z.; Meng, L.; Xi, W. Potential Regulatory Effects of Corticotropin-Releasing Factor on Tight Junction-Related Intestinal Epithelial Permeability are Partially Mediated by CK8 Upregulation. Cell. Physiol. Biochem. 2017, 44, 1161–1173. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Song, J.; Bai, T.; Qian, W.; Hou, X.-H. Stress induces more serious barrier dysfunction in follicle-associated epithelium than villus epithelium involving mast cells and protease-activated receptor-2. Sci. Rep. 2017, 7, 4950. [Google Scholar] [CrossRef]
- Yu, Y.; Liu, Z.-Q.; Liu, X.-Y.; Yang, L.; Geng, X.-R.; Yang, G.; Liu, Z.-G.; Zheng, P.-Y.; Yang, P.-C. Stress-Derived Corticotropin Releasing Factor Breaches Epithelial Endotoxin Tolerance. PLoS ONE 2013, 8, e65760. [Google Scholar] [CrossRef]
- Jizhong, S.; Qiaomin, W.; Chao, W.; Yanqing, L. Corticotropin-Releasing Factor and Toll-Like Receptor Gene Expression Is Associated with Low-Grade Inflammation in Irritable Bowel Syndrome Patients with Depression. Gastroenterol. Res. Pract. 2016, 2016, 7394924. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.; Yates, D.; Guilarte, M.; Vicario, M.; Alonso, C.; Perdue, M.H. Stress neuropeptides evoke epithelial responses via mast cell activation in the rat colon. Psychoneuroendocrinology 2008, 33, 1248–1256. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, J.M.; Baritaki, S.; Ruiz, J.J.; Sideri, A.; Pothoulakis, C. Corticotropin-Releasing Hormone Receptor 2 Signaling Promotes Mucosal Repair Responses after Colitis. Am. J. Pathol. 2016, 186, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Schoultz, I.; Keita, Å. Cellular and Molecular Therapeutic Targets in Inflammatory Bowel Disease—Focusing on Intestinal Barrier Function. Cells 2019, 8, 193. [Google Scholar] [CrossRef] [PubMed]
- Barreau, F.; Cartier, C.; Leveque, M.; Ferrier, L.; Moriez, R.; Laroute, V.; Rosztoczy, A.; Fioramonti, J.; Bueno, L. Pathways involved in gut mucosal barrier dysfunction induced in adult rats by maternal deprivation: Corticotrophin-releasing factor and nerve growth factor interplay. J. Physiol. 2007, 580, 347–356. [Google Scholar] [CrossRef]
- Cao, J.; Papadopoulou, N.; Kempuraj, D.; Boucher, W.S.; Sugimoto, K.; Cetrulo, C.L.; Theoharides, T.C. Human mast cells express corticotropin-releasing hormone (CRH) receptors and CRH leads to selective secretion of vascular endothelial growth factor. J. Immunol. 2005, 174, 7665–7675. [Google Scholar] [CrossRef]
- Ohman, L.; Isaksson, S.; Lundgren, A.; Simrén, M.; Sjövall, H. A controlled study of colonic immune activity and beta7+ blood T lymphocytes in patients with irritable bowel syndrome. Clin. Gastroenterol. Hepatol. 2005, 3, 980–986. [Google Scholar] [CrossRef]
- Gebhardt, T.; Lorentz, A.; Detmer, F.; Trautwein, C.; Bektas, H.; Manns, M.P.; Bischoff, S.C. Growth, phenotype, and function of human intestinal mast cells are tightly regulated by transforming growth factor beta1. Gut 2005, 54, 928–934. [Google Scholar] [CrossRef]
- Wallon, C.; Yang, P.-C.; Keita, A.V.; Ericson, A.-C.; McKay, D.M.; Sherman, P.M.; Perdue, M.H.; Söderholm, J.D. Corticotropin-releasing hormone (CRH) regulates macromolecular permeability via mast cells in normal human colonic biopsies in vitro. Gut 2008, 57, 50–58. [Google Scholar] [CrossRef]
- Saunders, P.R.; Santos, J.; Hanssen, N.P.M.; Yates, D.; Groot, J.A.; Perdue, M.H. Physical and psychological stress in rats enhances colonic epithelial permeability via peripheral CRH. Dig. Dis. Sci. 2002, 47, 208–215. [Google Scholar] [CrossRef]
- Overman, E.L.; Rivier, J.E.; Moeser, A.J. CRF induces intestinal epithelial barrier injury via the release of mast cell proteases and TNF-α. PLoS ONE 2012, 7, e39935. [Google Scholar] [CrossRef]
- Wilcz-Villega, E.M.; McClean, S.; O’Sullivan, M.A. Mast cell tryptase reduces junctional adhesion molecule-A (JAM-A) expression in intestinal epithelial cells: Implications for the mechanisms of barrier dysfunction in irritable bowel syndrome. Am. J. Gastroenterol. 2013, 108, 1140–1151. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.-Y.; Feng, B.-S.; Oluwole, C.; Struiksma, S.; Chen, X.; Li, P.; Tang, S.-G.; Yang, P.-C. Psychological stress induces eosinophils to produce corticotrophin releasing hormone in the intestine. Gut 2009, 58, 1473–1479. [Google Scholar] [CrossRef] [PubMed]
- Vanuytsel, T.; van Wanrooy, S.; Vanheel, H.; Vanormelingen, C.; Verschueren, S.; Houben, E.; Salim Rasoel, S.; Tόth, J.; Holvoet, L.; Farré, R.; et al. Psychological stress and corticotropin-releasing hormone increase intestinal permeability in humans by a mast cell-dependent mechanism. Gut 2014, 63, 1293–1299. [Google Scholar] [CrossRef] [PubMed]
- Cameron, H.L.; Perdue, M.H. Stress impairs murine intestinal barrier function: Improvement by glucagon-like peptide-2. J. Pharmacol. Exp. Ther. 2005, 314, 214–220. [Google Scholar] [CrossRef]
- Söderholm, J.D.; Perdue, M.H. Stress and gastrointestinal tract. II. Stress and intestinal barrier function. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 280, G7–G13. [Google Scholar] [CrossRef] [PubMed]
- Bale, T.L.; Giordano, F.J.; Hickey, R.P.; Huang, Y.; Nath, A.K.; Peterson, K.L.; Vale, W.W.; Lee, K.-F. Corticotropin-releasing factor receptor 2 is a tonic suppressor of vascularization. Proc. Natl. Acad. Sci. USA 2002, 99, 7734–7739. [Google Scholar] [CrossRef] [Green Version]
- Karalis, K.; Sano, H.; Redwine, J.; Listwak, S.; Wilder, R.L.; Chrousos, G.P. Autocrine or paracrine inflammatory actions of corticotropin-releasing hormone in vivo. Science 1991, 254, 421–423. [Google Scholar] [CrossRef]
- Simoncini, T.; Apa, R.; Reis, F.M.; Miceli, F.; Stomati, M.; Driul, L.; Lanzone, A.; Genazzani, A.R.; Petraglia, F. Human umbilical vein endothelial cells: A new source and potential target for corticotropin-releasing factor. J. Clin. Endocrinol. Metab. 1999, 84, 2802–2806. [Google Scholar] [CrossRef]
- Cantarella, G.; Lempereur, L.; Lombardo, G.; Chiarenza, A.; Pafumi, C.; Zappalà, G.; Bernardini, R. Divergent effects of corticotropin releasing hormone on endothelial cell nitric oxide synthase are associated with different expression of CRH type 1 and 2 receptors. Br. J. Pharmacol. 2001, 134, 837–844. [Google Scholar] [CrossRef] [Green Version]
- Porcher, C.; Juhem, A.; Peinnequin, A.; Sinniger, V.; Bonaz, B. Expression and effects of metabotropic CRF1 and CRF2 receptors in rat small intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G1091–G1103. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Cao, Q.; Cheng, Y.; Zhao, D.; Wang, Z.; Yang, H.; Wu, Q.; You, L.; Wang, Y.; Lin, Y.; et al. Chronic stress promotes colitis by disturbing the gut microbiota and triggering immune system response. Proc. Natl. Acad. Sci. USA 2018, 115, E2960–E2969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Zhang, M.; Chen, C.-C.; Gillilland, M.; Sun, X.; El-Zaatari, M.; Huffnagle, G.B.; Young, V.B.; Zhang, J.; Hong, S.-C.; et al. Stress-induced corticotropin-releasing hormone-mediated NLRP6 inflammasome inhibition and transmissible enteritis in mice. Gastroenterology 2013, 144, 1478–1487. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-M.; El-Zaatari, M.; Kao, J.Y. Does stress induce bowel dysfunction? Expert Rev. Gastroenterol. Hepatol. 2014, 8, 583–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Gao, J.; Gillilland, M.; Wu, X.; Song, I.; Kao, J.Y.; Owyang, C. Rifaximin alters intestinal bacteria and prevents stress-induced gut inflammation and visceral hyperalgesia in rats. Gastroenterology 2014, 146, 484–496. [Google Scholar] [CrossRef] [PubMed]
- Ait-Belgnaoui, A.; Han, W.; Lamine, F.; Eutamene, H.; Fioramonti, J.; Bueno, L.; Theodorou, V. Lactobacillus farciminis treatment suppresses stress induced visceral hypersensitivity: A possible action through interaction with epithelial cell cytoskeleton contraction. Gut 2006, 55, 1090–1094. [Google Scholar] [CrossRef] [PubMed]
- Eutamene, H.; Lamine, F.; Chabo, C.; Theodorou, V.; Rochat, F.; Bergonzelli, G.E.; Corthésy-Theulaz, I.; Fioramonti, J.; Bueno, L. Synergy between Lactobacillus paracasei and its bacterial products to counteract stress-induced gut permeability and sensitivity increase in rats. J. Nutr. 2007, 137, 1901–1907. [Google Scholar] [CrossRef]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 2015, 26, 26191. [Google Scholar] [CrossRef]
- Noor, S.O.; Ridgway, K.; Scovell, L.; Kemsley, E.K.; Lund, E.K.; Jamieson, C.; Johnson, I.T.; Narbad, A. Ulcerative colitis and irritable bowel patients exhibit distinct abnormalities of the gut microbiota. BMC Gastroenterol. 2010, 10, 134. [Google Scholar] [CrossRef]
- Moayyedi, P.; Ford, A.C.; Talley, N.J.; Cremonini, F.; Foxx-Orenstein, A.E.; Brandt, L.J.; Quigley, E.M.M. The efficacy of probiotics in the treatment of irritable bowel syndrome: A systematic review. Gut 2010, 59, 325–332. [Google Scholar] [CrossRef]
- Dai, C.; Zheng, C.-Q.; Jiang, M.; Ma, X.-Y.; Jiang, L.-J. Probiotics and irritable bowel syndrome. World J. Gastroenterol. 2013, 19, 5973–5980. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Kamada, K.; Mizushima, K.; Higashimura, Y.; Katada, K.; Uchiyama, K.; Handa, O.; Takagi, T.; Naito, Y.; Itoh, Y. Changes in Intestinal Motility and Gut Microbiota Composition in a Rat Stress Model. Digestion 2017, 95, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.R.; Osadchiy, V.; Kalani, A.; Mayer, E.A. The Brain-Gut-Microbiome Axis. Cell. Mol. Gastroenterol. Hepatol. 2018, 6, 133–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holzer, P.; Farzi, A. Neuropeptides and the microbiota–gut–brain axis. Adv. Exp. Med. Biol. 2014, 817, 195–219. [Google Scholar] [PubMed]
- Ducarouge, B.; Pelissier-Rota, M.; Lainé, M.; Cristina, N.; Vachez, Y.; Scoazec, J.-Y.; Bonaz, B.; Jacquier-Sarlin, M. CRF2 signaling is a novel regulator of cellular adhesion and migration in colorectal cancer cells. PLoS ONE 2013, 8, e79335. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.A.; Huerta-Yepez, S.; Law, I.K.M.; Baay-Guzman, G.J.; Tirado-Rodriguez, B.; Hoffman, J.M.; Iliopoulos, D.; Hommes, D.W.; Verspaget, H.W.; Chang, L.; et al. Diminished expression of CRHR2 in human colon cancer promotes tumor growth and EMT via persistent IL-6/Stat3 signaling. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 610–630. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.M.; Fusenig, N.E. Friends or foes—Bipolar effects of the tumour stroma in cancer. Nat. Rev. Cancer 2004, 4, 839–849. [Google Scholar] [CrossRef]
- Kageyama, K.; Hanada, K.; Nigawara, T.; Moriyama, T.; Terui, K.; Sakihara, S.; Suda, T. Urocortin induces interleukin-6 gene expression via cyclooxygenase-2 activity in aortic smooth muscle cells. Endocrinology 2006, 147, 4454–4462. [Google Scholar] [CrossRef]
- Jarnicki, A.; Putoczki, T.; Ernst, M. Stat3: Linking inflammation to epithelial cancer—More than a “gut” feeling? Cell Div. 2010, 5, 14. [Google Scholar] [CrossRef]
- Klampfer, L. The role of signal transducers and activators of transcription in colon cancer. Front. Biosci. 2008, 13, 2888–2899. [Google Scholar] [CrossRef]
- Waldner, M.J.; Foersch, S.; Neurath, M.F. Interleukin-6--a key regulator of colorectal cancer development. Int. J. Biol. Sci. 2012, 8, 1248–1253. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Hong, Y.; Dai, L.; Qian, Y.; Zhu, C.; Wu, B.; Li, S. CRH promotes human colon cancer cell proliferation via IL-6/JAK2/STAT3 signaling pathway and VEGF-induced tumor angiogenesis. Mol. Carcinog. 2017, 56, 2434–2445. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Fang, X.; Yuan, J.; Sun, Z.; Li, C.; Li, R.; Li, L.; Zhu, C.; Wan, R.; Guo, R.; et al. The role of corticotropin-releasing hormone receptor 1 in the development of colitis-associated cancer in mouse model. Endocr. Relat. Cancer 2014, 21, 639–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelissier-Rota, M.; Chartier, N.T.; Bonaz, B.; Jacquier-Sarlin, M.R. A crosstalk between muscarinic and CRF2 receptors regulates cellular adhesion properties of human colon cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1246–1259. [Google Scholar] [CrossRef] [PubMed]
- Ducarouge, B.; Pelissier-Rota, M.; Powell, R.; Buisson, A.; Bonaz, B.; Jacquier-Sarlin, M. Involvement of CRF2 signaling in enterocyte differentiation. World J. Gastroenterol. 2017, 23, 5127–5145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pothoulakis, C.; Torre-Rojas, M.; Duran-Padilla, M.A.; Gevorkian, J.; Zoras, O.; Chrysos, E.; Chalkiadakis, G.; Baritaki, S. CRHR2/Ucn2 signaling is a novel regulator of miR-7/YY1/Fas circuitry contributing to reversal of colorectal cancer cell resistance to Fas-mediated apoptosis. Int. J. Cancer 2018, 142, 334–346. [Google Scholar] [CrossRef]
- Zhang, N.; Li, X.; Wu, C.W.; Dong, Y.; Cai, M.; Mok, M.T.S.; Wang, H.; Chen, J.; Ng, S.S.M.; Chen, M.; et al. microRNA-7 is a novel inhibitor of YY1 contributing to colorectal tumorigenesis. Oncogene 2013, 32, 5078–5088. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baritaki, S.; de Bree, E.; Chatzaki, E.; Pothoulakis, C. Chronic Stress, Inflammation, and Colon Cancer: A CRH System-Driven Molecular Crosstalk. J. Clin. Med. 2019, 8, 1669. https://doi.org/10.3390/jcm8101669
Baritaki S, de Bree E, Chatzaki E, Pothoulakis C. Chronic Stress, Inflammation, and Colon Cancer: A CRH System-Driven Molecular Crosstalk. Journal of Clinical Medicine. 2019; 8(10):1669. https://doi.org/10.3390/jcm8101669
Chicago/Turabian StyleBaritaki, Stavroula, Eelco de Bree, Ekaterini Chatzaki, and Charalabos Pothoulakis. 2019. "Chronic Stress, Inflammation, and Colon Cancer: A CRH System-Driven Molecular Crosstalk" Journal of Clinical Medicine 8, no. 10: 1669. https://doi.org/10.3390/jcm8101669
APA StyleBaritaki, S., de Bree, E., Chatzaki, E., & Pothoulakis, C. (2019). Chronic Stress, Inflammation, and Colon Cancer: A CRH System-Driven Molecular Crosstalk. Journal of Clinical Medicine, 8(10), 1669. https://doi.org/10.3390/jcm8101669