Thrombin Induces Angiotensin II-Mediated Senescence in Atrial Endothelial Cells: Impact on Pro-Remodeling Patterns

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Isolation and Culture of Atrial Endothelial Cells
2.3. Determination of Senescence Associated-β-Galactosidase (SA-β-Gal) Activity by Flow Cytometry
2.4. Western Blot Analysis
2.5. Determination of Oxidative Stress
2.6. Determination of MMP Activity by Zymography
2.7. Statistical Analyses
3. Results
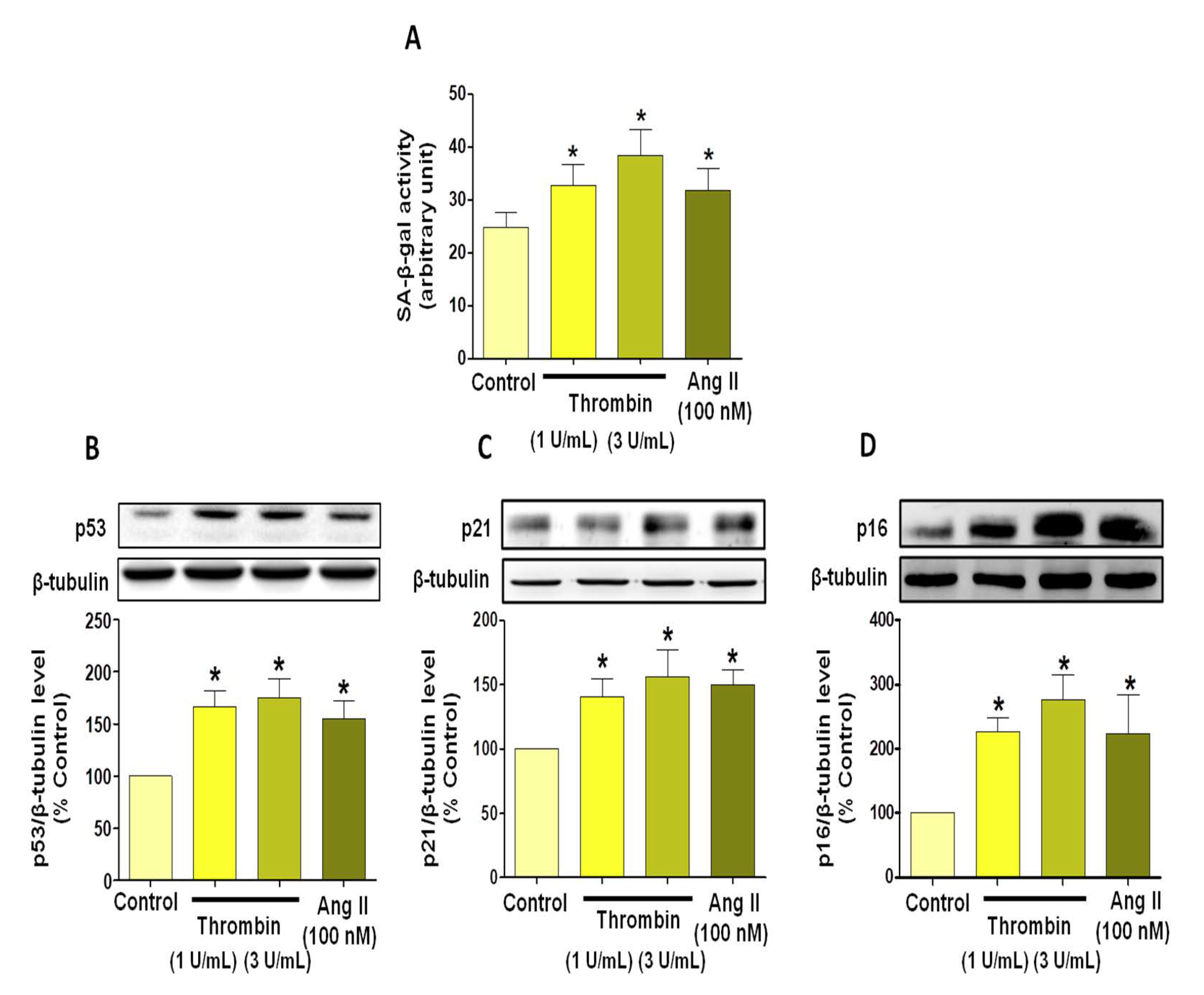
3.1. Thrombin Induces Atrial Endothelial Cells Senescence
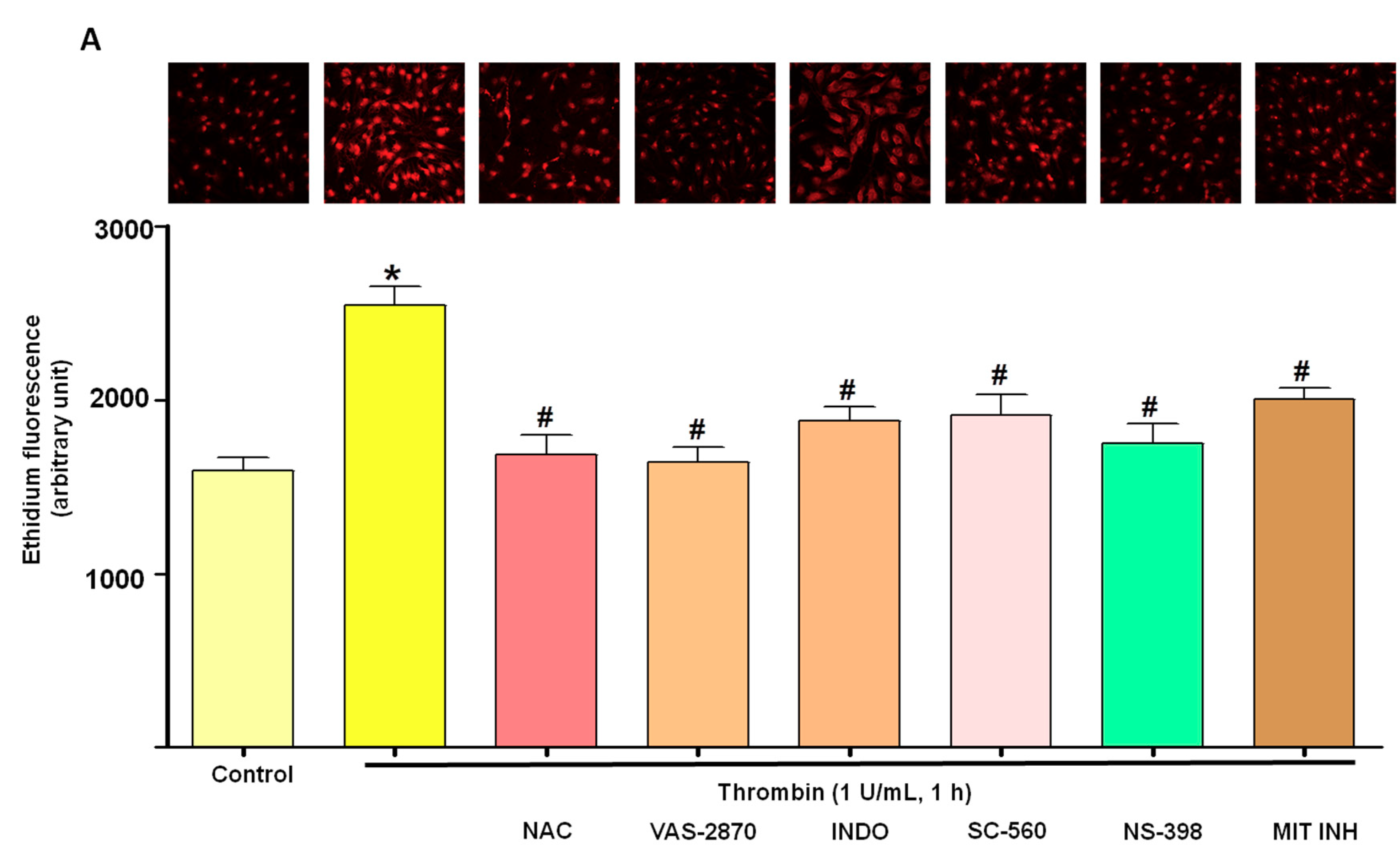
3.2. Thrombin Increases Oxidative Stress within Atrial Endothelial Cells
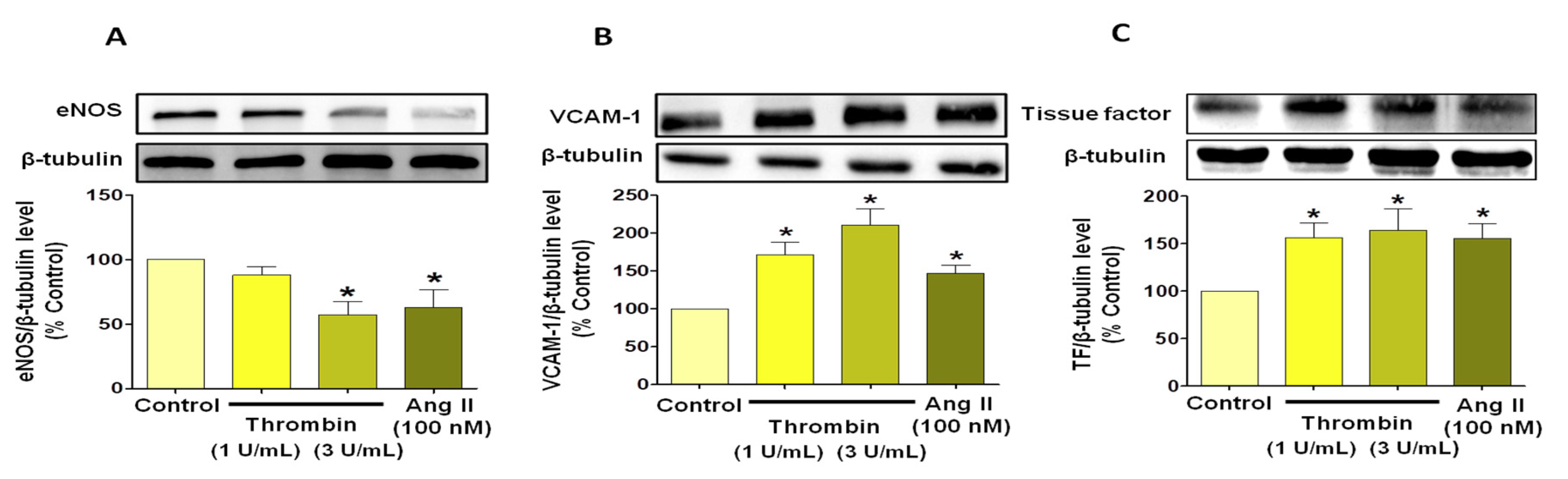
3.3. Thrombin Induces Endothelial Dysfunction Associated with Pro-Thrombotic and Pro-Inflammatory Patterns
3.4. Thrombin Increases Pro-Fibrotic and Pro-Remodeling Responses
3.5. Critical Role of the Local Angiotensin System in Thrombin-Induced Oxidative Stress in Atrial Endothelial Cells (ECs)
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Watson, T.; Shantsila, E.; Lip, G.Y. Mechanisms of thrombogenesis in atrial fibrillation: Virchow’s triad revisited. Lancet 2009, 373, 155–166. [Google Scholar] [CrossRef]
- Jesel, L.; Abbas, M.; Toti, F.; Cohen, A.; Arentz, T.; Morel, O. Microparticles in atrial fibrillation: A link between cell activation or apoptosis, tissue remodelling and thrombogenicity. Int. J. Cardiol. 2013, 168, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Ederhy, S.; Di Angelantonio, E.; Mallat, Z.; Hugel, B.; Janower, S.; Meuleman, C.; Boccara, F.; Freyssinet, J.-M.; Tedgui, A.; Cohen, A. Levels of Circulating Procoagulant Microparticles in Nonvalvular Atrial Fibrillation. Am. J. Cardiol. 2007, 100, 989–994. [Google Scholar] [CrossRef] [PubMed]
- Simak, J.; Gelderman, M.P.; Yu, H.; Wright, V.; Baird, A.E. Circulating endothelial microparticles in acute ischemic stroke: a link to severity, lesion volume and outcome. J. Thromb. Haemost. 2006, 4, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Morel, O.; Florence, T.; Bénédicte, H.; Babé, B.; Laurence, C.J.; Françoise, D.G.; Freyssinet, J.M. Disrupting the vascular homeostasis equation? Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2594–2604. [Google Scholar] [CrossRef] [PubMed]
- Altieri, P.; Bertolotto, M.; Fabbi, P.; Sportelli, E.; Balbi, M.; Santini, F.; Brunelli, C.; Canepa, M.; Montecucco, F.; Ameri, P. Thrombin induces protease-activated receptor 1 signaling and activation of human atrial fibroblasts and dabigatran prevents these effects. Int. J. Cardiol. 2018, 271, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Jumeau, C.; Rupin, A.; Chieng-Yane, P.; Mougenot, N.; Zahr, N.; David-Dufilho, M.; Hatem, S.N. Direct Thrombin Inhibitors Prevent Left Atrial Remodeling Associated with Heart Failure in Rats. JACC: Basic Transl. Sci. 2016, 1, 328–339. [Google Scholar]
- Chang, C.-J.; Chen, Y.-C.; Kao, Y.-H.; Lin, Y.-K.; Chen, S.-A.; Chen, Y.-J. Dabigatran and Thrombin Modulate Electrophysiological Characteristics of Pulmonary Vein and Left Atrium. Circ. Arrhythmia Electrophysiol. 2012, 5, 1176–1183. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Henri, M.H.S.; Anne, M.D.J.; Sander, V.; Hetty, C.D.B.; Alexander, H.M.; Dennis, H.L.; Michiel, R.; Arne, V.H.; Marion, K.; Stijn, L.; et al. Hypercoagulability causes atrial fibrosis and promotes atrial fibrillation. Eur. Heart J. 2017, 38, 38–50. [Google Scholar]
- Phillip, C.D.; Ingrid, J.A.; Shufang, G.; Katy, S.; Yoshiyuki, H.; Linda, M.M.; Peter, C.L. Thrombin-dependent NF-kB activation and monocyte/endothelial adhesion are mediated by the CARMA3.Bcl10. MALT1 signalosome. J. Biol. Chem. 2010, 285, 41432–41442. [Google Scholar]
- Carlquist, J.F.; Knight, S.; Cawthon, R.M.; Le, V.T.; Bunch, T.J.; Horne, B.D.; Rollo, J.S.; Huntinghouse, J.A.; Muhlestein, J.B.; Anderson, J.L. Shortened telomere length is associated with paroxysmal atrial fibrillation among cardiovascular patients enrolled in the Intermountain Heart Collaborative Study. Hear. Rhythm. 2016, 13, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Chen, Y.; Hu, C.; Pan, Q.; Wang, B.; Li, X.; Geng, J.; Xu, B. Premature senescence of cardiac fibroblasts and atrial fibrosis in patients with atrial fibrillation. Oncotarget 2017, 8, 57981–57990. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Kawamoto, S.; Ohtani, N.; Hara, E. Impact of senescence-associated secretory phenotype and its potential as a therapeutic target for senescence-associated diseases. Cancer Sci. 2017, 108, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Pickering, J.G. Cellular Senescence and Vascular Disease: Novel Routes to Better Understanding and Therapy. Can. J. Cardiol. 2016, 32, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Abbas, M.; Jesel, L.; Auger, C.; Amoura, L.; Messas, N.; Manin, G.; Rumig, C.; León-González, A.J.; Ribeiro, T.P.; Silva, G.C.; et al. Endothelial Microparticles from Acute Coronary Syndrome Patients Induce Premature Coronary Artery Endothelial Cell Aging and Thrombogenicity: Role of the Ang II/AT1 Receptor/NADPH Oxidase-Mediated Activation of MAPKs and PI3-Kinase Pathways. Circulation 2017, 135, 280–296. [Google Scholar] [CrossRef] [PubMed]
- Mann, K.G.; Brummel, K.; Butenas, S. What is all that thrombin for? J. Thromb. Haemost. 2003, 1, 1504–1514. [Google Scholar] [CrossRef]
- Cotgreave, I.; Moldeus, P.; Schuppe, I. The metabolism of N-acetylcysteine by human endothelial cells. Biochem. Pharmacol. 1991, 42, 13–16. [Google Scholar] [CrossRef]
- Kurz, D.J.; Decary, S.; Hong, Y.; Erusalimsky, J.D. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J. Cell Sci. 2000, 113 Pt 20, 3613–3622. [Google Scholar]
- Sonia, K.B.; Noureddine, I.K.; Thais, P.R.; Grazielle, C.S.; Malak, A.; Marouane, K.; Jung, L.; Florence, T.; Cyril, A.; Valérie, B.; et al. The Redox-sensitive Induction of the Local Angiotensin System Promotes Both Premature and Replicative Endothelial Senescence: Preventive Effect of a Standardized Crataegus Extract. J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2016, 71, 1581–1590. [Google Scholar]
- Hu, X.; Beeton, C. Detection of functional matrix metalloproteinases by zymography. J. Vis. Exp. 2010. [Google Scholar] [CrossRef]
- Polyakova, V.; Miyagawa, S.; Szalay, Z.; Risteli, J.; Kostin, S. Atrial extracellular matrix remodelling in patients with atrial fibrillation. J. Cell. Mol. Med. 2008, 12, 189–208. [Google Scholar] [CrossRef] [PubMed]
- Anné, W.; Willems, R.; Roskams, T.; Sergeant, P.; Herijgers, P.; Holemans, P.; Ector, H.; Heidbüchel, H. Matrix metalloproteinases and atrial remodeling in patients with mitral valve disease and atrial fibrillation. Cardiovasc. Res. 2005, 67, 655–666. [Google Scholar] [CrossRef]
- Bukowska, A.; Zacharias, I.; Weinert, S.; Skopp, K.; Hartmann, C.; Huth, C.; Goette, A. Coagulation factor Xa induces an inflammatory signalling by activation of protease-activated receptors in human atrial tissue. Eur. J. Pharmacol. 2013, 718, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Park, T.H.; McHowat, J.; A Wolf, R.; Corr, P.B. Increased lysophosphatidylcholine content induced by thrombin receptor stimulation in adult rabbit cardiac ventricular myocytes. Cardiovasc. Res. 1994, 28, 1263–1268. [Google Scholar] [CrossRef] [PubMed]
- Burger, D.; Turner, M.; Munkonda, M.N.; Touyz, R.M. Endothelial Microparticle-Derived Reactive Oxygen Species: Role in Endothelial Signaling and Vascular Function. Oxidative Med. Cell. Longev. 2016, 2016, 1–10. [Google Scholar] [CrossRef]
- Sanada, F.; Taniyama, Y.; Muratsu, J.; Otsu, R.; Iwabayashi, M.; Carracedo, M.; Rakugi, H.; Morishita, R. Activated Factor X Induces Endothelial Cell Senescence Through IGFBP-5. Sci. Rep. 2016, 6, 35580. [Google Scholar] [CrossRef]
- Yang, Z.; Arnet, U.; Bauer, E.; Von Segesser, L.; Siebenmann, R.; Turina, M.; Luscher, T.F. Thrombin-induced endothelium-dependent inhibition and direct activation of platelet-vessel wall interaction. Role of prostacyclin, nitric oxide, and thromboxane A2. Circ. 1994, 89, 2266–2272. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.W.; Choi, S.Y.; Cho, M.K.; Lee, C.H.; Kim, S.G. Thrombin Induces Nitric-oxide Synthase via Galpha 12/13-coupled Protein Kinase C-dependent I-kappa Balpha Phosphorylation and JNK-mediated I-kappa Balpha Degradation. J. Boil. Chem. 2003, 278, 17368–17378. [Google Scholar] [CrossRef] [PubMed]
- Akar, J.G.; Jeske, W.; Wilber, D.J. Acute onset human atrial fibrillation is associated with local cardiac platelet activation and endothelial dysfunction. J. Am. Coll. Cardiol. 2008, 51, 1790–1793. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, F.; Deng, L.; Lin, K.; Shi, X.; Zhaoliang, S.; Wang, Y. Febuxostat attenuates paroxysmal atrial fibrillation-induced regional endothelial dysfunction. Thromb. Res. 2017, 149, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Spronk, H.M.; De Jong, A.M.; Crijns, H.J.; Schotten, U.; Van Gelder, I.C.; Cate, H.T. Pleiotropic effects of factor Xa and thrombin: what to expect from novel anticoagulants. Cardiovasc. Res. 2014, 101, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Jansen, H.J.; Mackasey, M.; Moghtadaei, M.; Belke, D.D.; Egom, E.E.; Tuomi, J.M.; Rafferty, S.A.; Kirkby, A.W.; Rose, R.A. Distinct patterns of atrial electrical and structural remodeling in angiotensin II mediated atrial fibrillation. J. Mol. Cell. Cardiol. 2018, 124, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Harling, L.; Lambert, J.; Ashrafian, H.; Darzi, A.; Gooderham, N.J.; Athanasiou, T. Pre-operative serum VCAM-1 as a biomarker of atrial fibrillation after coronary artery bypass grafting. J. Cardiothorac. Surg. 2017, 12, 70. [Google Scholar] [CrossRef] [PubMed]
- Verdejo, H.; Roldán, J.; Garcia, L.; Del Campo, A.; Becerra, E.; Chiong, M.; Mellado, R.; Garcia, A.; Zalaquett, R.; Braun, S.; et al. Systemic vascular cell adhesion molecule-1 predicts the occurrence of post-operative atrial fibrillation. Int. J. Cardiol. 2011, 150, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Polejaeva, I.A.; Ranjan, R.; Davies, C.J.; Regouski, M.; Hall, J.; Olsen, A.L.; Meng, Q.; Rutigliano, H.M.1; Dosdall, D.J.; Angel, N.A.; et al. Increased Susceptibility to Atrial Fibrillation Secondary to Atrial Fibrosis in Transgenic Goats Expressing Transforming Growth Factor-beta1. J. Cardiovasc. Electrophysiol. 2016, 27, 1220–1229. [Google Scholar] [CrossRef]
- Verheule, S.; Sato, T.; Everett, T.; Engle, S.K.; Otten, D.; Der Lohe, M.R.-V.; Nakajima, H.O.; Nakajima, H.; Field, L.J.; Olgin, J.E. Increased vulnerability to atrial fibrillation in transgenic mice with selective atrial fibrosis caused by overexpression of TGF-beta1. Circ. Res. 2004, 94, 1458–1465. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Low, B.; Rutherford, S.A.; Hao, Q. Thrombin induces endocytosis of endoglin and type-II TGF-beta receptor and down-regulation of TGF-beta signaling in endothelial cells. Blood 2005, 105, 1977–1985. [Google Scholar] [CrossRef] [PubMed]
- Abe, I.; Teshima, Y.; Kondo, H.; Kaku, H.; Kira, S.; Ikebe, Y.; Saito, S.; Fukui, A.; Shinohara, T.; Yufu, K.; et al. Association of fibrotic remodeling and cytokines/chemokines content in epicardial adipose tissue with atrial myocardial fibrosis in patients with atrial fibrillation. Hear. Rhythm. 2018, 15, 1717–1727. [Google Scholar] [CrossRef]
- Kaireviciute, D.; Lip, G.Y.H.; Balakrishnan, B.; Uzdavinys, G.; Norkunas, G.; Kalinauskas, G.; Sirvydis, V.; Aidietis, A.; Zanetto, U.; Sihota, H.; et al. Intracardiac expression of markers of endothelial damage/dysfunction, inflammation, thrombosis, and tissue remodeling, and the development of postoperative atrial fibrillation. J. Thromb. Haemost. 2011, 9, 2345–2352. [Google Scholar] [CrossRef]
- Xu, J.; Cui, G.; Esmailian, F.; Plunkett, M.; Marelli, D.; Ardehali, A.; Odim, J.; Laks, H.; Sen, L. Atrial Extracellular Matrix Remodeling and the Maintenance of Atrial Fibrillation. Circ. 2004, 109, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Sonin, D.L.; Wakatsuki, T.; Routhu, K.V.; Harmann, L.M.; Petersen, M.; Meyer, J.; Strande, J.L. Protease-activated receptor 1 inhibition by SCH79797 attenuates left ventricular remodeling and profibrotic activities of cardiac fibroblasts. J. Cardiovasc. Pharmacol. Ther. 2013, 18, 460–475. [Google Scholar] [CrossRef] [PubMed]
- Watson, T.; Karthikeyan, V.J.; Lip, G.Y.; Beevers, D.G. Atrial fibrillation in primary aldosteronism. J. Renin-Angiotensin-Aldosterone Syst. 2009, 10, 190–194. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasan, H.; Park, S.-H.; Auger, C.; Belcastro, E.; Matsushita, K.; Marchandot, B.; Lee, H.-H.; Qureshi, A.W.; Kauffenstein, G.; Ohlmann, P.; et al. Thrombin Induces Angiotensin II-Mediated Senescence in Atrial Endothelial Cells: Impact on Pro-Remodeling Patterns. J. Clin. Med. 2019, 8, 1570. https://doi.org/10.3390/jcm8101570
Hasan H, Park S-H, Auger C, Belcastro E, Matsushita K, Marchandot B, Lee H-H, Qureshi AW, Kauffenstein G, Ohlmann P, et al. Thrombin Induces Angiotensin II-Mediated Senescence in Atrial Endothelial Cells: Impact on Pro-Remodeling Patterns. Journal of Clinical Medicine. 2019; 8(10):1570. https://doi.org/10.3390/jcm8101570
Chicago/Turabian StyleHasan, Hira, Sin-Hee Park, Cyril Auger, Eugenia Belcastro, Kensuke Matsushita, Benjamin Marchandot, Hyun-Ho Lee, Abdul Wahid Qureshi, Gilles Kauffenstein, Patrick Ohlmann, and et al. 2019. "Thrombin Induces Angiotensin II-Mediated Senescence in Atrial Endothelial Cells: Impact on Pro-Remodeling Patterns" Journal of Clinical Medicine 8, no. 10: 1570. https://doi.org/10.3390/jcm8101570
APA StyleHasan, H., Park, S.-H., Auger, C., Belcastro, E., Matsushita, K., Marchandot, B., Lee, H.-H., Qureshi, A. W., Kauffenstein, G., Ohlmann, P., Schini-Kerth, V. B., Jesel, L., & Morel, O. (2019). Thrombin Induces Angiotensin II-Mediated Senescence in Atrial Endothelial Cells: Impact on Pro-Remodeling Patterns. Journal of Clinical Medicine, 8(10), 1570. https://doi.org/10.3390/jcm8101570