Epigenetic Modifications in Thyroid Cancer Cells Restore NIS and Radio-Iodine Uptake and Promote Cell Death


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Cell Culture
2.3. Sample Collection
2.4. Real-Time Cell Viability Analysis
2.5. RNA Isolation and Quantitative Real Time RT-PCR
2.6. Protein Isolation and Western Blotting
2.7. In Vitro Radioiodine Up-Take
2.8. Statistical Analysis
3. Results
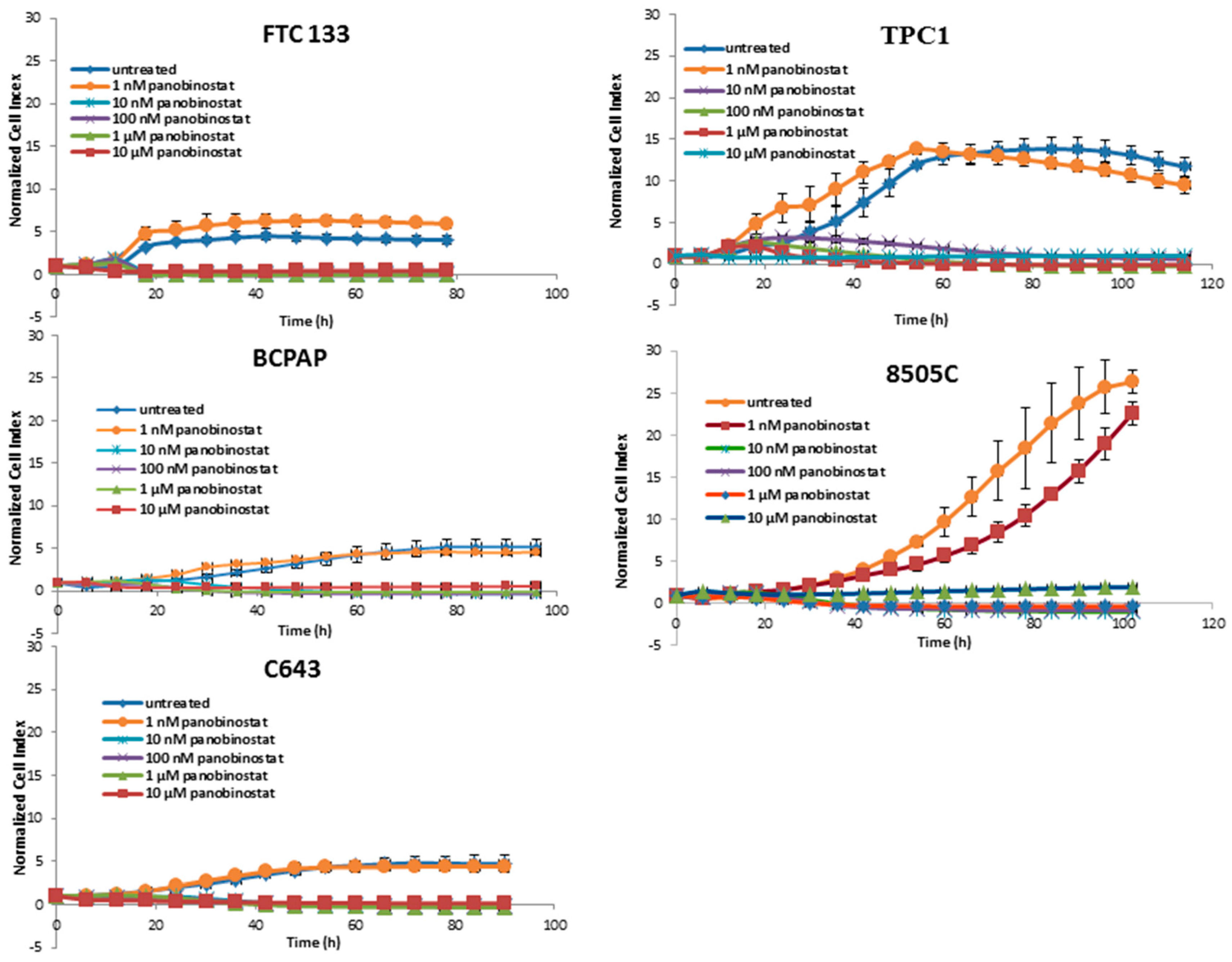
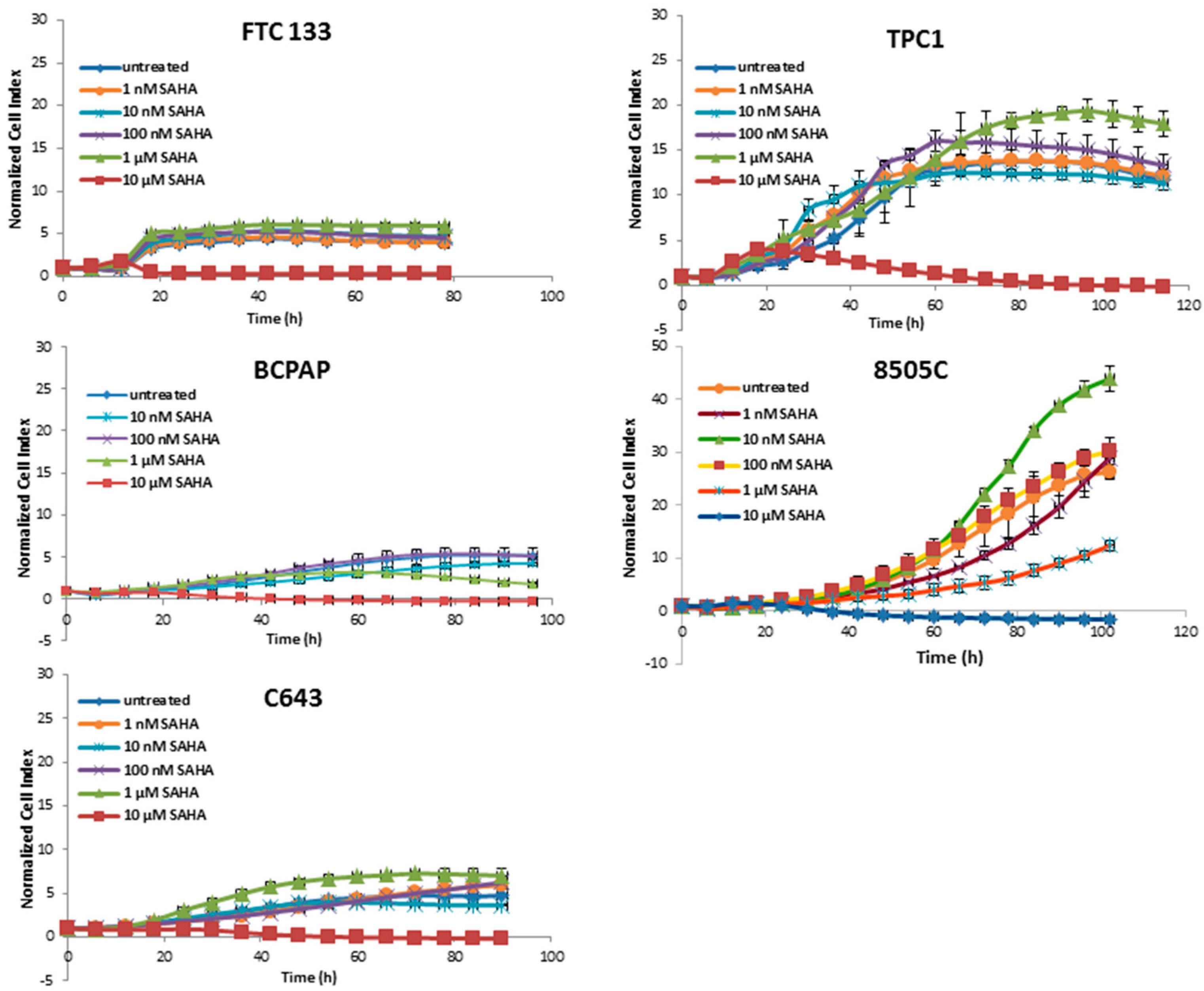
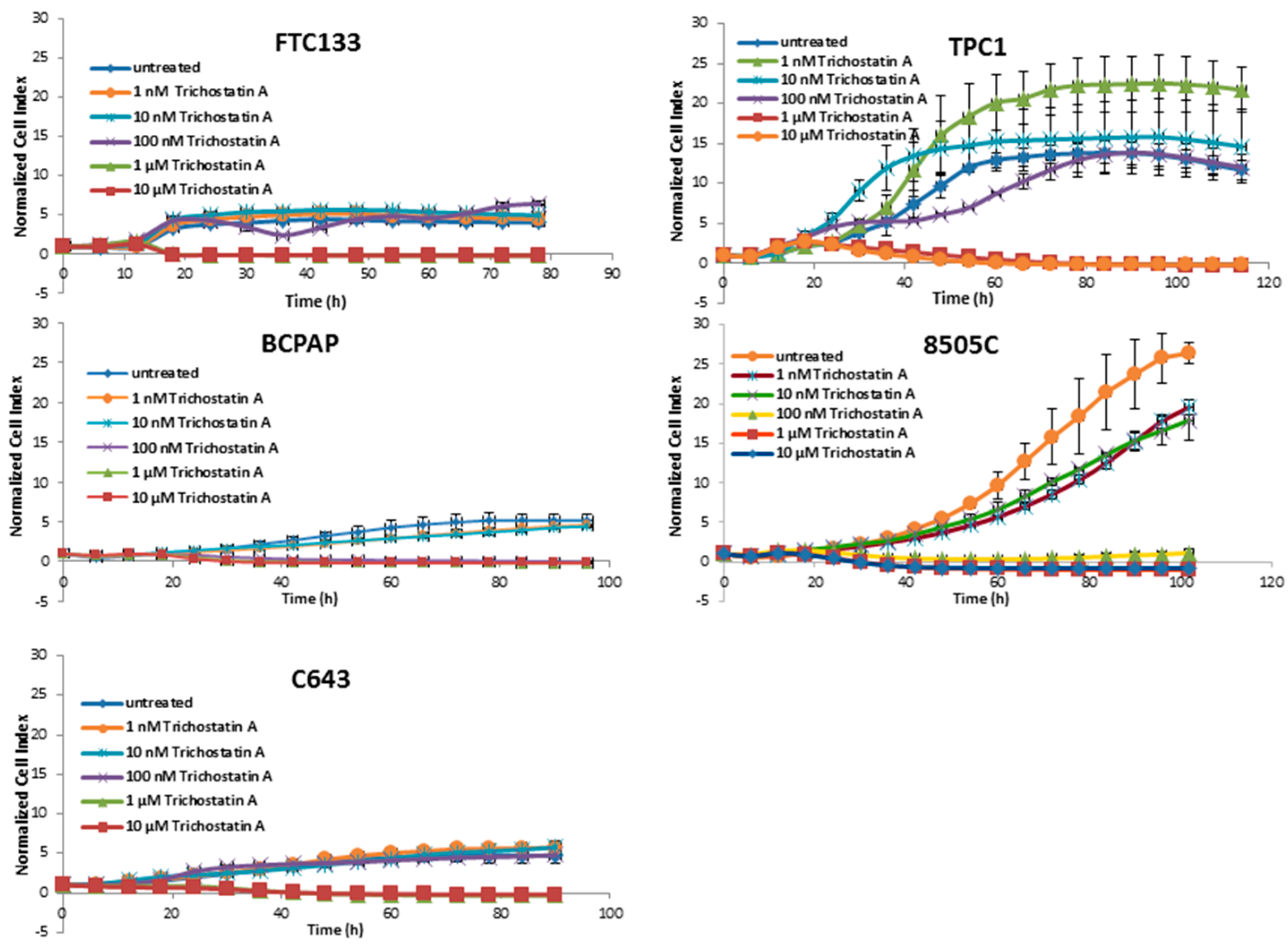
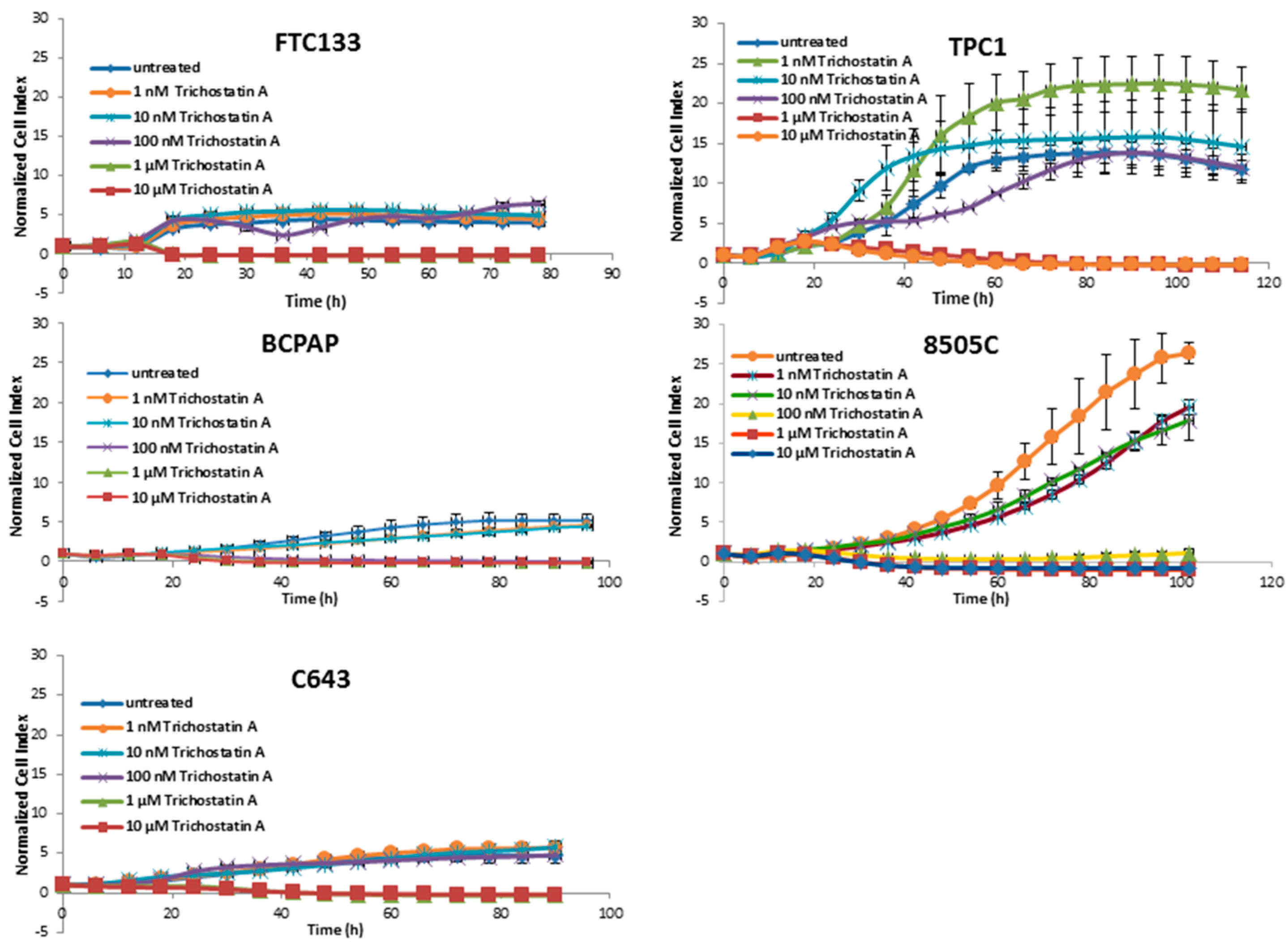
3.1. Cytotoxic Effects of Deacetylase Inhibitors in Thyroid Cancer Cells
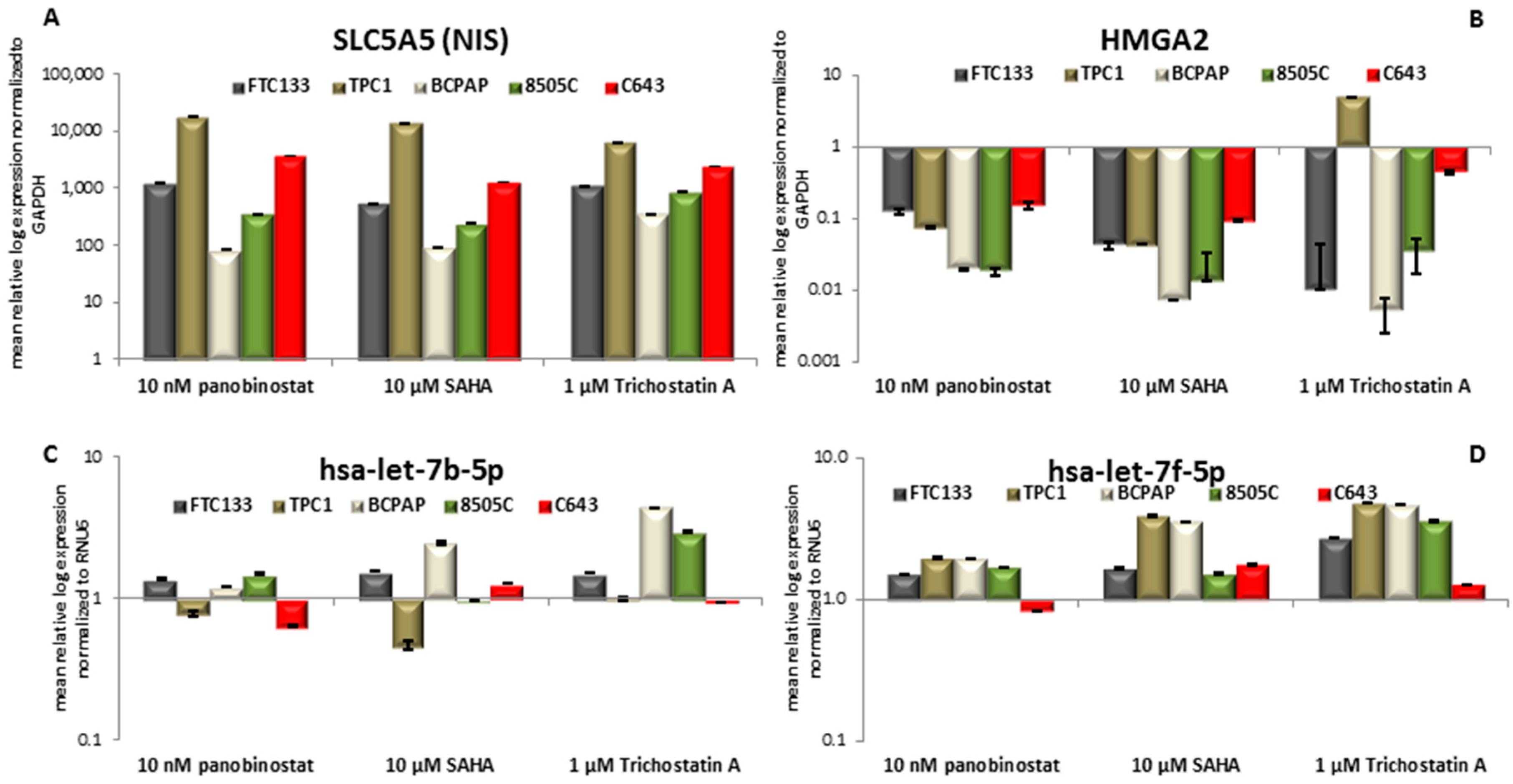
3.2. Expression of SLC5A5 (NIS), HMGA2, and Their Regulatory miRNAs Hsa-let-7b-5p Hsa-let-7f-5p
3.3. NIS Protein Modulation after Treatment with HDACi
3.4. Radioiodine Up-take after HDACi Treatment
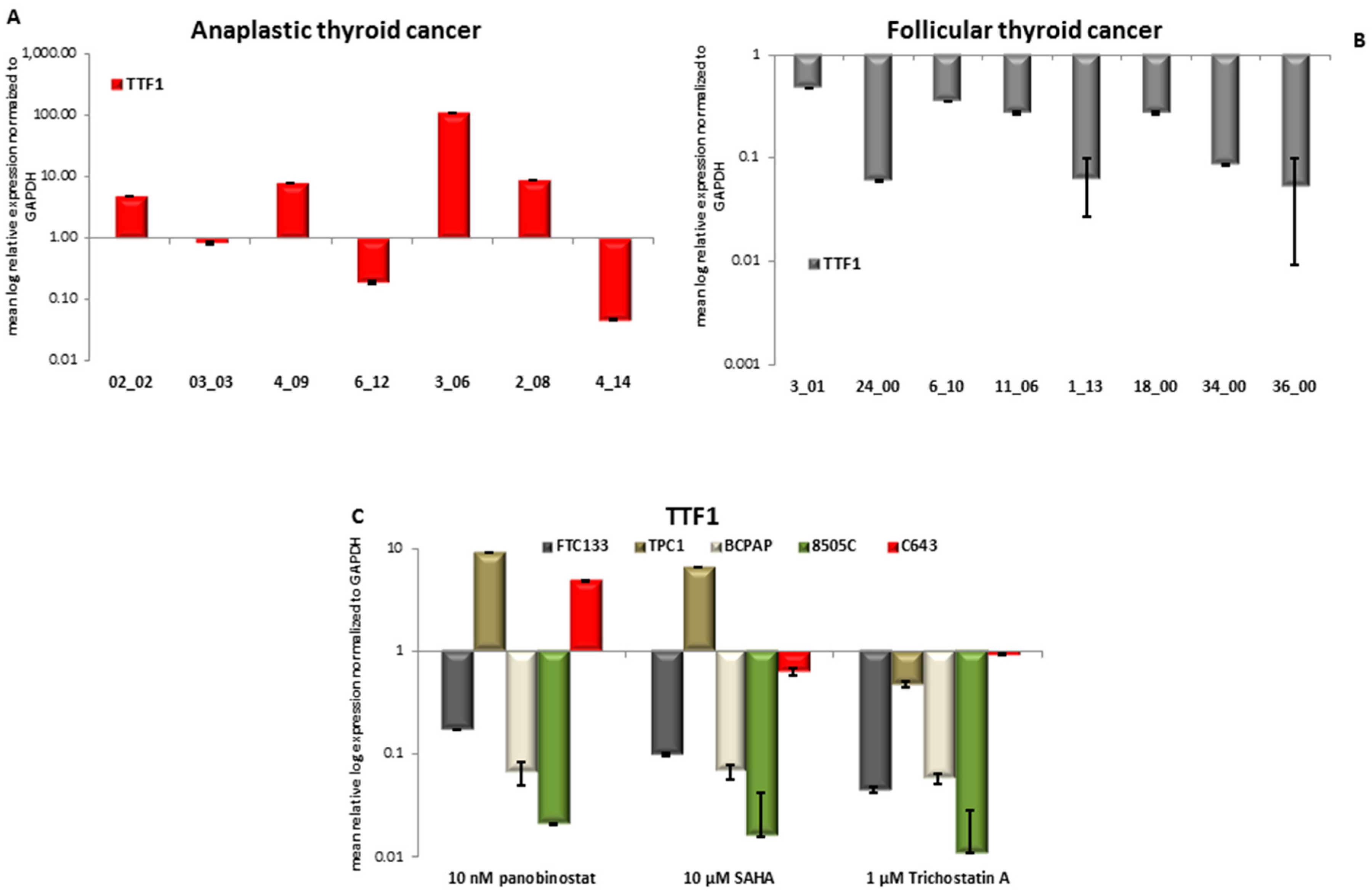
3.5. TTF1 Expression in Primary Thyroid Tumor Samples and in Vitro after Treatment with HDACi
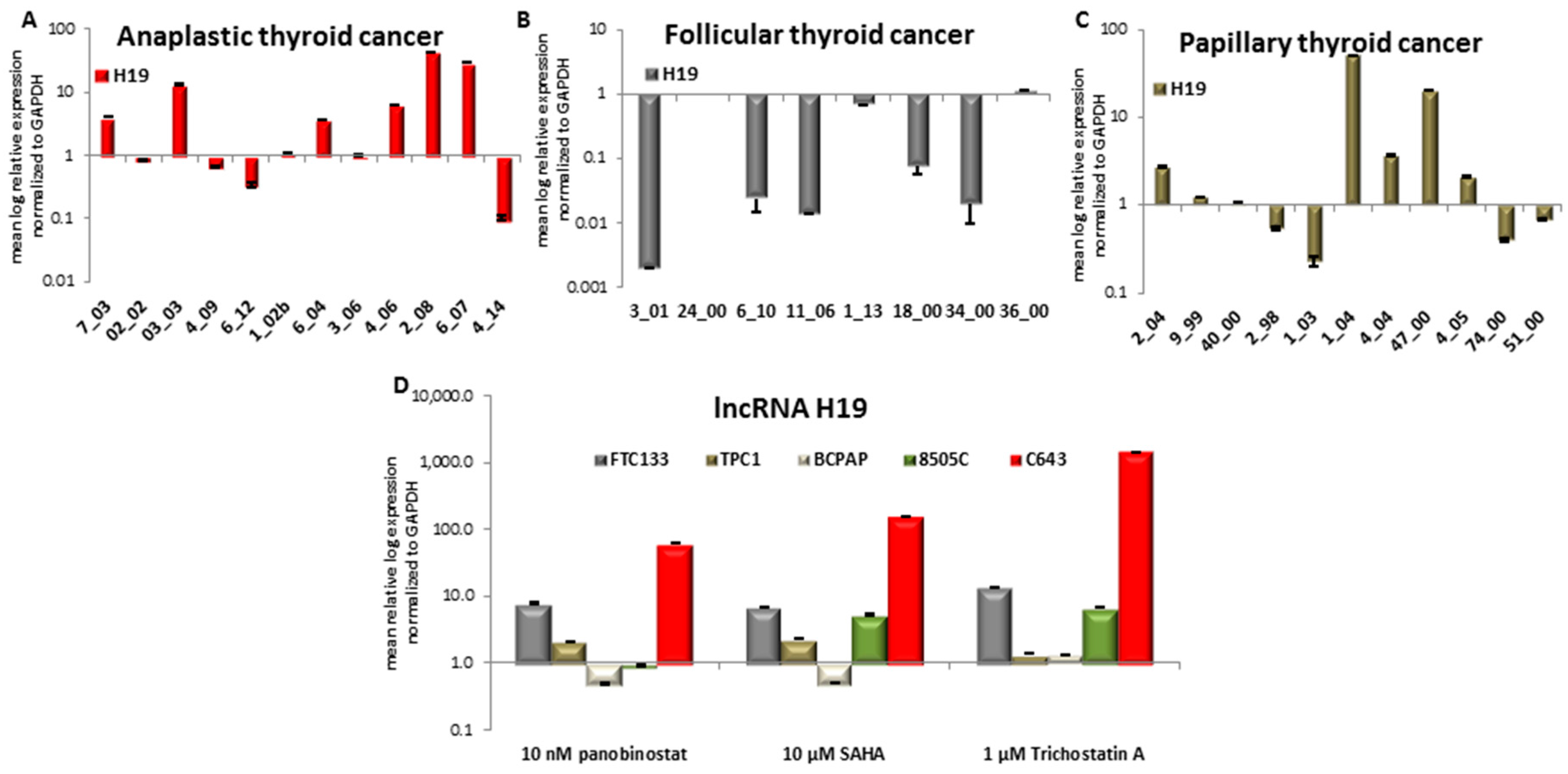
3.6. Expression of Long Non-Coding RNA H19 in Primary Thyroid Tumor Samples and In Vitro after Treatment with HDACi
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hsu, K.T.; Yu, X.M.; Audhya, A.W.; Jaume, J.C.; Lloyd, R.V.; Miyamoto, S.; Prolla, T.A.; Chen, H. Novel approaches in anaplastic thyroid cancer therapy. Oncologist 2014, 19, 1148–1155. [Google Scholar] [CrossRef] [PubMed]
- Dohan, O.; Carrasco, N. Advances in Na+/I- symporter (NIS) research in the thyroid and beyond. Mol. Cell. Endocrinol. 2003, 213, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Dohan, O.; De la Vieja, A.; Paroder, V.; Riedel, C.; Artani, M.; Reed, M.; Ginter, C.S.; Carrasco, N. The sodium/iodide symporter (NIS): Characterization, regulation, and medical significance. Endocr. Rev. 2003, 24, 48–77. [Google Scholar] [CrossRef] [PubMed]
- Wapnir, I.L.; van de Rijn, M.; Nowels, K.; Amenta, P.S.; Walton, K.; Montgomery, K.; Greco, R.S.; Dohan, O.; Carrasco, N. Immunohistochemical profile of the sodium/iodide symporter in thyroid, breast, and other carcinomas using high density tissue microarrays and conventional sections. J. Clin. Endocrinol. Metab. 2003, 88, 1880–1888. [Google Scholar] [CrossRef] [PubMed]
- Schmohl, K.A.; Dolp, P.; Schug, C.; Knoop, K.; Klutz, K.; Schwenk, N.; Bartenstein, P.; Nelson, P.J.; Ogris, M.; Wagner, E.; et al. Reintroducing the sodium-iodide symporter to anaplastic thyroid carcinoma. Thyroid 2017, 27, 1534–1543. [Google Scholar] [CrossRef] [PubMed]
- Damanakis, A.I.; Eckhardt, S.; Wunderlich, A.; Roth, S.; Wissniowski, T.T.; Bartsch, D.K.; Di Fazio, P. MicroRNAs let7 expression in thyroid cancer: Correlation with their deputed targets HMGA2 and SLC5A5. J. Cancer Res. Clin. Oncol. 2016, 142, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Pallante, P.; Sepe, R.; Puca, F.; Fusco, A. High mobility group a proteins as tumor markers. Front. Med. (Lausanne) 2015, 2, 15. [Google Scholar] [CrossRef] [PubMed]
- Fedele, M.; Palmieri, D.; Fusco, A. HMGA2: A pituitary tumour subtype-specific oncogene? Mol. Cell. Endocrinol. 2010, 326, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Fedele, M.; Visone, R.; De Martino, I.; Troncone, G.; Palmieri, D.; Battista, S.; Ciarmiello, A.; Pallante, P.; Arra, C.; Melillo, R.M.; et al. HMGA2 induces pituitary tumorigenesis by enhancing E2F1 activity. Cancer Cell 2006, 9, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Di Fazio, P.; Montalbano, R.; Neureiter, D.; Alinger, B.; Schmidt, A.; Merkel, A.L.; Quint, K.; Ocker, M. Downregulation of HMGA2 by the pan-deacetylase inhibitor panobinostat is dependent on hsa-let-7b expression in liver cancer cell lines. Exp. Cell Res. 2012, 318, 1832–1843. [Google Scholar] [CrossRef] [PubMed]
- Peiling Yang, S.; Ngeow, J. Familial non-medullary thyroid cancer: Unraveling the genetic maze. Endocr. Relat. Cancer 2016, 23, R577–R595. [Google Scholar] [CrossRef] [PubMed]
- Ngan, E.S.W.; Lang, B.H.H.; Liu, T.; Shum, C.K.Y.; So, M.-T.; Lau, D.K.C.; Leon, T.Y.Y.; Cherny, S.S.; Tsai, S.Y.; Lo, C.-Y.; et al. A germline mutation (A339V) in thyroid transcription factor-1 (TITF-1/NKX2.1) in patients with multinodular goiter and papillary thyroid carcinoma. JNCI J. Natl. Cancer Inst. 2009, 101, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Moraes, L.; Galrao, A.L.; Rubio, I.; Cerutti, J.M. Transcriptional regulation of the potential tumor suppressor ABI3 gene in thyroid carcinomas: Interplay between methylation and NKX2-1 availability. Oncotarget 2016, 7, 25960–25970. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.T.; Pan, H.; Xia, G.F.; Qiu, C.; Zhu, Z.M. Prognostic and clinicopathological significance of long noncoding RNA H19 overexpression in human solid tumors: Evidence from a meta-analysis. Oncotarget 2016, 7, 83177–83186. [Google Scholar] [CrossRef] [PubMed]
- Murugan, A.K.; Munirajan, A.K.; Alzahrani, A.S. Long noncoding RNAs: Emerging players in thyroid cancer pathogenesis. Endocr. Relat. Cancer 2018, 25, R59–R82. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.W.; Chen, C.; Loh, E.W.; Chu, C.C.; Wang, M.Y.; Ouyang, H.J.; Chang, Y.T.; Zhuang, W.Z.; Chou, C.W.; Huang, D.J.; et al. Tyrosine kinase inhibitors for advanced or metastatic thyroid cancer: A meta-analysis of randomized controlled trials. Curr. Med. Res. Opin. 2017, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gild, M.L.; Topliss, D.J.; Learoyd, D.; Parnis, F.; Tie, J.; Hughes, B.; Walsh, J.P.; McLeod, D.S.A.; Clifton-Bligh, R.J.; Robinson, B.G. Clinical guidance for radioiodine refractory differentiated thyroid cancer. Clin. Endocrinol. (Oxf.) 2018, 88, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Gryder, B.E.; Sodji, Q.H.; Oyelere, A.K. Targeted cancer therapy: Giving histone deacetylase inhibitors all they need to succeed. Future Med. Chem. 2012, 4, 505–524. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.M.; Ahn, B.C. Redifferentiation of radioiodine refractory differentiated thyroid cancer for reapplication of I-131 therapy. Front. Endocrinol. (Lausanne) 2017, 8, 260. [Google Scholar] [CrossRef] [PubMed]
- Baldan, F.; Lavarone, E.; Di Loreto, C.; Filetti, S.; Russo, D.; Damante, G.; Puppin, C. Histone post-translational modifications induced by histone deacetylase inhibition in transcriptional control units of NIS gene. Mol. Biol. Rep. 2014, 41, 5257–5265. [Google Scholar] [CrossRef] [PubMed]
- Kelkar, M.G.; Senthilkumar, K.; Jadhav, S.; Gupta, S.; Ahn, B.C.; De, A. Enhancement of human sodium iodide symporter gene therapy for breast cancer by HDAC inhibitor mediated transcriptional modulation. Sci. Rep. 2016, 6, 19341. [Google Scholar] [CrossRef] [PubMed]
- Damaskos, C.; Garmpis, N.; Valsami, S.; Spartalis, E.; Antoniou, E.A.; Tomos, P.; Karamaroudis, S.; Zoumpou, T.; Pergialiotis, V.; Stergios, K.; et al. Histone deacetylase inhibitors: A novel therapeutic weapon against medullary thyroid cancer? Anticancer Res. 2016, 36, 5019–5024. [Google Scholar] [CrossRef] [PubMed]
- Ngamphaiboon, N.; Dy, G.K.; Ma, W.W.; Zhao, Y.; Reungwetwattana, T.; DePaolo, D.; Ding, Y.; Brady, W.; Fetterly, G.; Adjei, A.A. A phase I study of the histone deacetylase (HDAC) inhibitor entinostat, in combination with sorafenib in patients with advanced solid tumors. Investig. New Drugs 2015, 33, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Bonaterra, G.A.; Then, H.; Oezel, L.; Schwarzbach, H.; Ocker, M.; Thieme, K.; Di Fazio, P.; Kinscherf, R. Morphological alterations in gastrocnemius and soleus muscles in male and female mice in a fibromyalgia model. PLoS ONE 2016, 11, e0151116. [Google Scholar] [CrossRef] [PubMed]
- Montalbano, R.; Waldegger, P.; Quint, K.; Jabari, S.; Neureiter, D.; Illig, R.; Ocker, M.; Di Fazio, P. Endoplasmic reticulum stress plays a pivotal role in cell death mediated by the pan-deacetylase inhibitor panobinostat in human hepatocellular cancer cells. Transl. Oncol. 2013, 6, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Di Fazio, P.; Schneider-Stock, R.; Neureiter, D.; Okamoto, K.; Wissniowski, T.; Gahr, S.; Quint, K.; Meissnitzer, M.; Alinger, B.; Montalbano, R.; et al. The pan-deacetylase inhibitor panobinostat inhibits growth of hepatocellular carcinoma models by alternative pathways of apoptosis. Cell. Oncol. 2010, 32, 285–300. [Google Scholar] [PubMed]
- Zopf, S.; Ocker, M.; Neureiter, D.; Alinger, B.; Gahr, S.; Neurath, M.F.; Di Fazio, P. Inhibition of DNA methyltransferase activity and expression by treatment with the pan-deacetylase inhibitor panobinostat in hepatocellular carcinoma cell lines. BMC Cancer 2012, 12, 386. [Google Scholar] [CrossRef] [PubMed]
- Henrici, A.; Montalbano, R.; Neureiter, D.; Krause, M.; Stiewe, T.; Slater, E.P.; Quint, K.; Ocker, M.; Di Fazio, P. The pan-deacetylase inhibitor panobinostat suppresses the expression of oncogenic miRNAs in hepatocellular carcinoma cell lines. Mol. Carcinog. 2015, 54, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Ishizaka, Y.; Itoh, F.; Tahira, T.; Ikeda, I.; Ogura, T.; Sugimura, T.; Nagao, M. Presence of aberrant transcripts of ret proto-oncogene in a human papillary thyroid carcinoma cell line. Jpn. J. Cancer Res. 1989, 80, 1149–1152. [Google Scholar] [CrossRef] [PubMed]
- Heldin, N.E.; Westermark, B. The molecular biology of the human anaplastic thyroid carcinoma cell. Thyroidology 1991, 3, 127–131. [Google Scholar] [PubMed]
- Ito, T.; Seyama, T.; Iwamoto, K.S.; Hayashi, T.; Mizuno, T.; Tsuyama, N.; Dohi, K.; Nakamura, N.; Akiyama, M. In vitro irradiation is able to cause RET oncogene rearrangement. Cancer Res. 1993, 53, 2940–2943. [Google Scholar] [PubMed]
- Wright, P.A.; Lemoine, N.R.; Goretzki, P.E.; Wyllie, F.S.; Bond, J.; Hughes, C.; Roher, H.D.; Williams, E.D.; Wynford-Thomas, D. Mutation of the p53 gene in a differentiated human thyroid carcinoma cell line, but not in primary thyroid tumours. Oncogene 1991, 6, 1693–1697. [Google Scholar] [PubMed]
- Bernhart, E.; Stuendl, N.; Kaltenegger, H.; Windpassinger, C.; Donohue, N.; Leithner, A.; Lohberger, B. Histone deacetylase inhibitors vorinostat and panobinostat induce G1 cell cycle arrest and apoptosis in multidrug resistant sarcoma cell lines. Oncotarget 2017, 8, 77254–77267. [Google Scholar] [CrossRef] [PubMed]
- Wilson, P.M.; Labonte, M.J.; Martin, S.C.; Kuwahara, S.T.; El-Khoueiry, A.; Lenz, H.J.; Ladner, R.D. Sustained inhibition of deacetylases is required for the antitumor activity of the histone deactylase inhibitors panobinostat and vorinostat in models of colorectal cancer. Investig. New Drugs 2013, 31, 845–857. [Google Scholar] [CrossRef] [PubMed]
- Shao, W.; Growney, J.D.; Feng, Y.; O’Connor, G.; Pu, M.; Zhu, W.; Yao, Y.M.; Kwon, P.; Fawell, S.; Atadja, P. Activity of deacetylase inhibitor panobinostat (LBH589) in cutaneous T-cell lymphoma models: Defining molecular mechanisms of resistance. Int. J. Cancer 2010, 127, 2199–2208. [Google Scholar] [CrossRef] [PubMed]
- Richon, V.M.; Sandhoff, T.W.; Rifkind, R.A.; Marks, P.A. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc. Natl. Acad. Sci. USA 2000, 97, 10014–10019. [Google Scholar] [CrossRef] [PubMed]
- Furuya, F.; Shimura, H.; Suzuki, H.; Taki, K.; Ohta, K.; Haraguchi, K.; Onaya, T.; Endo, T.; Kobayashi, T. Histone deacetylase inhibitors restore radioiodide uptake and retention in poorly differentiated and anaplastic thyroid cancer cells by expression of the sodium/iodide symporter thyroperoxidase and thyroglobulin. Endocrinology 2004, 145, 2865–2875. [Google Scholar] [CrossRef] [PubMed]
- Dai, G.; Levy, O.; Carrasco, N. Cloning and characterization of the thyroid iodide transporter. Nature 1996, 379, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Raveh, E.; Matouk, I.J.; Gilon, M.; Hochberg, A. The H19 long non-coding RNA in cancer initiation, progression and metastasis—A proposed unifying theory. Mol. Cancer 2015, 14, 184. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, F.; Yuan, J.H.; Yuan, S.X.; Zhou, W.P.; Huo, X.S.; Xu, D.; Bi, H.S.; Wang, F.; Sun, S.H. Epigenetic activation of the MiR-200 family contributes to H19-mediated metastasis suppression in hepatocellular carcinoma. Carcinogenesis 2013, 34, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Pedone, P.V.; Pikaart, M.J.; Cerrato, F.; Vernucci, M.; Ungaro, P.; Bruni, C.B.; Riccio, A. Role of histone acetylation and DNA methylation in the maintenance of the imprinted expression of the H19 and Igf2 genes. FEBS Lett. 1999, 458, 45–50. [Google Scholar] [CrossRef]
- Wang, P.; Liu, G.; Xu, W.; Liu, H.; Bu, Q.; Sun, D. Long noncoding RNA H19 inhibits cell viability, migration, and invasion via downregulation of IRS-1 in thyroid cancer cells. Technol. Cancer Res. Treat. 2017, 16, 1102–1112. [Google Scholar] [CrossRef] [PubMed]
- De Souza, C.; Chatterji, B.P. HDAC inhibitors as novel anti-cancer therapeutics. Recent Pat. Anticancer Drug Discov. 2015, 10, 145–162. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FTC133 | TPC1 | BCPAP | 8505C | C643 | |
|---|---|---|---|---|---|
| Origin | follicular | papillary | papillary | poorly differentiated | anaplastic |
| TP53 | mut | mut | mut | ||
| HRAS | mut | ||||
| Ret/PTC | mut | ||||
| BRAF | mut; V600 | mut; V600 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wächter, S.; Damanakis, A.I.; Elxnat, M.; Roth, S.; Wunderlich, A.; Verburg, F.A.; Fellinger, S.A.; Bartsch, D.K.; Di Fazio, P. Epigenetic Modifications in Thyroid Cancer Cells Restore NIS and Radio-Iodine Uptake and Promote Cell Death. J. Clin. Med. 2018, 7, 61. https://doi.org/10.3390/jcm7040061
Wächter S, Damanakis AI, Elxnat M, Roth S, Wunderlich A, Verburg FA, Fellinger SA, Bartsch DK, Di Fazio P. Epigenetic Modifications in Thyroid Cancer Cells Restore NIS and Radio-Iodine Uptake and Promote Cell Death. Journal of Clinical Medicine. 2018; 7(4):61. https://doi.org/10.3390/jcm7040061
Chicago/Turabian StyleWächter, Sabine, Alexander I. Damanakis, Moritz Elxnat, Silvia Roth, Annette Wunderlich, Frederik A. Verburg, Sebastian A. Fellinger, Detlef K. Bartsch, and Pietro Di Fazio. 2018. "Epigenetic Modifications in Thyroid Cancer Cells Restore NIS and Radio-Iodine Uptake and Promote Cell Death" Journal of Clinical Medicine 7, no. 4: 61. https://doi.org/10.3390/jcm7040061
APA StyleWächter, S., Damanakis, A. I., Elxnat, M., Roth, S., Wunderlich, A., Verburg, F. A., Fellinger, S. A., Bartsch, D. K., & Di Fazio, P. (2018). Epigenetic Modifications in Thyroid Cancer Cells Restore NIS and Radio-Iodine Uptake and Promote Cell Death. Journal of Clinical Medicine, 7(4), 61. https://doi.org/10.3390/jcm7040061