Natural Compound Modulates the Cervical Cancer Microenvironment—A Pharmacophore Guided Molecular Modelling Approaches

Abstract
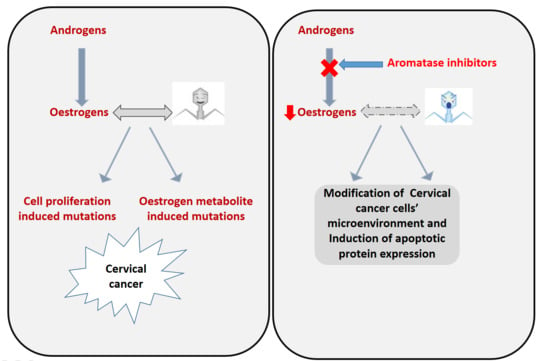
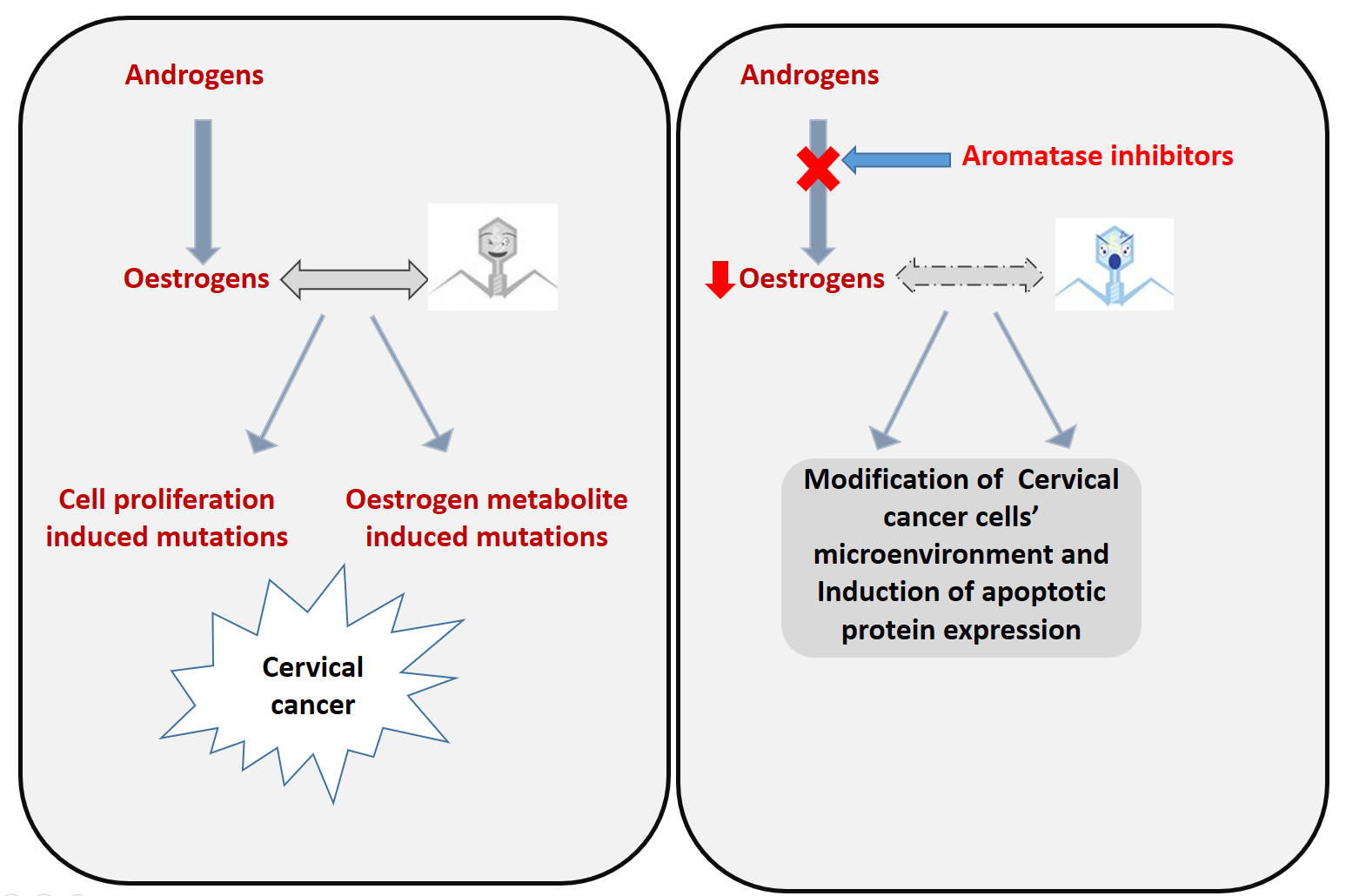
1. Introduction
2. Methods
2.1. Structure Based Pharmacophore Generation
2.2. Validation of the Generated Pharmacophore Model
2.3. Virtual Screening of Natural Compounds Database Using Two Pharmacophore Models
2.4. Molecular Docking Studies
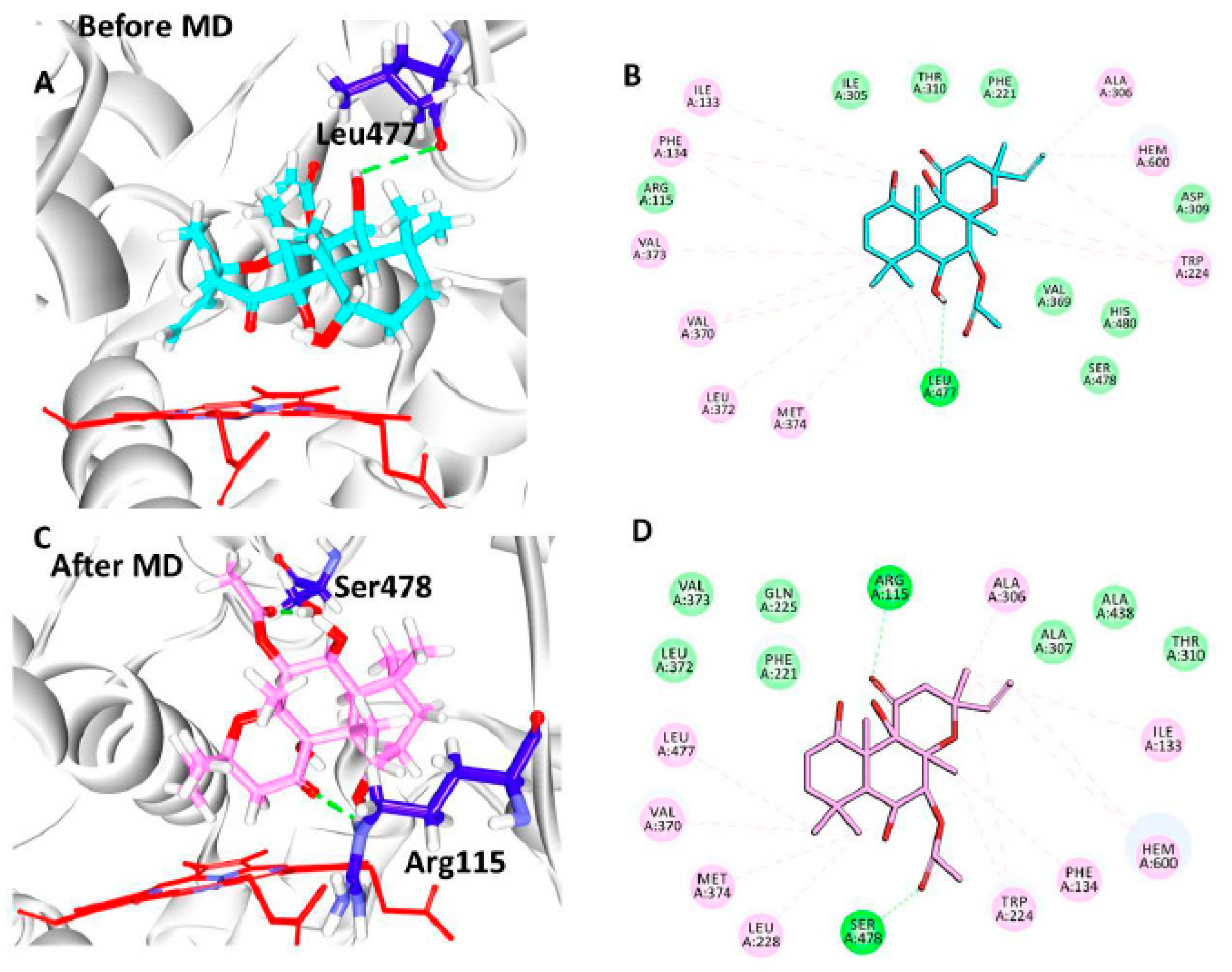
2.5. Molecular Dynamics Simulations to Elucidate the Binding Mode of the Compounds
2.6. In Vitro Assay for Evaluating the Effect of the Drug on Cancer Physiology/Micro Environment
2.7. Procurement of the Material
2.8. Maintenance of Mammalian Cell Culture
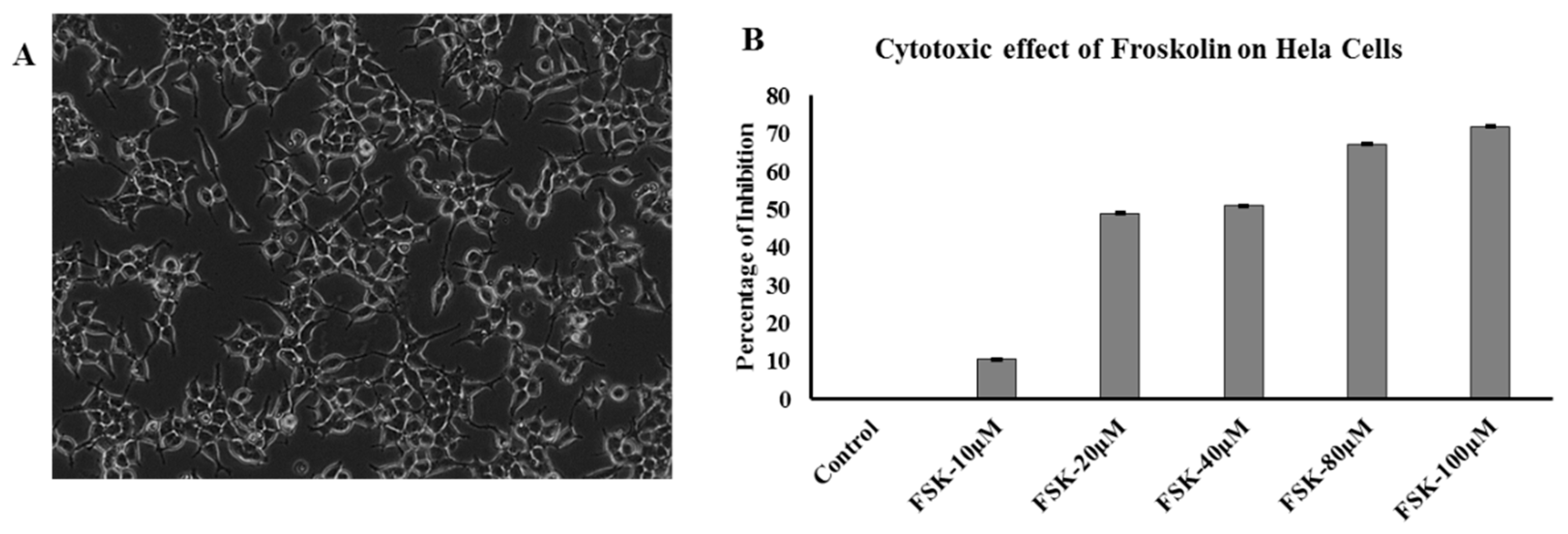
2.9. Cytotoxicity Assay
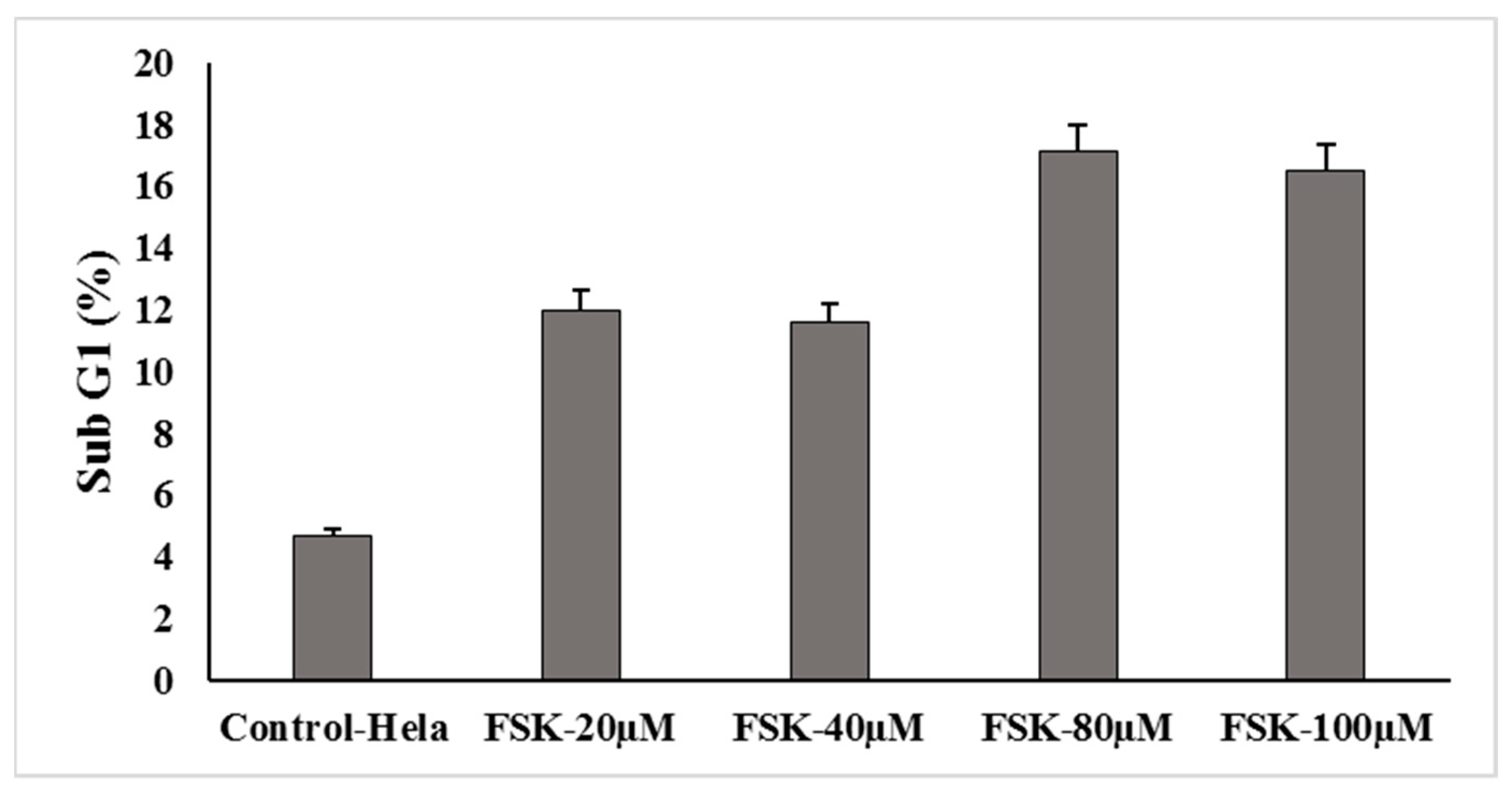
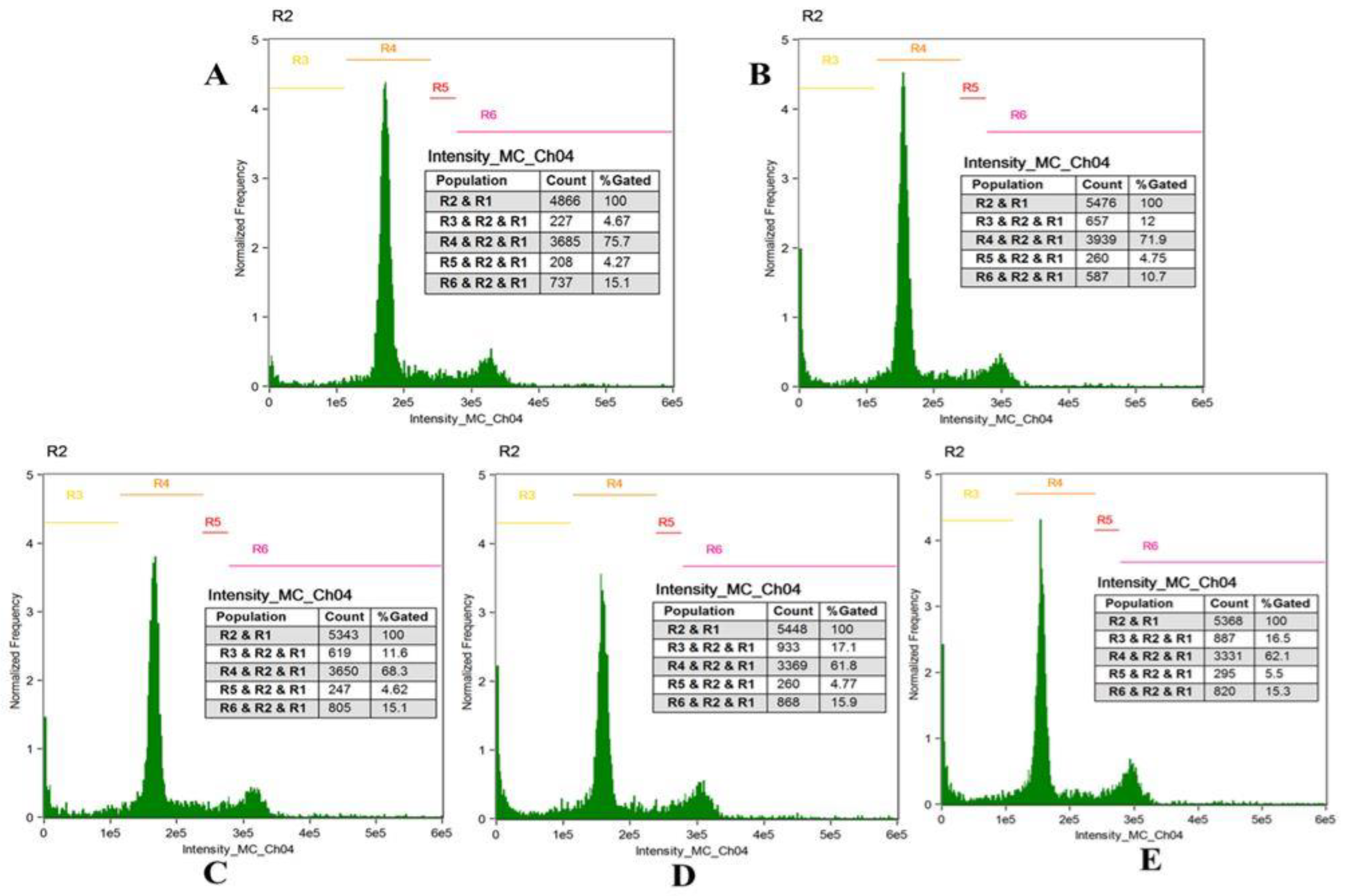
2.10. Cell Cycle Analysis
2.11. Western Blot Analysis
2.12. Statistical Analysis
3. Results
3.1. Structure-Based Pharmacophore Generation
3.2. Pharmacophore Validation
3.3. Retrieving the Compounds with the Pharmacophore Features
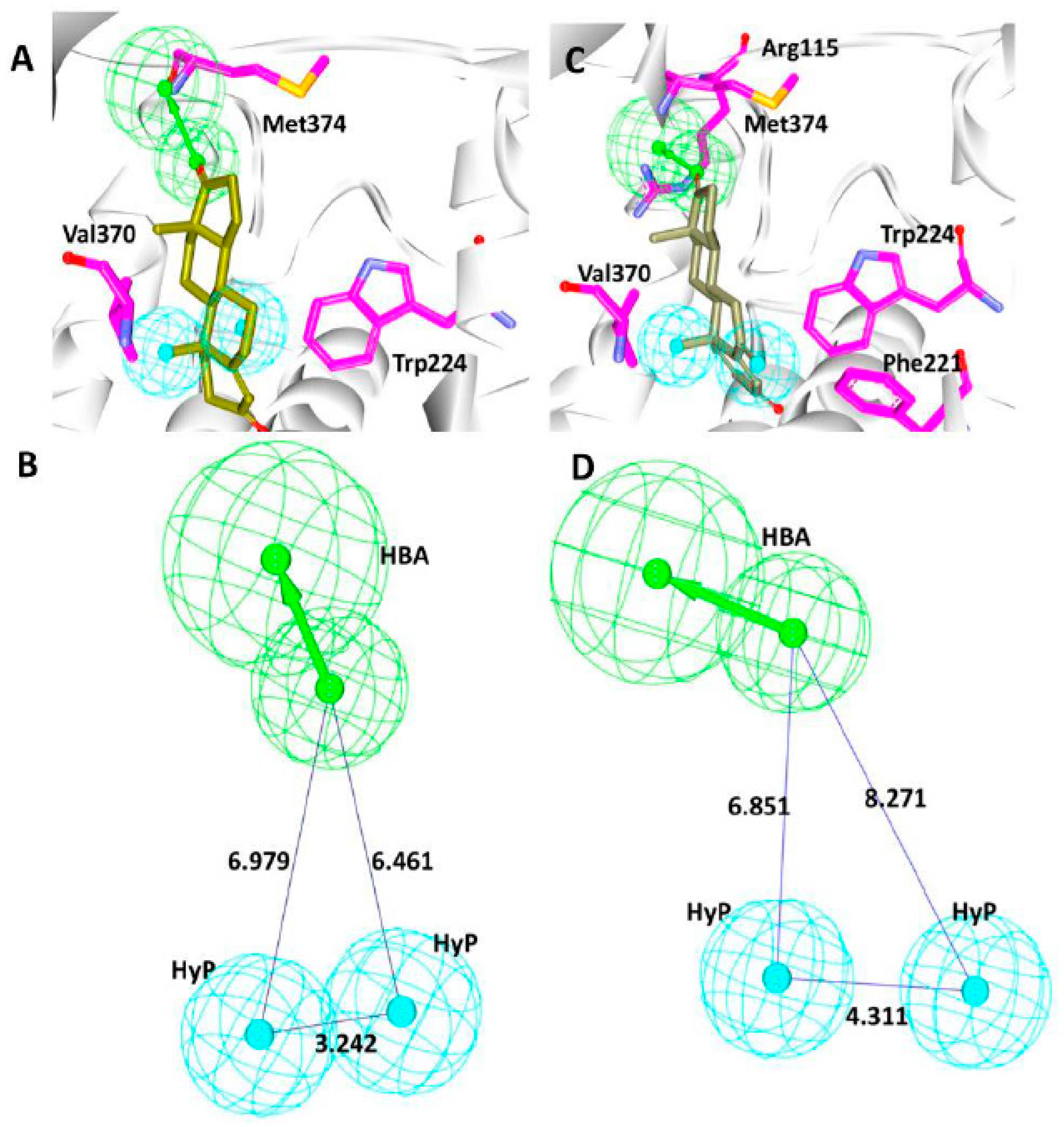
3.4. Screening by Molecular Docking Studies
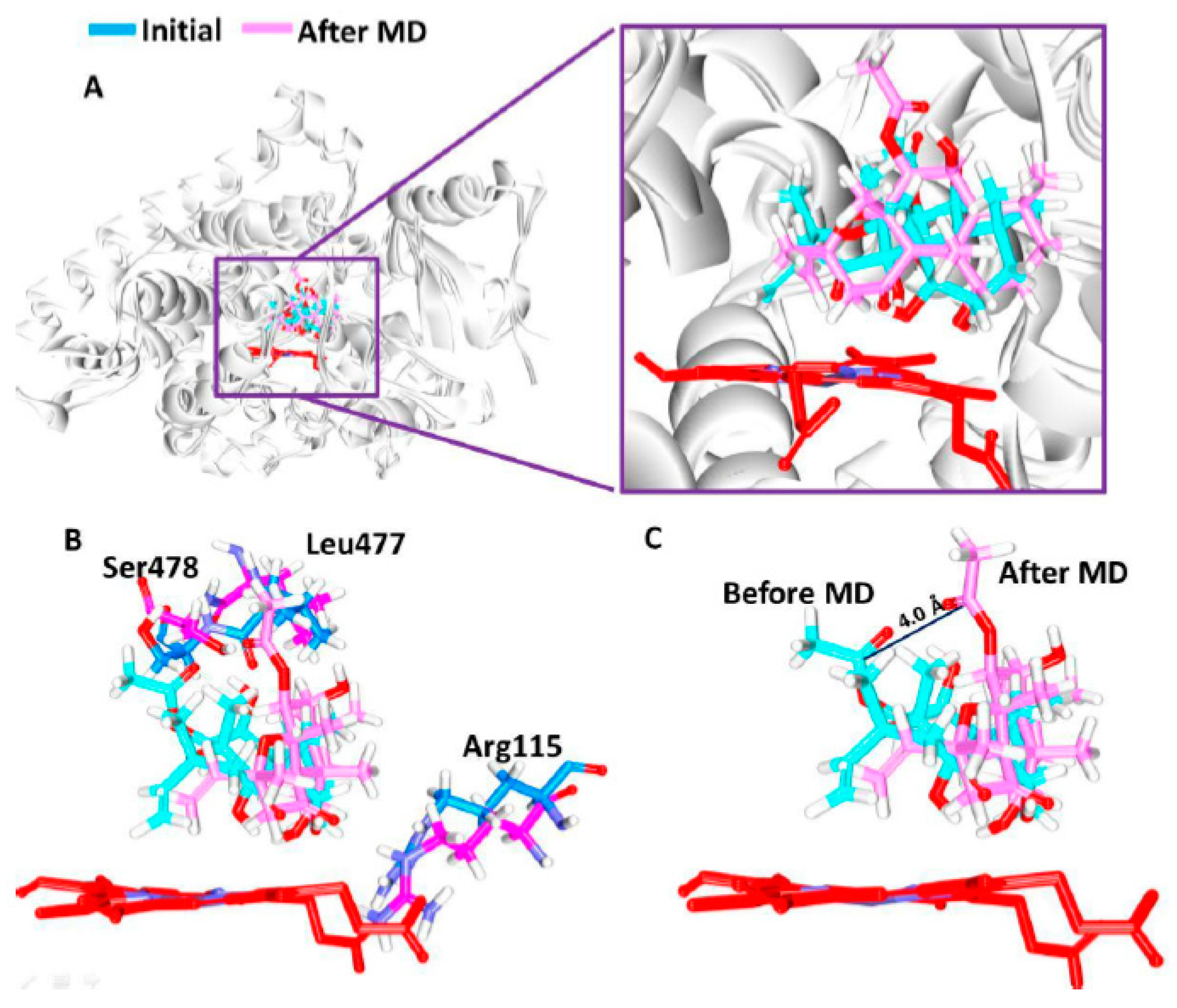
3.5. Elucidation of the Binding Mode by Molecular Dynamics Studies
3.6. Maintenance of Mammalian Cell Culture
3.7. Effect of FSK on Hela Cells (Cytotoxicity Assay)
3.8. Cell Cycle Analysis
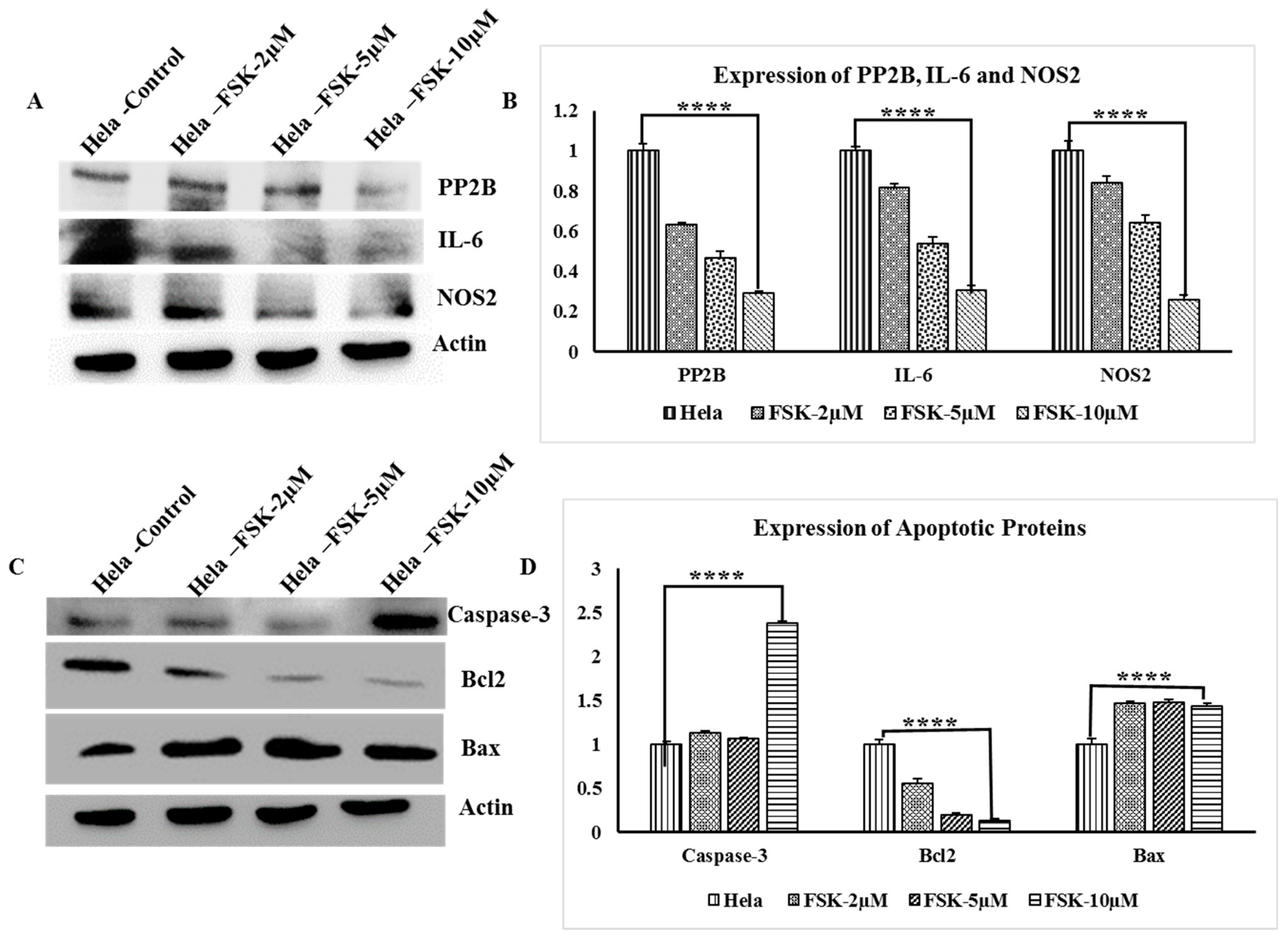
3.9. Western Blot Analysis of PP2B, IL-6 and NOS2 Genes
3.10. Analysis Apoptotic Proteins
4. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Mwaka, A.D.; Orach, C.G.; Were, E.M.; Lyratzopoulos, G.; Wabinga, H.; Roland, M. Awareness of cervical cancer risk factors and symptoms: Cross-sectional community survey in post-conflict northern Uganda. Health Expect. 2016, 19, 854–867. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.-H.; Lambert, P.F. Prevention and treatment of cervical cancer in mice using estrogen receptor antagonists. Proc. Natl. Acad. Sci. USA 2009. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.H.; Franceschi, S.; Lambert, P.F. Estrogen and ERα: Culprits in cervical cancer? Trends Endocrinol. Metab. 2010, 21, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H.; Meinhof, W.; Scheiber, W.; Bornkamm, G.W. Attempts to detect virus-specific DNA in human tumors. I. Nucleic acid hybridizations with complementary RNA of human wart virus. Int. J. Cancer 1974, 13, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, F.; Foppoli, C.; Coccia, R.; Perluigi, M. Antioxidants in cervical cancer: Chemopreventive and chemotherapeutic effects of polyphenols. Biochim. Biophys. Acta-Mol. Basis Dis. 2012, 1822, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Hamid, N.A.; Brown, C.; Gaston, K. The regulation of cell proliferation by the papillomavirus early proteins. Cell. Mol. Life Sci. 2009, 66, 1700–1717. [Google Scholar] [CrossRef]
- Nair, H.B.; Luthra, R.; Kirma, N.; Liu, Y.G.; Flowers, L.; Evans, D.; Tekmal, R.R. Induction of aromatase expression in cervical carcinomas: Effects of endogenous estrogen on cervical cancer cell proliferation. Cancer Res. 2005, 65, 11164–11173. [Google Scholar] [CrossRef]
- Mitrani-Rosenbaum, S.; Tsvieli, R.; Tur-Kaspa, R. Oestrogen stimulates differential transcription of human papillomavirus type 16 in SiHa cervical carcinoma cells. J. Gen. Virol. 1989, 70, 2227–2232. [Google Scholar] [CrossRef]
- Nelson, L.R.; Bulun, S.E. Estrogen production and action. J. Am. Acad. Dermatol. 2001, 45, S116–S124. [Google Scholar] [CrossRef]
- Cline, J.M. Neoplasms of the reproductive tract: The role of hormone exposure. ILAR J. 2004, 45, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Moreno, V.; Bosch, F.X.; Muñoz, N.; Meijer, C.J.L.M.; Shah, K.V.; Walboomers, J.M.M.; Herrero, R.; Franceschi, S. Effect of oral contraceptives on risk of cervical cancer in women with human papillomavirus infection: The IARC multicentric case-control study. Lancet 2002, 359, 1085–1092. [Google Scholar] [CrossRef]
- Brake, T.; Lambert, P.F. Estrogen contributes to the onset, persistence, and malignant progression of cervical cancer in a human papillomavirus-transgenic mouse model. Proc. Natl. Acad. Sci. USA 2005, 102, 2490–2495. [Google Scholar] [CrossRef] [PubMed]
- Au, W.W.; Abdou-Salama, S.; Al-Hendy, A. Inhibition of growth of cervical cancer cells using a dominant negative estrogen receptor gene. Gynecol. Oncol. 2007, 104, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Spurgeon, M.E.; den Boon, J.A.; Horswill, M.; Barthakur, S.; Forouzan, O.; Rader, J.S.; Beebe, D.J.; Roopra, A.; Ahlquist, P.; Lambert, P.F. Human papillomavirus oncogenes reprogram the cervical cancer microenvironment independently of and synergistically with estrogen. Proc. Natl. Acad. Sci. USA 2017, 114, E9076–E9085. [Google Scholar] [CrossRef] [PubMed]
- Hilborn, E.; Stål, O.; Jansson, A. Estrogen and androgen-converting enzymes 17β-hydroxysteroid dehydrogenase and their involvement in cancer: With a special focus on 17β-hydroxysteroid dehydrogenase type 1, 2, and breast cancer. Oncotarget 2017, 8, 30552–30562. [Google Scholar] [CrossRef] [PubMed]
- Simpson, E.R. Sources of estrogen and their importance. J. Steroid Biochem. Mol. Biol. 2003, 86, 225–230. [Google Scholar] [CrossRef]
- Fowler, K.A.; Gill, K.; Kirma, N.; Dillehay, D.L.; Tekmal, R.R. Overexpression of aromatase leads to development of testicular Leydig cell tumors: An in vivo model for hormone-mediated testicular cancer. Am. J. Pathol. 2000, 156, 347–353. [Google Scholar] [CrossRef]
- Kirma, N.; Gill, K.; Mandava, U.; Tekmal, R.R. Overexpression of aromatase leads to hyperplasia and changes in the expression of genes involved in apoptosis, cell cycle, growth, and tumor suppressor functions in the mammary glands of transgenic mice. Cancer Res. 2001, 61, 1910–1918. [Google Scholar]
- Hata, S.; Miki, Y.; Saito, R.; Ishida, K.; Watanabe, M.; Sasano, H. Aromatase in human liver and its diseases. Cancer Med. 2013, 2, 305–315. [Google Scholar] [CrossRef]
- Zhou, Y.; Zheng, J.; Li, Y.; Xu, D.P.; Li, S.; Chen, Y.M.; Li, H.B. Natural polyphenols for prevention and treatment of cancer. Nutrients 2016, 8, 515. [Google Scholar] [CrossRef] [PubMed]
- Phuah, N.H.; Azmi, M.N.; Awang, K.; Nagoor, N.H. Suppression of microRNA-629 enhances sensitivity of cervical cancer cells to 1′S-1′-acetoxychavicol acetate via regulating RSU1. Onco Targets. Ther. 2017, 10, 1695–1705. [Google Scholar] [CrossRef] [PubMed]
- Mamgain, S.; Sharma, P.; Pathak, R.K.; Baunthiyal, M. Computer aided screening of natural compounds targeting the E6 protein of HPV using molecular docking. Bioinformation 2015, 11, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Khor, T.O.; Shu, L.; Su, Z.; Fuentes, F.; Lee, J.-H.; Kong, A.-N.T. Plants vs. cancer: A review on natural phytochemicals in preventing and treating cancers and their druggability. Anticancer Agents Med. Chem. 2012, 12, 1281–1305. [Google Scholar] [CrossRef] [PubMed]
- Bharti, A.C.; Shukla, S.; Mahata, S.; Hedau, S.; Das, B.C. Anti-human papillomavirus therapeutics: Facts and future. Indian J. Med. Res. 2009, 130, 296. [Google Scholar] [PubMed]
- Aung, T.N.; Qu, Z.; Kortschak, R.D.; Adelson, D.L. Understanding the effectiveness of natural compound mixtures in cancer through their molecular mode of action. Int. J. Mol. Sci. 2017, 18, 656. [Google Scholar] [CrossRef] [PubMed]
- Stephens, F.O. The rising incidence of breast cancer in women and prostate cancer in men. Dietary influences: A possible preventive role for nature’s sex hormone modifiers—The phytoestrogens (review). Oncol. Rep. 1999, 6, 865–935. [Google Scholar] [CrossRef]
- Kumar, S.; Jena, L.; Galande, S.; Daf, S.; Mohod, K.; Varma, A.K. Elucidating molecular interactions of natural inhibitors with HPV-16 E6 oncoprotein through docking analysis. Genom. Inform. 2014, 12, 64–70. [Google Scholar] [CrossRef]
- Morris, G.M.; Lim-Wilby, M. Molecular docking. Methods Mol. Biol. 2008, 443, 365–382. [Google Scholar] [CrossRef]
- Wu, G.; Robertson, D.H.; Brooks, C.L.; Vieth, M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER—A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef]
- Rampogu, S.; Baek, A.; Zeb, A.; Lee, K.W. Exploration for novel inhibitors showing back-to-front approach against VEGFR-2 kinase domain (4AG8) employing molecular docking mechanism and molecular dynamics simulations. BMC Cancer 2018, 18, 264. [Google Scholar] [CrossRef] [PubMed]
- Rampogu, S.; Son, M.; Baek, A.; Park, C.; Rana, R.M.; Zeb, A.; Parameswaran, S.; Lee, K.W. Targeting natural compounds against HER2 kinase domain as potential anticancer drugs applying pharmacophore based molecular modelling approaches. Comput. Biol. Chem. 2018, 74, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Lopes, P.E.M.; Mackerell, A.D. Recent developments and applications of the CHARMM force fields. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 167–185. [Google Scholar] [CrossRef] [PubMed]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef] [PubMed]
- Rampogu, S.; Baek, A.; Son, M.; Zeb, A.; Park, C.; Kumar, R.; Lee, G.; Kim, D.; Choi, Y.; Cho, Y.; et al. Computational exploration for lead compounds that can reverse the nuclear morphology in Progeria. BioMed Res. Int. 2017, 2017, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, M. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manua; Cold Spring Harbor Laboratory Press: LongIsland, NY, USA, 1989. [Google Scholar]
- Awasthi, M.; Singh, S.; Pandey, V.P.; Dwivedi, U.N. Molecular docking and 3D-QSAR-based virtual screening of flavonoids as potential aromatase inhibitors against estrogen-dependent breast cancer. J. Biomol. Struct. Dyn. 2015, 33, 804–819. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Lao, K.; Hu, J.; Pang, T.; Jiang, Z.; Yuan, H.; Miao, J.; Chen, X.; Ning, S.; Xiang, H.; et al. Discovery of novel aromatase inhibitors using a homogeneous time-resolved fluorescence assay. Acta Pharmacol. Sin. 2014, 35, 1082–1092. [Google Scholar] [CrossRef] [PubMed]
- Brun, M.; Godbout, R. Activation of calcineurin in cancer: Many paths, one hub. Transl. Cancer Res. 2016, 3, S497–S506. [Google Scholar] [CrossRef]
- Peuker, K.; Muff, S.; Wang, J.; Künzel, S.; Bosse, E.; Zeissig, Y.; Luzzi, G.; Basic, M.; Strigli, A.; Ulbricht, A.; et al. Epithelial calcineurin controls microbiota-dependent intestinal tumor development. Nat. Med. 2016, 22, 506. [Google Scholar] [CrossRef] [PubMed]
- Padma, S.; Sowjanya, A.P.; Poli, U.R.; Jain, M.; Rao, B.N.; Ramakrishna, G. Downregulation of calcineurin activity in cervical carcinoma. Cancer Cell Int. 2005, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.-H.; Kuo, M.-L.; Chen, C.-A.; Cheng, W.-F.; Cheng, S.-P.; Hsieh, F.-J.; Hsieh, C.-Y. Interleukin-6 in cervical cancer: The relationship with vascular endothelial growth factor. Gynecol. Oncol. 2001, 82, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Lin, Y.; Ye, X.; Feng, C.; Lu, Y.; Yang, G.; Dong, C. Expression of IL-1α and IL-6 is associated with progression and prognosis of human cervical cancer. Med. Sci. Monit. 2016, 22, 4475–4481. [Google Scholar] [CrossRef]
- Heinecke, J.L.; Ridnour, L.A.; Cheng, R.Y.S.; Switzer, C.H.; Lizardo, M.M.; Khanna, C.; Glynn, S.A.; Hussain, S.P.; Young, H.A.; Ambs, S.; et al. Tumor microenvironment-based feed-forward regulation of NOS2 in breast cancer progression. Proc. Natl. Acad. Sci. USA 2014, 111, 6323–6328. [Google Scholar] [CrossRef]
- Ekmekcioglu, S.; Ellerhorst, J.; Smid, C.M.; Prieto, V.G.; Munsell, M.; Buzaid, A.C.; Grimm, E.A. Inducible nitric oxide synthase and nitrotyrosine in human metastatic melanoma tumors correlate with poor survival. Clin. Cancer Res. 2000, 6, 4768–4775. [Google Scholar]
- Wang, J.; He, P.; Gaida, M.; Yang, S.; Schetter, A.J.; Gaedcke, J.; Ghadimi, B.M.; Ried, T.; Yfantis, H.; Lee, D.; et al. Inducible nitric oxide synthase enhances disease aggressiveness in pancreatic cancer. Oncotarget 2016, 7, 52993. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Pharm 1 | Pharm 2 |
|---|---|---|
| Total number of molecules in database (D) | 1500 | 1500 |
| Total number of actives in database (A) | 20 | 20 |
| Total number of hit molecules from the database (Ht) | 14 | 16 |
| Total number of active molecules in hit list (Ha) | 13 | 13 |
| % Yield of active (Ha/Ht) | 92 | 81 |
| % Ratio of actives ((Ha/A) × 100) | 65 | 65 |
| Enrichment Factor (EF) | 69.64 | 60.93 |
| False negatives (A-Ha) | 7 | 7 |
| False positives (Ht–Ha) | 1 | 3 |
| Goodness of fit score (GF) | 0.73 | 0.75 |
| Docking Results | Hydrogen Bond Interactions | van der Waals Interactions | π-π/π-alkyl Interactions |
|---|---|---|---|
| Before MD | Leu477: O-H53 (2.9) | Arg115, Phe221, Ile305, Asp309, Thr310, Val369, His480, Ser478 | Ile133, Phe134, Trp224, Ala306, Val370, Leu372, Val373, Met374, |
| After MD | Arg115: HE-O6 (2.0) Ser478:HG1-O7 (1.9) | Phe221, Gln225, Ala307, Thr310, Leu372, Val373, Ala438 | Ile133, Phe134, Tpr224, Leu228, Ala306, Val370, Met374, Leu477 |
| Reference | Arg115:HH11-O1 (2.5) Met374:HN-O1 (1.9) | Phe134, Ile305, Ala306, Asp309, Thr310, Leu477, Ser478 | Ile133, Phe221, Val370, Val373 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rampogu, S.; Ravinder, D.; Pawar, S.C.; Lee, K.W. Natural Compound Modulates the Cervical Cancer Microenvironment—A Pharmacophore Guided Molecular Modelling Approaches. J. Clin. Med. 2018, 7, 551. https://doi.org/10.3390/jcm7120551
Rampogu S, Ravinder D, Pawar SC, Lee KW. Natural Compound Modulates the Cervical Cancer Microenvironment—A Pharmacophore Guided Molecular Modelling Approaches. Journal of Clinical Medicine. 2018; 7(12):551. https://doi.org/10.3390/jcm7120551
Chicago/Turabian StyleRampogu, Shailima, Doneti Ravinder, Smita C. Pawar, and Keun Woo Lee. 2018. "Natural Compound Modulates the Cervical Cancer Microenvironment—A Pharmacophore Guided Molecular Modelling Approaches" Journal of Clinical Medicine 7, no. 12: 551. https://doi.org/10.3390/jcm7120551
APA StyleRampogu, S., Ravinder, D., Pawar, S. C., & Lee, K. W. (2018). Natural Compound Modulates the Cervical Cancer Microenvironment—A Pharmacophore Guided Molecular Modelling Approaches. Journal of Clinical Medicine, 7(12), 551. https://doi.org/10.3390/jcm7120551