Systematic Analysis of Transcriptomic Profile of Chondrocytes in Osteoarthritic Knee Using Next-Generation Sequencing and Bioinformatics


Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. RNA Sequencing
2.3. Ingenuity Pathway Analysis (IPA)
2.4. Database for Annotation, Visualization and Integrated Discovery (DAVID) Bioinformatics Resources
2.5. Gene Expression Omnibus (GEO) Database
2.6. miRmap Database
2.7. Statistical Analysis
3. Results
3.1. The Sequencing Quality and Mapped Reads for RNA and Small RNA Sequencing of Chondrocytes
3.2. The Differentially Expressed Genes in Osteoarthritic Knee Chondrocytes were Associated with Osteoarthritis Pathway, Cell Adhesion and Extracellular Matrix Organization
3.3. Identification of Dysregulated Genes Related to Joint Structural Damage in OA
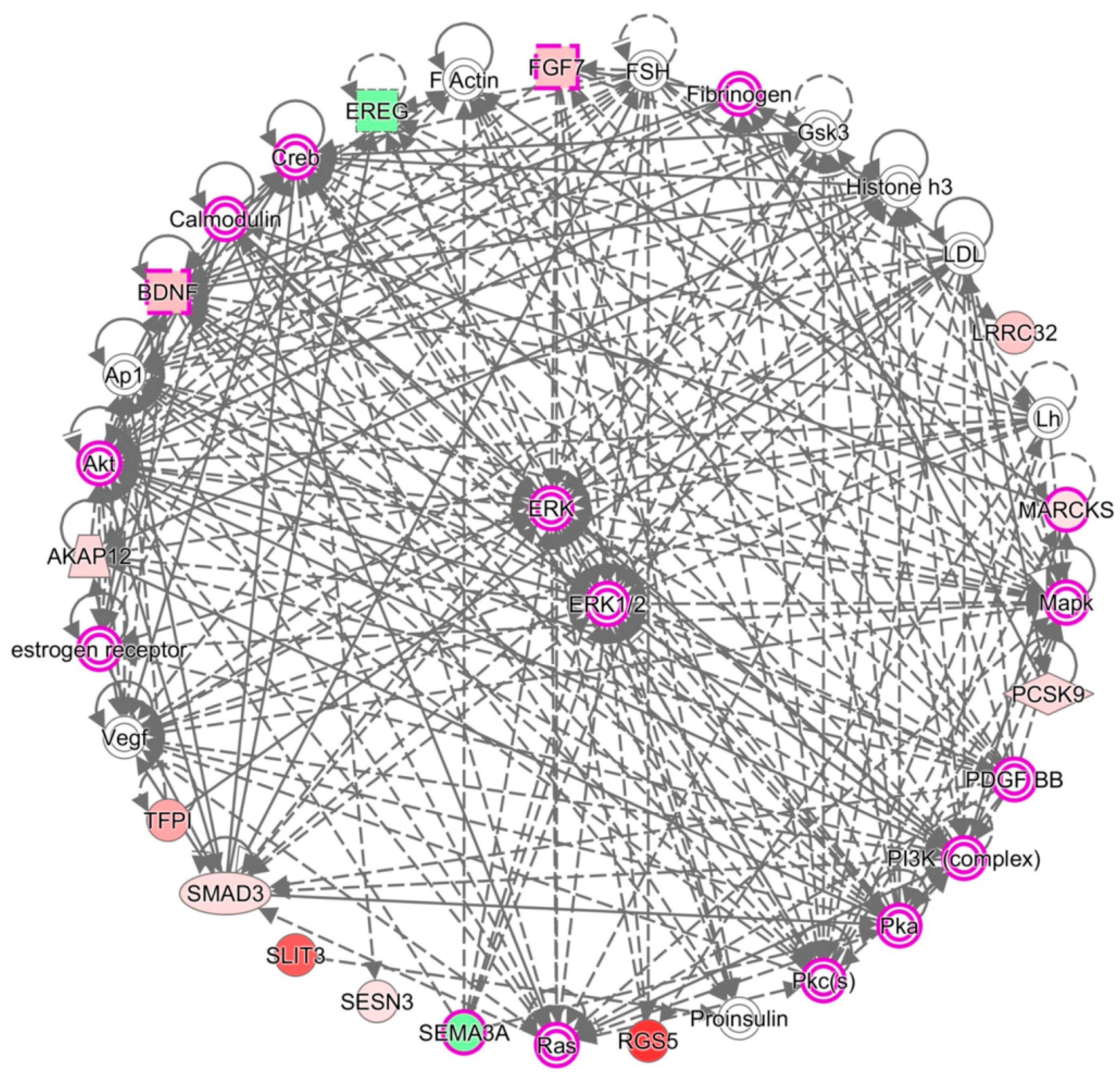
3.4. SMAD3 and WNT5A were Involved in Growth of Blood Vessel and Cell Aggregation
3.5. Identification of Differentially Expressed miRNAs and Potential miRNA–mRNA Interactions between Normal and OA Knee Chondrocytes
3.6. Analysis of Candidate Genes with Potential miRNA-mRNA Interactions in Gene Expression Omnibus (GEO) Database and Identification of Potential Molecular Signatures in OA Knee Joint Microenvironment
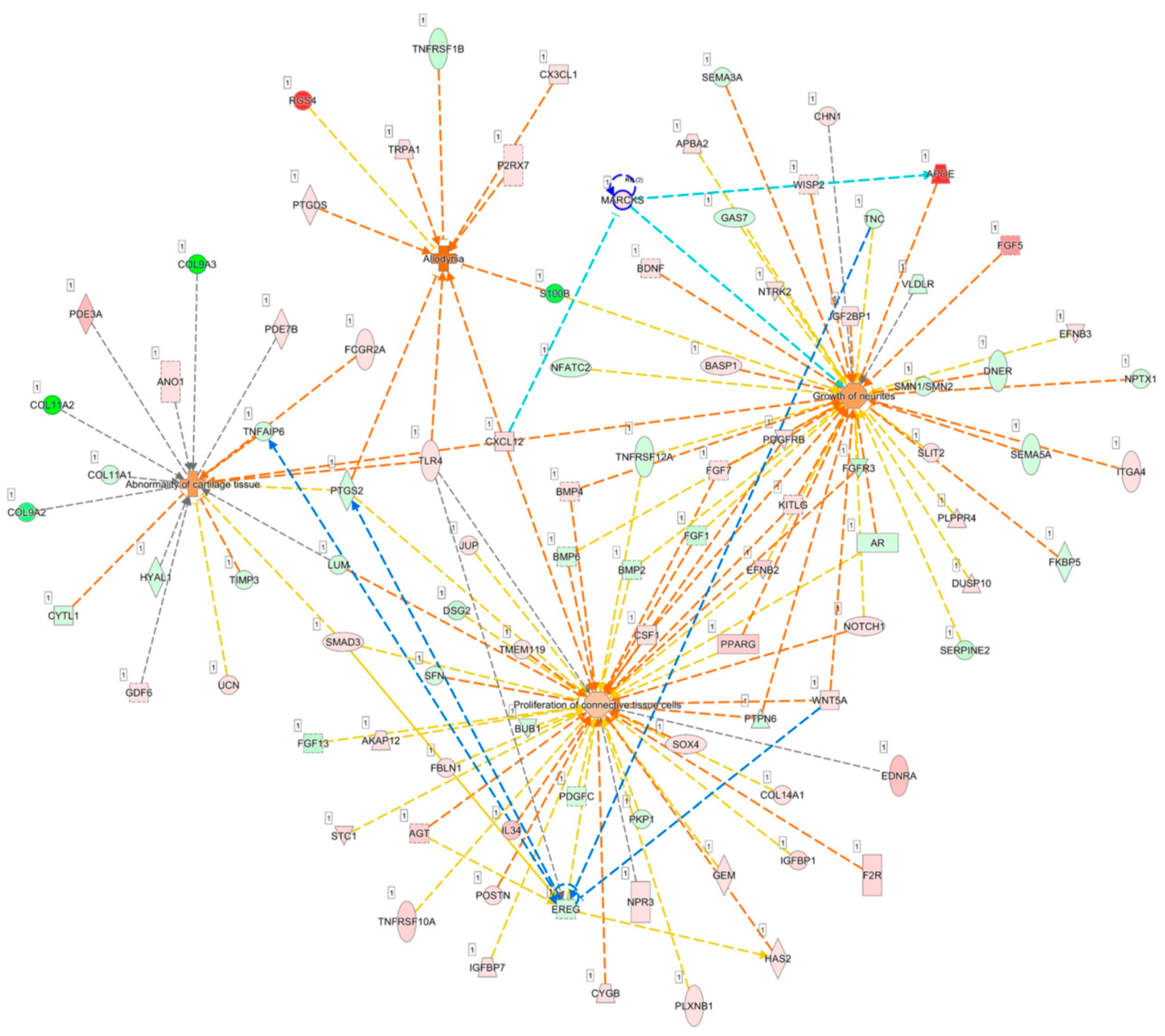
3.7. Identification of Potential miRNA-mRNA Interactions of LRRC15, MARCKS, and EREG in OA Knee Chondrocytes
3.8. MARCKS and EREG were Potentially Involved in the Pathogenesis of Arthritic Knee Joint Pain
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Johnson, V.L.; Hunter, D.J. The epidemiology of osteoarthritis. Best Pract. Res. Clin. Rheumatol. 2014, 28, 5–15. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, K.V.; Sanmartin, C.; Langlois, K.; Marshall, D.A. Symptom onset, diagnosis and management of osteoarthritis. Health Rep. 2014, 25, 10–17. [Google Scholar] [PubMed]
- Cross, M.; Smith, E.; Hoy, D.; Nolte, S.; Ackerman, I.; Fransen, M.; Bridgett, L.; Williams, S.; Guillemin, F.; Hill, C.L.; et al. The global burden of hip and knee osteoarthritis: Estimates from the global burden of disease 2010 study. Ann. Rheum. Dis. 2014, 73, 1323–1230. [Google Scholar] [CrossRef] [PubMed]
- Findlay, D.M.; Atkins, G.J. Osteoblast-chondrocyte interactions in osteoarthritis. Curr Osteoporos Rep. 2014, 12, 127–134. [Google Scholar] [CrossRef]
- Zhong, L.; Huang, X.; Karperien, M.; Post, J.N. The Regulatory Role of Signaling Crosstalk in Hypertrophy of MSCs and Human Articular Chondrocytes. Int J. Mol. Sci. 2015, 16, 19225–19247. [Google Scholar] [CrossRef]
- Pesesse, L.; Sanchez, C.; Delcour, J.P.; Bellahcene, A.; Baudouin, C.; Msika, P.; Henrotin, Y. Consequences of chondrocyte hypertrophy on osteoarthritic cartilage: Potential effect on angiogenesis. Osteoarthr. Cartil. 2013, 21, 1913–1923. [Google Scholar] [CrossRef]
- Schroeppel, J.P.; Crist, J.D.; Anderson, H.C.; Wang, J. Molecular regulation of articular chondrocyte function and its significance in osteoarthritis. Histol. Histopathol. 2011, 26, 377–394. [Google Scholar] [CrossRef]
- Lories, R.J.; Luyten, F.P. The bone-cartilage unit in osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 43–49. [Google Scholar] [CrossRef]
- Yuan, X.L.; Meng, H.Y.; Wang, Y.C.; Peng, J.; Guo, Q.Y.; Wang, A.Y.; Lu, S.B. Bone-cartilage interface crosstalk in osteoarthritis: Potential pathways and future therapeutic strategies. Osteoarthr. Cartil. 2014, 22, 1077–1089. [Google Scholar] [CrossRef]
- Sondag, G.R.; Haqqi, T.M. The Role of MicroRNAs and Their Targets in Osteoarthritis. Curr. Rheumatol. Rep. 2016, 18, 56. [Google Scholar] [CrossRef]
- Miyaki, S.; Asahara, H. Macro view of microRNA function in osteoarthritis. Nat. Rev. Rheumatol. 2012, 8, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Nugent, M. MicroRNAs: Exploring new horizons in osteoarthritis. Osteoarthr. Cartil. 2016, 24, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Liu, D.; Qu, H. Systematic review of next-generation sequencing simulators: Computational tools, features and perspectives. Brief Funct. Genomics 2017, 16, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Lewallen, E.A.; Bonin, C.A.; Li, X.; Smith, J.; Karperien, M.; Larson, A.N.; Lewallen, D.G.; Cool, S.M.; Westendorf, J.J.; Krych, A.J.; et al. The synovial microenvironment of osteoarthritic joints alters RNA-seq expression profiles of human primary articular chondrocytes. Gene 2016, 591, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Asahara, H. Current Status and Strategy of microRNA Research for Cartilage Development and Osteoarthritis Pathogenesis. J. Bone Metab. 2016, 23, 121–127. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Bonchev, D. A survey of current software for network analysis in molecular biology. Hum. Genomics 2010, 4, 353–360. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Clough, E.; Barrett, T. The Gene Expression Omnibus Database. Methods Mol. Biol. 2016, 1418, 93–110. [Google Scholar] [CrossRef] [PubMed]
- Vejnar, C.E.; Blum, M.; Zdobnov, E.M. miRmap web: Comprehensive microRNA target prediction online. Nucleic Acids Res. 2013, 41, W165–W168. [Google Scholar] [CrossRef] [PubMed]
- Vejnar, C.E.; Zdobnov, E.M. MiRmap: Comprehensive prediction of microRNA target repression strength. Nucleic Acids Res. 2012, 40, 11673–11683. [Google Scholar] [CrossRef] [PubMed]
- Stokowy, T.; Eszlinger, M.; Świerniak, M.; Fujarewicz, K.; Jarząb, B.; Paschke, R.; Krohn, K. Analysis options for high-throughput sequencing in miRNA expression profiling. BMC Res. Notes. 2014, 7, 144. [Google Scholar] [CrossRef] [PubMed]
- Hart, T.; Komori, H.K.; LaMere, S.; Podshivalova, K.; Salomon, D.R. Finding the active genes in deep RNA-seq gene expression studies. BMC Genomics 2013, 14, 778. [Google Scholar] [CrossRef] [PubMed]
- Baras, A.S.; Mitchell, C.J.; Myers, J.R.; Gupta, S.; Weng, L.C.; Ashton, J.M.; Cornish, T.C.; Pandey, A.; Halushka, M.K. miRge—A Multiplexed Method of Processing Small RNA-Seq Data to Determine MicroRNA Entropy. PLoS ONE 2015, 10, e0143066. [Google Scholar] [CrossRef] [PubMed]
- Krämer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef]
- García-Campos, M.A.; Espinal-Enríquez, J.; Hernández-Lemus, E. Pathway Analysis: State of the Art. Front. Physiol. 2015, 6, 383. [Google Scholar] [CrossRef]
- Noble, W.S. How dose multiple testing correction work? Nat. Biotechnol. 2009, 27, 1135–1137. [Google Scholar] [CrossRef]
- Streiner, D.L. Best (but oft-forgotten) practices: The multiple problems of multiplicity-whether and how to correct for many statistical tests. Am. J. Clin Nutr. 2015, 102, 721–728. [Google Scholar] [CrossRef]
- Goldring, M.B.; Marcu, K.B. Epigenomic and microRNA-mediated regulation in cartilage development, homeostasis, and osteoarthritis. Trends Mol. Med. 2012, 18, 109–118. [Google Scholar] [CrossRef]
- Bioinformatics & Evolutionary Genomics. Available online: http://bioinformatics.psb.ugent.be/webtools/Venn/ (accessed on 27 December 2017).
- Chen, Y.; Wang, T.; Guan, M.; Zhao, W.; Leung, F.K.; Pan, H.; Cao, X.; Guo, X.E.; Lu, W.W. Bone turnover and articular cartilage differences localized to subchondral cysts in knees with advanced osteoarthritis. Osteoarthr. Cartil. 2015, 23, 2174–2183. [Google Scholar] [CrossRef] [PubMed]
- Suri, S.; Gill, S.E.; Massena de Camin, S.; Wilson, D.; McWilliams, D.F.; Walsh, D.A. Neurovascular invasion at the osteochondral junction and in osteophytes in osteoarthritis. Ann. Rheum Dis. 2007, 66, 1423–1428. [Google Scholar] [CrossRef] [PubMed]
- Jeffries, M.A.; Donica, M.; Baker, L.W.; Stevenson, M.E.; Annan, A.C.; Beth Humphrey, M.; James, J.A.; Sawalha, A.H. Genome-Wide DNA Methylation Study Identifies Significant Epigenomic Changes in Osteoarthritic Subchondral Bone and Similarity to Overlying Cartilage. Arthritis Rheumatol. 2016, 68, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Miyaki, S.; Lotz, M.K. Extracellular vesicles in cartilage homeostasis and osteoarthritis. Curr. Opin. Rheumatol. 2018, 30, 129–135. [Google Scholar] [CrossRef]
- Lin, Z.; Rodriguez, N.E.; Zhao, J.; Ramey, A.N.; Hyzy, S.L.; Boyan, B.D.; Schwartz, Z. Selective enrichment of microRNAs in extracellular matrix vesicles produced by growth plate chondrocytes. Bone 2016, 88, 47–55. [Google Scholar] [CrossRef]
- Kato, T.; Miyaki, S.; Ishitobi, H.; Nakamura, Y.; Nakasa, T.; Lotz, M.K.; Ochi, M. Exosomes from IL-1beta stimulated synovial fibroblasts induce osteoarthritic changes in articular chondrocytes. Arthritis Res. Ther. 2014, 16, R163. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, A.K. Articular cartilage vesicles and calcium crystal deposition diseases. Curr. Opin. Rheumatol. 2016, 28, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Mapp, P.I.; Walsh, D.A. Mechanisms and targets of angiogenesis and nerve growth in osteoarthritis. Nat. Rev. Rheumatol. 2012, 8, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Madej, W.; van Caam, A.; Blaney Davidson, E.N.; van der Kraan, P.M.; Buma, P. Physiological and excessive mechanical compression of articular cartilage activates Smad2/3P signaling. Osteoarthr. Cartil. 2014, 22, 1018–1025. [Google Scholar] [CrossRef]
- Tardif, G.; Pelletier, J.P.; Fahmi, H.; Hum, D.; Zhang, Y.; Kapoor, M.; Martel-Pelletier, J. NFAT3 and TGF-beta/SMAD3 regulate the expression of miR-140 in osteoarthritis. Arthritis Res. Ther. 2013, 15, R197. [Google Scholar] [CrossRef]
- Aref-Eshghi, E.; Liu, M.; Razavi-Lopez, S.B.; Hirasawa, K.; Harper, P.E.; Martin, G.; Furey, A.; Green, R.; Sun, G.; Rahman, P.; et al. SMAD3 Is Upregulated in Human Osteoarthritic Cartilage Independent of the Promoter DNA Methylation. J. Rheumatol. 2016, 43, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Guo, L.W.; Seedial, S.M.; Si, Y.; Wang, B.; Takayama, T.; Suwanabol, P.A.; Ghosh, S.; DiRenzo, D.; Liu, B.; et al. TGF-beta/Smad3 inhibit vascular smooth muscle cell apoptosis through an autocrine signaling mechanism involving VEGF-A. Cell. Death Dis. 2014, 5, e1317. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.; Katoh, M. STAT3-induced WNT5A signaling loop in embryonic stem cells, adult normal tissues, chronic persistent inflammation, rheumatoid arthritis and cancer (Review). Int J. Mol. Med. 2007, 19, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Yuasa, T.; Otani, T.; Koike, T.; Iwamoto, M.; Enomoto-Iwamoto, M. Wnt/beta-catenin signaling stimulates matrix catabolic genes and activity in articular chondrocytes: its possible role in joint degeneration. Lab. Invest. 2008, 88, 264–274. [Google Scholar] [CrossRef]
- Li, Y.; Xiao, W.; Sun, M.; Deng, Z.; Zeng, C.; Li, H.; Yang, T.; Li, L.; Luo, W.; Lei, G. The Expression of Osteopontin and Wnt5a in Articular Cartilage of Patients with Knee Osteoarthritis and Its Correlation with Disease Severity. Biomed. Res. Int. 2016, 2016, 9561058. [Google Scholar] [CrossRef]
- Huang, G.; Chubinskaya, S.; Liao, W.; Loeser, R.F. Wnt5a induces catabolic signaling and matrix metalloproteinase production in human articular chondrocytes. Osteoarthr. Cartil. 2017, 25, 1505–1515. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Schivo, S.; Huang, X.; Leijten, J.; Karperien, M.; Post, J.N. Nitric Oxide Mediates Crosstalk between Interleukin 1beta and WNT Signaling in Primary Human Chondrocytes by Reducing DKK1 and FRZB Expression. Int J. Mol. Sci. 2017, 18, E2491. [Google Scholar] [CrossRef]
- Jones, S.W.; Watkins, G.; Le Good, N.; Roberts, S.; Murphy, C.L.; Brockbank, S.M.; Needham, M.R.; Read, S.J.; Newham, P. The identification of differentially expressed microRNA in osteoarthritic tissue that modulate the production of TNF-alpha and MMP13. Osteoarthr. Cartil. 2009, 17, 464–472. [Google Scholar] [CrossRef]
- Wu, C.; Tian, B.; Qu, X.; Liu, F.; Tang, T.; Qin, A.; Zhu, Z.; Dai, K. MicroRNAs play a role in chondrogenesis and osteoarthritis (review). Int J. Mol. Med. 2014, 34, 13–23. [Google Scholar] [CrossRef]
- Miyaki, S.; Sato, T.; Inoue, A.; Otsuki, S.; Ito, Y.; Yokoyama, S.; Kato, Y.; Takemoto, F.; Nakasa, T.; Yamashita, S.; et al. MicroRNA-140 plays dual roles in both cartilage development and homeostasis. Genes Dev. 2010, 24, 1173–1185. [Google Scholar] [CrossRef]
- Miyaki, S.; Nakasa, T.; Otsuki, S.; Grogan, S.P.; Higashiyama, R.; Inoue, A.; Kato, Y.; Sato, T.; Lotz, M.K.; Asahara, H. MicroRNA-140 is expressed in differentiated human articular chondrocytes and modulates interleukin-1 responses. Arthritis Rheum. 2009, 60, 2723–2730. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.M.; Suen, W.C.; Lin, S.; Wu, X.M.; Li, G.; Pan, X.H. Dysregulation of both miR-140-3p and miR-140-5p in synovial fluid correlate with osteoarthritis severity. Bone Joint Res. 2017, 6, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Eitner, A.; Hofmann, G.O.; Schaible, H.G. Mechanisms of Osteoarthritic Pain. Studies in Humans and Experimental Models. Front. Mol. Neurosci. 2017, 10, 349. [Google Scholar] [CrossRef] [PubMed]
- Schaible, H.G. Mechanisms of chronic pain in osteoarthritis. Curr. Rheumatol. Rep. 2012, 14, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Grassel, S.G. The role of peripheral nerve fibers and their neurotransmitters in cartilage and bone physiology and pathophysiology. Arthritis Res. Ther. 2014, 16, 485. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, S.; Wibberley, H.; Mapp, P.I.; Hill, R.; Wilson, D.; Walsh, D.A. Increased vascular penetration and nerve growth in the meniscus: a potential source of pain in osteoarthritis. Ann. Rheum. Dis. 2011, 70, 523–529. [Google Scholar] [CrossRef]
- Jimenez-Andrade, J.M.; Mantyh, P.W. Sensory and sympathetic nerve fibers undergo sprouting and neuroma formation in the painful arthritic joint of geriatric mice. Arthritis Res. Ther. 2012, 14, R101. [Google Scholar] [CrossRef]
- Tatsumi, S.; Mabuchi, T.; Katano, T.; Matsumura, S.; Abe, T.; Hidaka, H.; Suzuki, M.; Sasaki, Y.; Minami, T.; Ito, S. Involvement of Rho-kinase in inflammatory and neuropathic pain through phosphorylation of myristoylated alanine-rich C-kinase substrate (MARCKS). Neuroscience 2005, 131, 491–498. [Google Scholar] [CrossRef]
- Tenti, S.; Pascarelli, N.A.; Cheleschi, S.; Guidelli, G.M.; Fioravanti, A. The Emerging Role of Bradykinin in the Pathogenesis of Osteoarthritis and its Possible Clinical Implications. Curr. Rheumatol. Rev. 2016, 12, 177–184. [Google Scholar] [CrossRef]
- Tanabe, A.; Shiraishi, M.; Negishi, M.; Saito, N.; Tanabe, M.; Sasaki, Y. MARCKS dephosphorylation is involved in bradykinin-induced neurite outgrowth in neuroblastoma SH-SY5Y cells. J. Cell. Physiol. 2012, 227, 618–629. [Google Scholar] [CrossRef]
- Gruber, H.E.; Hoelscher, G.L.; Bullock, L.; Ingram, J.A.; Norton, H.J.; Hanley, E.N., Jr. Human annulus signaling cues for nerve outgrowth: In vitro studies. J. Orthop. Res. 2016, 34, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Grimsholm, O.; Guo, Y.; Ny, T.; Forsgren, S. Expression patterns of neurotrophins and neurotrophin receptors in articular chondrocytes and inflammatory infiltrates in knee joint arthritis. Cells Tissues Organs. 2008, 188, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Yan, F.; Deng, Q.; Li, F.; Lu, Z.; Liu, M.; Wang, L.; Conklin, D.J.; McCracken, J.; Srivastava, S.; et al. Modulation of tumorigenesis by the pro-inflammatory microRNA miR-301a in mouse models of lung cancer and colorectal cancer. Cell. Discov. 2015, 1, 15005. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Yin, K.; Zhu, H.; Tian, J.; Shen, D.; Yi, L.; Rui, K.; Ma, J.; Xu, H.; Wang, S. Correlation Between the Expression of MicroRNA-301a-3p and the Proportion of Th17 Cells in Patients with Rheumatoid Arthritis. Inflammation. 2016, 39, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Qi, J.; Bi, Q.; Zhang, S. Suppression of miR-301a alleviates LPS-induced inflammatory injury in ATDC5 chondrogenic cells by targeting Sirt1. Int J. Clin Exp. Pathol. 2017, 10, 8991–9000. [Google Scholar]
- Riese Ⅱ, D.J.; Cullum, R.L. Epiregulin: Roles in normal physiology and cancer. Semin. Cell Dev. Biol. 2014, 28, 49–56. [Google Scholar] [CrossRef]
- Martin, L.J.; Smith, S.B.; Khoutorsky, A.; Magnussen, C.A.; Samoshkin, A.; Sorge, R.E.; Cho, C.; Yosefpour, N.; Sivaselvachandran, S.; Tohyama, S.; et al. Epiregulin and EGFR interactions are involved in pain processing. J. Clin. Invest. 2017, 127, 3353–3366. [Google Scholar] [CrossRef] [PubMed]
- Lahoti, T.S.; Hughes, J.M.; Kusnadi, A.; John, K.; Zhu, B.; Murray, I.A.; Gowda, K.; Peters, J.M.; Amin, S.G.; Perdew, G.H. Aryl hydrocarbon receptor antagonism attenuates growth factor expression, proliferation, and migration in fibroblast-like synoviocytes from patients with rheumatoid arthritis. J. Pharmacol. Exp. Ther. 2014, 348, 236–245. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | No. of Genes | p-Value |
|---|---|---|
| Top canonical pathways | ||
| Osteoarthritis Pathway | 23 | 4.72 × 10−10 |
| Hepatic Fibrosis/Hepatic Stellate Cell Activation | 20 | 8.99 × 10−9 |
| Axonal Guidance Signaling | 31 | 4.32 × 10−8 |
| Atherosclerosis Signaling | 15 | 1.77 × 10−7 |
| GP6 Signaling Pathway | 13 | 1.06 × 10−5 |
| Top predicted upstream regulators | ||
| TNF | 2.20 × 10−22 | |
| TGFB1 | 1.01 × 10−21 | |
| Dexamethasone | 1.57 × 10−16 | |
| IFNG | 6.75 × 10−16 | |
| CTNNB1 | 1.50 × 10−15 |
| Biological Process | Count | p-Value | Up-Regulated Genes | Down-Regulated Genes | Fold Enrichment |
|---|---|---|---|---|---|
| Positive regulation of bone mineralization | 7 | 2.66 × 10−4 | BMP4, GPM6B, P2RX7, SMAD3, TMEM119 | BMP2, BMP6 | 7.62 |
| Positive regulation of chondrocyte differentiation | 5 | 1.32 × 10−3 | ACVRL1, GDF6,SMAD3 | BMP6, SOX9 | 10.02 |
| Positive regulation of cartilage development | 4 | 7.76 × 10−3 | BMP4, WNT5A | BMP2, SOX9 | 9.52 |
| Chondrocyte differentiation | 5 | 1.86 × 10−2 | BMP4 | BMP2, COL11A2, CYTL1, FGFR3 | 4.88 |
| Endochondral ossification | 4 | 2.97 × 10−2 | ALPL, BMP4 | BMP6, FGFR3 | 5.86 |
| Gene Symbol | Gene Name | HC-OA FPKM | HC FPKM | Fold-Change (HC-OA/HC) |
|---|---|---|---|---|
| AKAP12 | A-kinase anchoring protein 12 | 94.69 | 32.02 | 2.96 |
| BASP1 | brain abundant, membrane attached signal protein 1 | 80.88 | 38.09 | 2.12 |
| BDNF | brain-derived neurotrophic factor | 8.28 | 1.76 | 4.70 |
| FGF7 | fibroblast growth factor 7 | 37.21 | 8.52 | 4.37 |
| GREM2 | gremlin 2, DAN family BMP antagonist | 9.66 | 3.54 | 2.73 |
| LMOD1 | leiomodin 1 | 26.03 | 1.78 | 14.65 |
| LRRC15 | leucine rich repeat containing 15 | 6.47 | 2.56 | 2.53 |
| LRRC32 | leucine rich repeat containing 32 | 9.64 | 2.30 | 4.19 |
| MARCKS | myristoylated alanine-rich protein kinase C substrate | 278.54 | 133.99 | 2.08 |
| PCSK9 | proprotein convertase subtilisin/kexin type 9 | 1.51 | 0.53 | 2.86 |
| PDE3A | phosphodiesterase 3A | 19.30 | 1.88 | 10.25 |
| PDE7B | phosphodiesterase 7B | 4.98 | 1.53 | 3.27 |
| PPM1L | protein phosphatase, Mg2+/Mn2+ dependent 1L | 4.44 | 0.97 | 4.59 |
| RALGPS2 | Ral GEF with PH domain and SH3 binding motif 2 | 48.94 | 18.20 | 2.69 |
| RGS5 | regulator of G-protein signaling 5 | 90.50 | 6.24 | 14.50 |
| RNF152 | ring finger protein 152 | 15.71 | 3.83 | 4.10 |
| RPP25 | ribonuclease P/MRP 25kDa subunit | 4.69 | 2.02 | 2.32 |
| SESN3 | sestrin 3 | 14.57 | 6.95 | 2.10 |
| SLIT3 | slit guidance ligand 3 | 29.08 | 2.46 | 11.80 |
| SMAD3 | SMAD family member 3 | 408.29 | 166.01 | 2.46 |
| TFPI | tissue factor pathway inhibitor | 56.10 | 8.72 | 6.43 |
| THSD4 | thrombospondin type 1 domain containing 4 | 3.44 | 0.65 | 5.31 |
| KIAA1644 | KIAA1644 | 22.55 | 61.04 | 0.37 |
| SEMA3A | semaphorin 3A | 12.52 | 39.96 | 0.31 |
| EREG | epiregulin | 1.50 | 4.65 | 0.32 |
| SDK2 | sidekick cell adhesion molecule 2 | 2.04 | 15.99 | 0.13 |
| GEO Accession Number | GSE114007 | GSE51588 | GSE55235 | GSE55457 | |
|---|---|---|---|---|---|
| Specimen | Cartilage | Subchondral bone | Synovial tissue | ||
| Normal/OA | Normal/OA | Normal/OA | Normal/OA | ||
| Medial | Lateral | ||||
| Numbers | 18/20 | 5/20 | 5/20 | 10/10 | 10/10 |
| Up-Regulated mRNA * | |||||
| AKAP12 | DOWN | n.s. † | n.s. | n.s. | n.s. |
| BASP1 | UP | n.s. | DOWN | n.s. | n.s. |
| BDNF | UP | n.s. | n.s. | n.s. | n.s. |
| FGF7 | UP | n.s. | n.s.† | n.s. | n.s. |
| GREM2 | n.s. | n.s. | n.s. | n.s. | n.s. |
| LMOD1 | n.s. | n.s. | n.s. | DOWN | n.s. |
| LRRC15 | UP | UP | n.s. | UP | UP |
| LRRC32 | n.s. | n.s. | n.s. | n.s. | n.s. |
| MARCKS | UP | UP | UP | UP | n.s. † |
| PCSK9 | UP | UP | n.s. | -- | -- |
| PDE3A | UP | UP | UP | n.s. | n.s. |
| PDE7B | n.s. | n.s. | n.s. | DOWN | n.s. |
| PPM1L | n.s. | n.s. | n.s. | -- | -- |
| RALGPS2 | n.s. | n.s. | n.s. | n.s. | n.s. |
| RGS5 | n.s. | n.s. | n.s. | n.s. | n.s. |
| RNF152 | n.s. | n.s. | n.s. | -- | -- |
| RPP25 | n.s. | UP | n.s. | n.s. | n.s. |
| SESN3 | n.s. | n.s. | n.s. | -- | -- |
| SLIT3 | n.s. | UP | n.s. † | n.s. † | n.s. † |
| SMAD3 | DOWN | UP | UP | n.s. † | n.s. † |
| TFPI | n.s. | n.s. † | n.s. | DOWN | n.s. |
| THSD4 | DOWN | UP | UP | n.s. | n.s. |
| Down-Regulated mRNA * | |||||
| KIAA1644 | UP | n.s. † | n.s. | UP | n.s. |
| SEMA3A | n.s. | n.s. | n.s. | UP | n.s. |
| EREG | n.s. | DOWN | DOWN | DOWN | n.s. |
| SDK2 | n.s. | UP | UP | n.s. | n.s. |
| Down-Regulated miRNA | Fold-Change | Predicted Target Up-Regulated mRNA | miRmap Score | TargetScan | miRDB |
| hsa-miR-140-5p | −3.11 | LRRC15 | 99.03 | − | − |
| hsa-miR-140-3p | −2.87 | MARCKS | 99.27 | + | + |
| hsa-miR-495-3p | −4.80 | PDE3A | 99.93 | − | + |
| Up-Regulated miRNA | Fold-Change | Predicted Target Down-Regulated mRNA | miRmap Score | TargetScan | miRDB |
| hsa-miR-301a-3p | 2.45 | EREG | 99.06 | + | + |
| Biological Process | p-Value | Related Genes | Fold Enrichment |
|---|---|---|---|
| Negative regulation of TORC1 signaling | 0.013 | RNF152, SESN3 | 149.26 |
| Cytokine-mediated signaling pathway | 0.015 | EREG, LRRC15, GREM2 | 15.38 |
| Axon extension involved in axon guidance | 0.017 | SEMA3A, SLIT3 | 111.95 |
| cAMP catabolic process | 0.021 | PDE7B, PDE3A | 89.56 |
| Axon guidance | 0.021 | BDNF, SEMA3A, SLIT3 | 12.67 |
| Oocyte maturation | 0.025 | EREG, PDE3A | 74.63 |
| Positive regulation of GTPase activity | 0.045 | RALGPS2, FGF7, EREG, RGS5 | 4.76 |
| Negative chemotaxis | 0.048 | SEMA3A, SLIT3 | 39.51 |
| MAPK cascade | 0.053 | FGF7, EREG, PPM1L | 7.69 |
| Positive regulation of cell division | 0.065 | FGF7, EREG | 28.58 |
| Synapse assembly | 0.084 | BDNF, SDK2 | 22.02 |
| Top Diseases and Functions | Score | Focus Molecules | Molecules in Network | |
|---|---|---|---|---|
| 1 | Cellular Movement, Cardiovascular System Development and Function, Organismal Development | 32 | 13 | ↑AKAP12, Akt, Ap1, ↑BDNF, Calmodulin, Creb, ↓EREG, ERK,ERK1/2, estrogen receptor, F Actin, ↑FGF7, Fibrinogen, FSH, Gsk3, Histone h3, LDL, Lh, ↑LRRC32, Mapk, ↑MARCKS, ↑PCSK9, PDGF BB, PI3K (complex), Pka, Pkc(s), Proinsulin, Ras, ↑RGS5, ↓SEMA3A, ↑SESN3, ↑SLIT3, ↑SMAD3, ↑TFPI, Vegf |
| 2 | Cardiovascular Disease, Organismal Injury and Abnormalities, Cardiovascular System Development and Function | 15 | 7 | 26s Proteasome, BMP2, CLU, F10, G protein alphai, GAS6, ↑GREM2, HNF4A, Jnk, LPA, LRPAP1, ↑LRRC15, MAGI3, MAP2K5, MAP3K, MAPK9, mir-25, mir-181, MMP9, MMP12, MZB1, Neurotrophin, NFkB (complex), NRG (family), P38 MAPK, PP1/PP2A, Pp2c, ↑PPM1L, PTPN13, ↑RALGPS2, ↑RNF152, SAA, ↓SDK2, ↑THSD4, tyrosine kinase |
| 3 | Gastrointestinal Disease, Hepatic System Disease, Organismal Injury and Abnormalities | 12 | 6 | ABCC4, AR, ↑BASP1, C2CD5, CENPE, CENPH, CEPT1, EGR3, ELAVL1, GADD45GIP1, INPP5K, ↓KIAA1644, KIF11, KIFC3, LMNB1, ↑LMOD1, miR-149-3p, miR-185-5p, MRRF, NONO, Pde, ↑PDE3A, PDE4A, PDE5A, ↑PDE7B, PFKFB3, PITX3, PLBD2, PRMT1, PSMF1, Rb, RNF14, ↑RPP25, TAF12, ZNF281 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-J.; Chang, W.-A.; Wu, L.-Y.; Hsu, Y.-L.; Chen, C.-H.; Kuo, P.-L. Systematic Analysis of Transcriptomic Profile of Chondrocytes in Osteoarthritic Knee Using Next-Generation Sequencing and Bioinformatics. J. Clin. Med. 2018, 7, 535. https://doi.org/10.3390/jcm7120535
Chen Y-J, Chang W-A, Wu L-Y, Hsu Y-L, Chen C-H, Kuo P-L. Systematic Analysis of Transcriptomic Profile of Chondrocytes in Osteoarthritic Knee Using Next-Generation Sequencing and Bioinformatics. Journal of Clinical Medicine. 2018; 7(12):535. https://doi.org/10.3390/jcm7120535
Chicago/Turabian StyleChen, Yi-Jen, Wei-An Chang, Ling-Yu Wu, Ya-Ling Hsu, Chia-Hsin Chen, and Po-Lin Kuo. 2018. "Systematic Analysis of Transcriptomic Profile of Chondrocytes in Osteoarthritic Knee Using Next-Generation Sequencing and Bioinformatics" Journal of Clinical Medicine 7, no. 12: 535. https://doi.org/10.3390/jcm7120535
APA StyleChen, Y.-J., Chang, W.-A., Wu, L.-Y., Hsu, Y.-L., Chen, C.-H., & Kuo, P.-L. (2018). Systematic Analysis of Transcriptomic Profile of Chondrocytes in Osteoarthritic Knee Using Next-Generation Sequencing and Bioinformatics. Journal of Clinical Medicine, 7(12), 535. https://doi.org/10.3390/jcm7120535