Pterostilbene Attenuates Hexavalent Chromium-Induced Allergic Contact Dermatitis by Preventing Cell Apoptosis and Inhibiting IL-1β-Related NLRP3 Inflammasome Activation

 ,
,
Abstract
1. Introduction
2. Experimental Section
2.1. Reagents
2.2. Mice
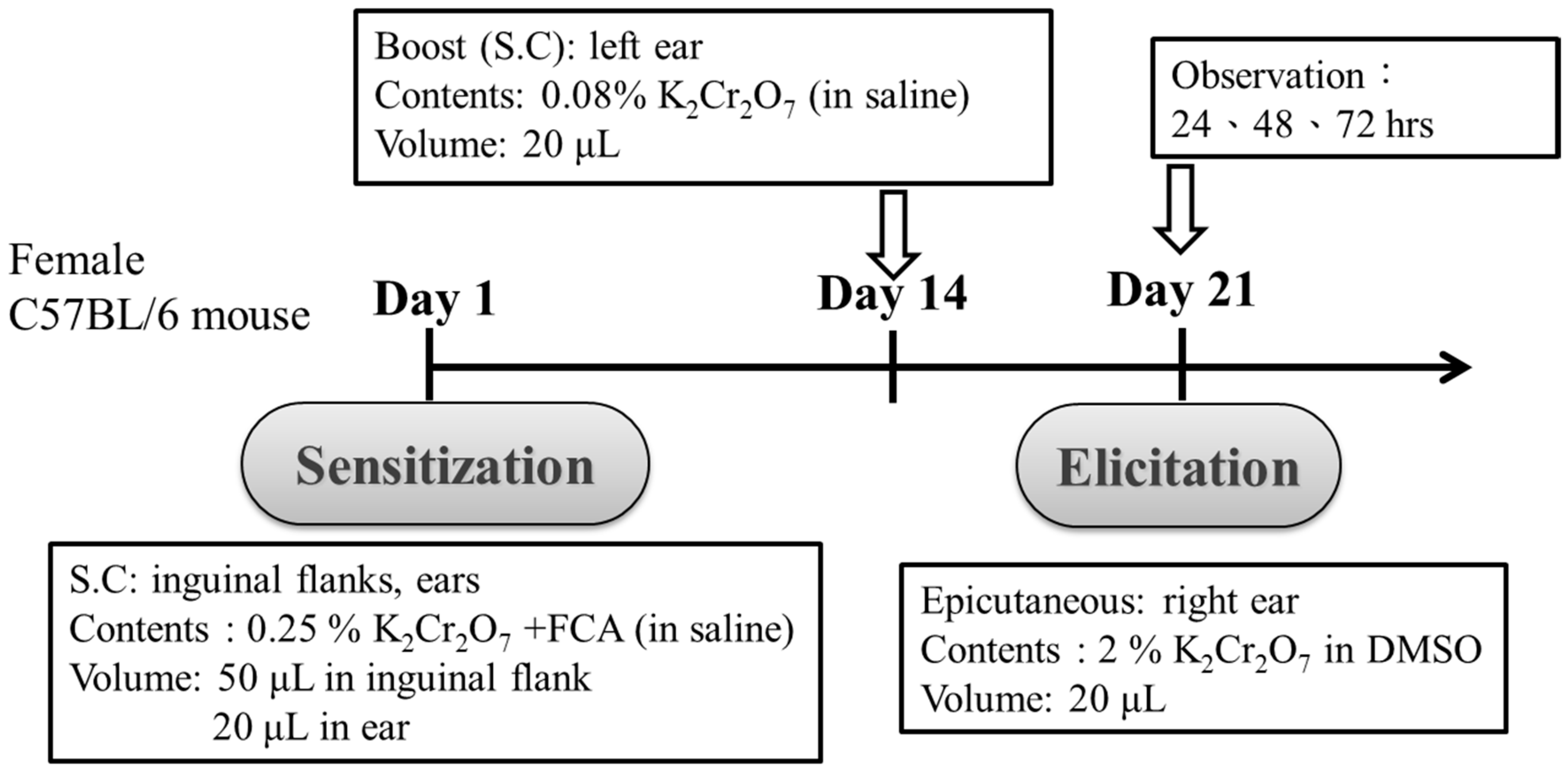
2.3. Administration of PT and Sensitization of Mice
2.4. Challenge for Recall and the Epicutaneous Elicitation Test
2.5. Immunohistochemical (IHC) Staining Analysis
2.6. Cells Culture
2.7. MTT Cell Viability Assay
2.8. Cell Cycle Assay
2.9. Determination of Apoptosis Using Annexin V Staining
2.10. ROS Analysis
2.11. 2,2-Diphenyl-1-Picrylhydrazyl (DPPH) Radical Scavenging Activity
2.12. ER Stress Measurements
2.13. Western Blot Analysis
2.14. Real-Time Quantitative Polymerase Chain Reaction (qPCR)
2.15. Statistical Analysis
3. Results
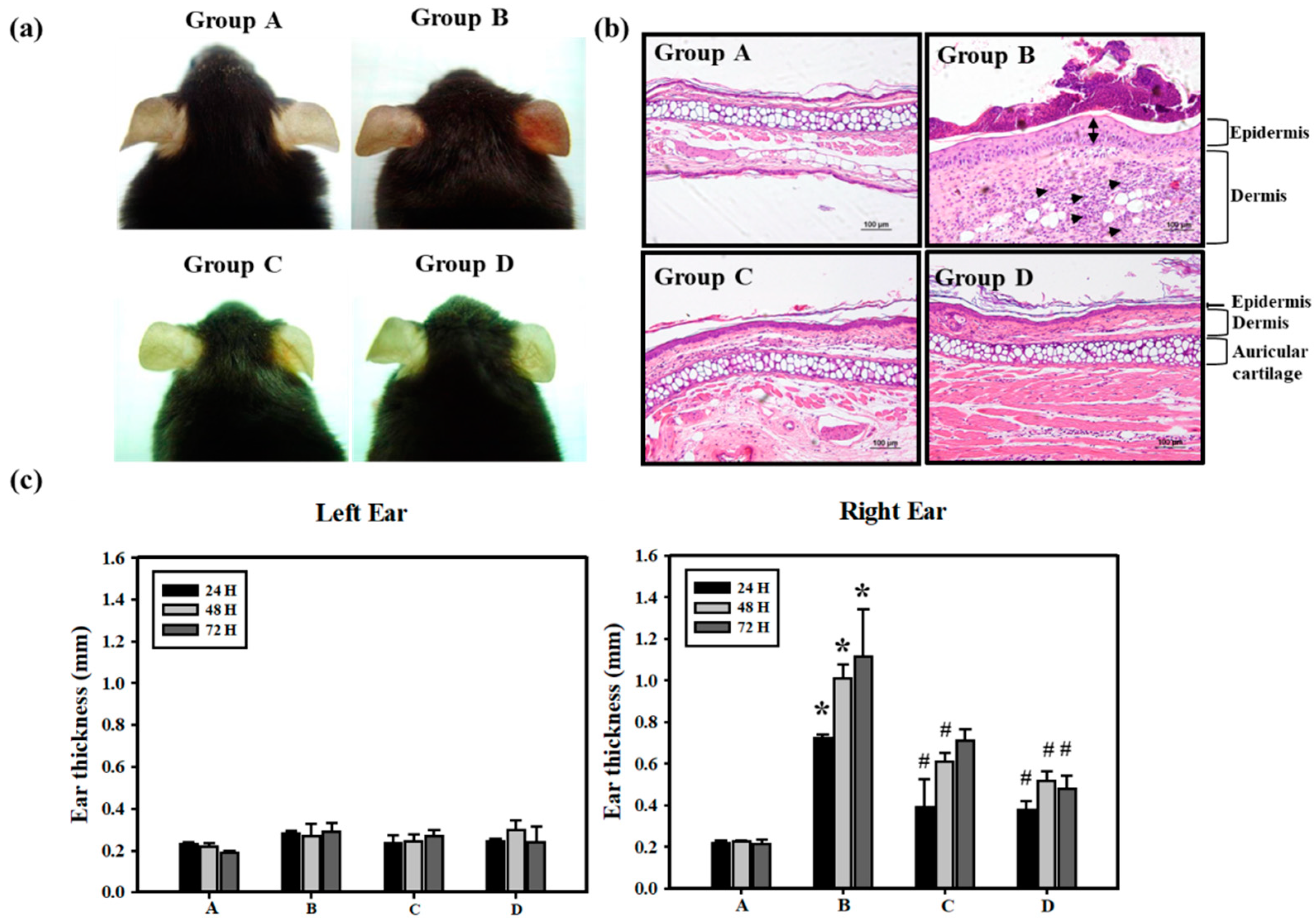
3.1. Pterostilbene (PT) Prevented Hexavalent Chromium (Cr(VI))-Induced Allergic Contact Dermatitis
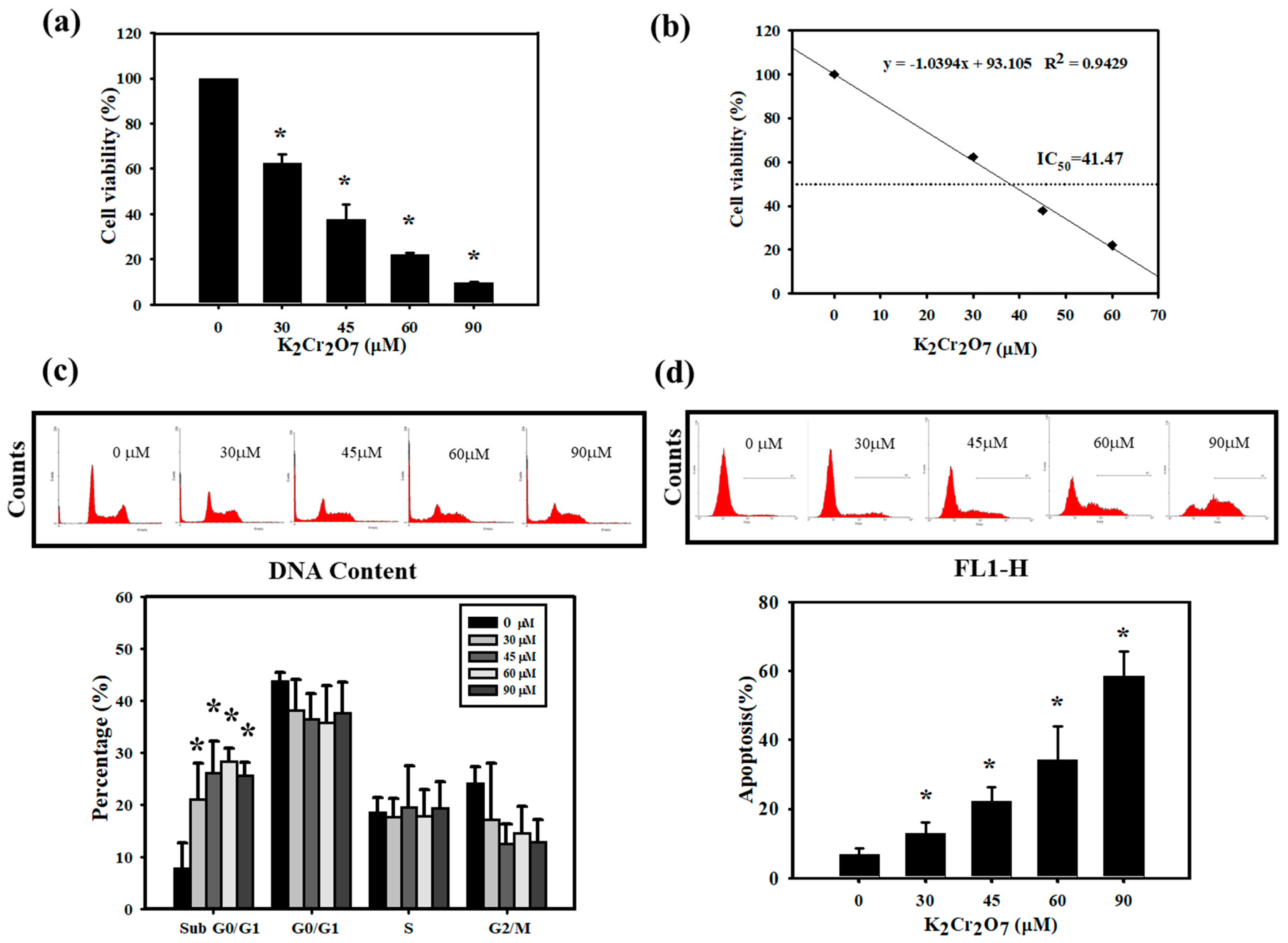
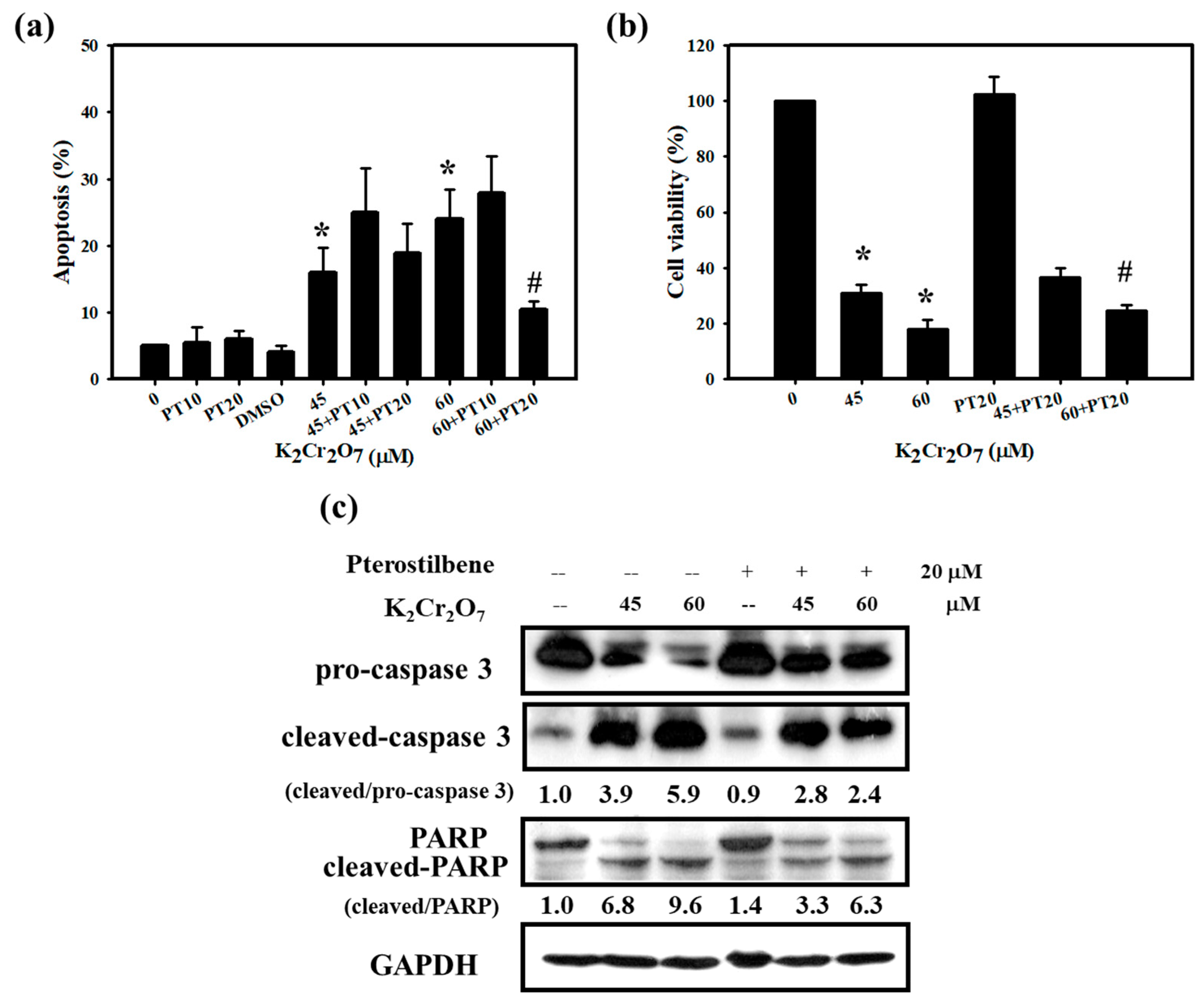
3.2. Cr(VI)-Triggered Apoptosis was Eliminated by PT in HaCaT Cells
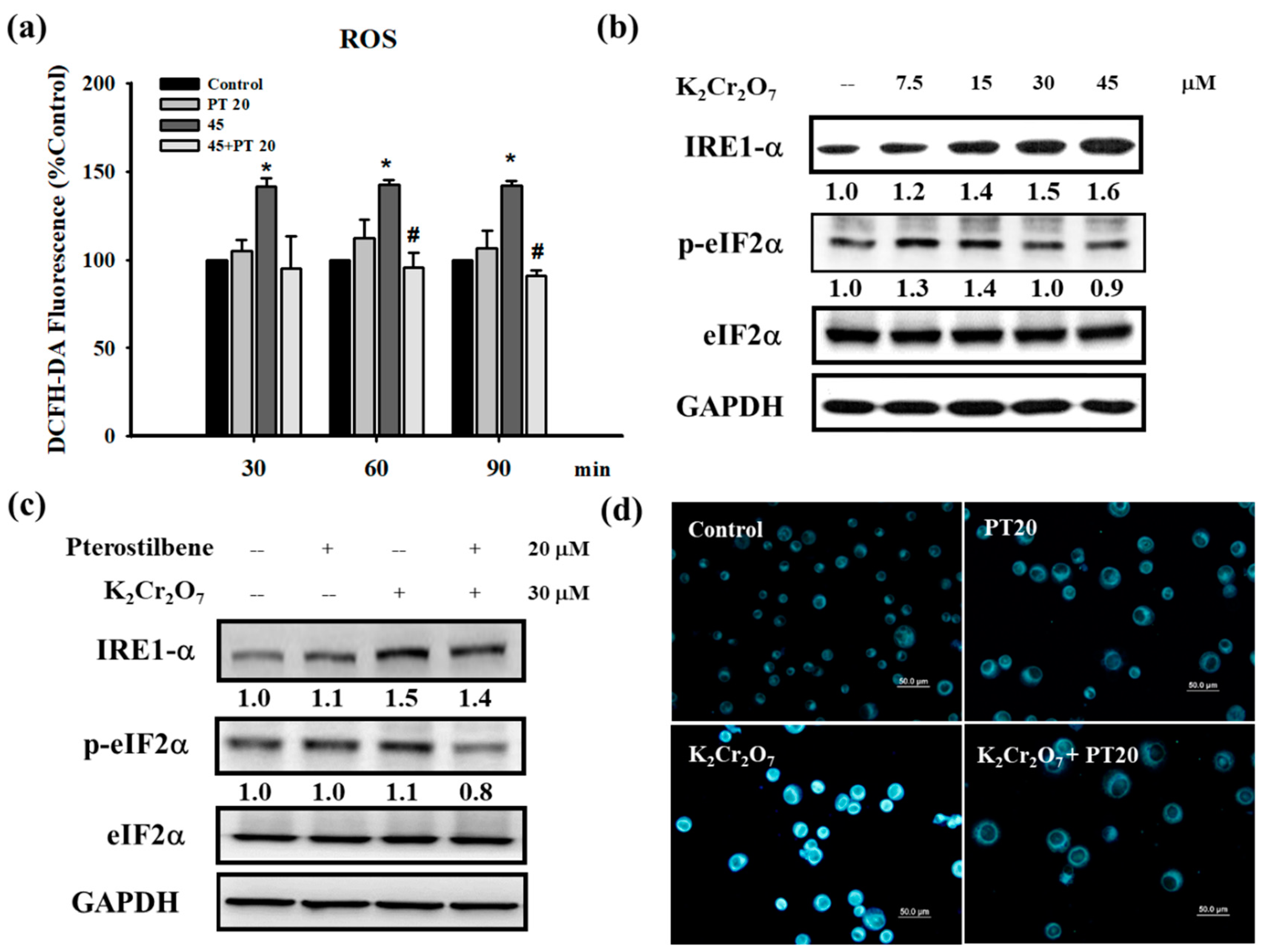
3.3. Cr(VI) Treatment Induced Reactive Oxygen Species (ROS) Generation and Endoplasmic Reticulum (ER) Stress in HaCaT Cells, Which were Attenuated by PT
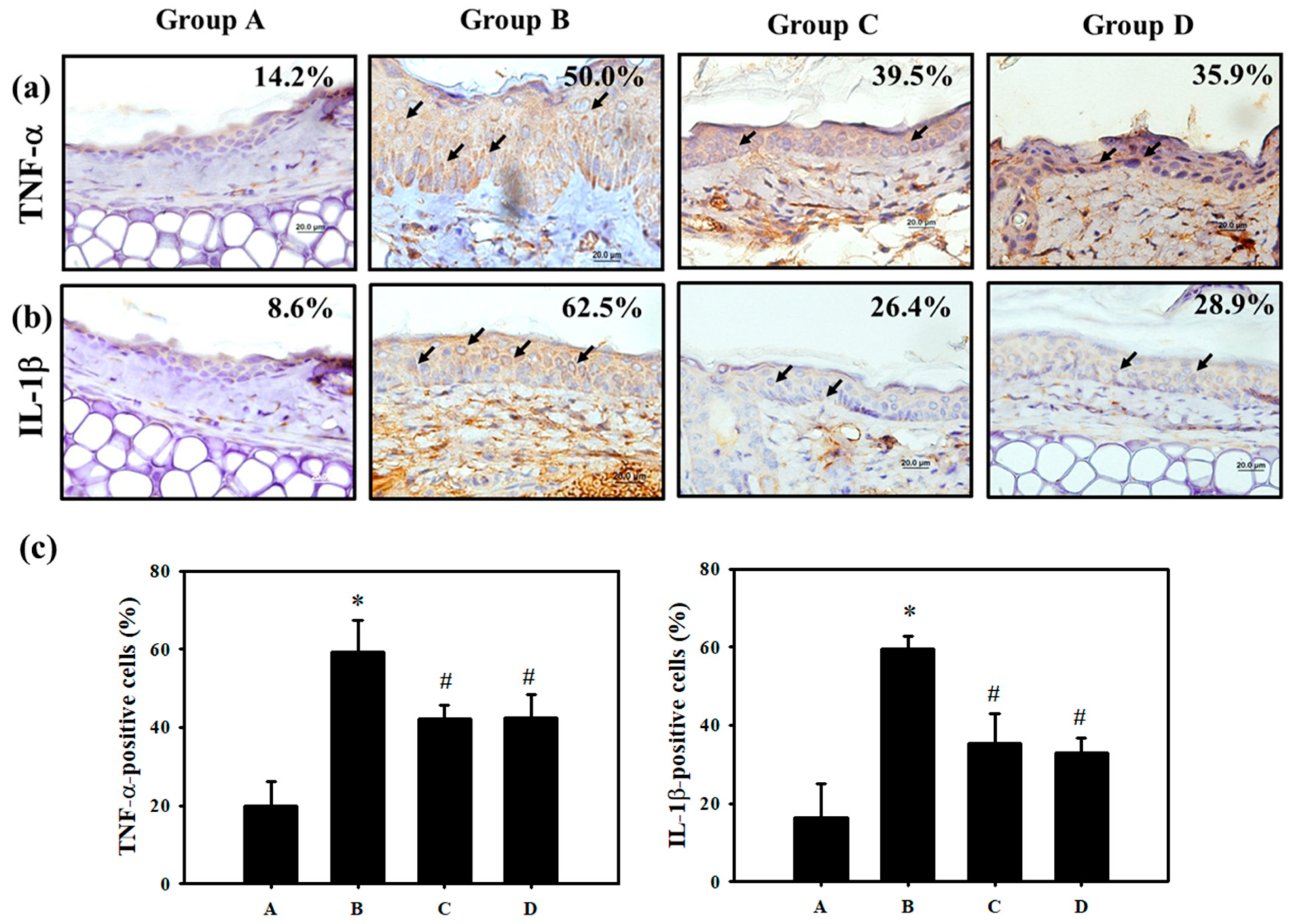
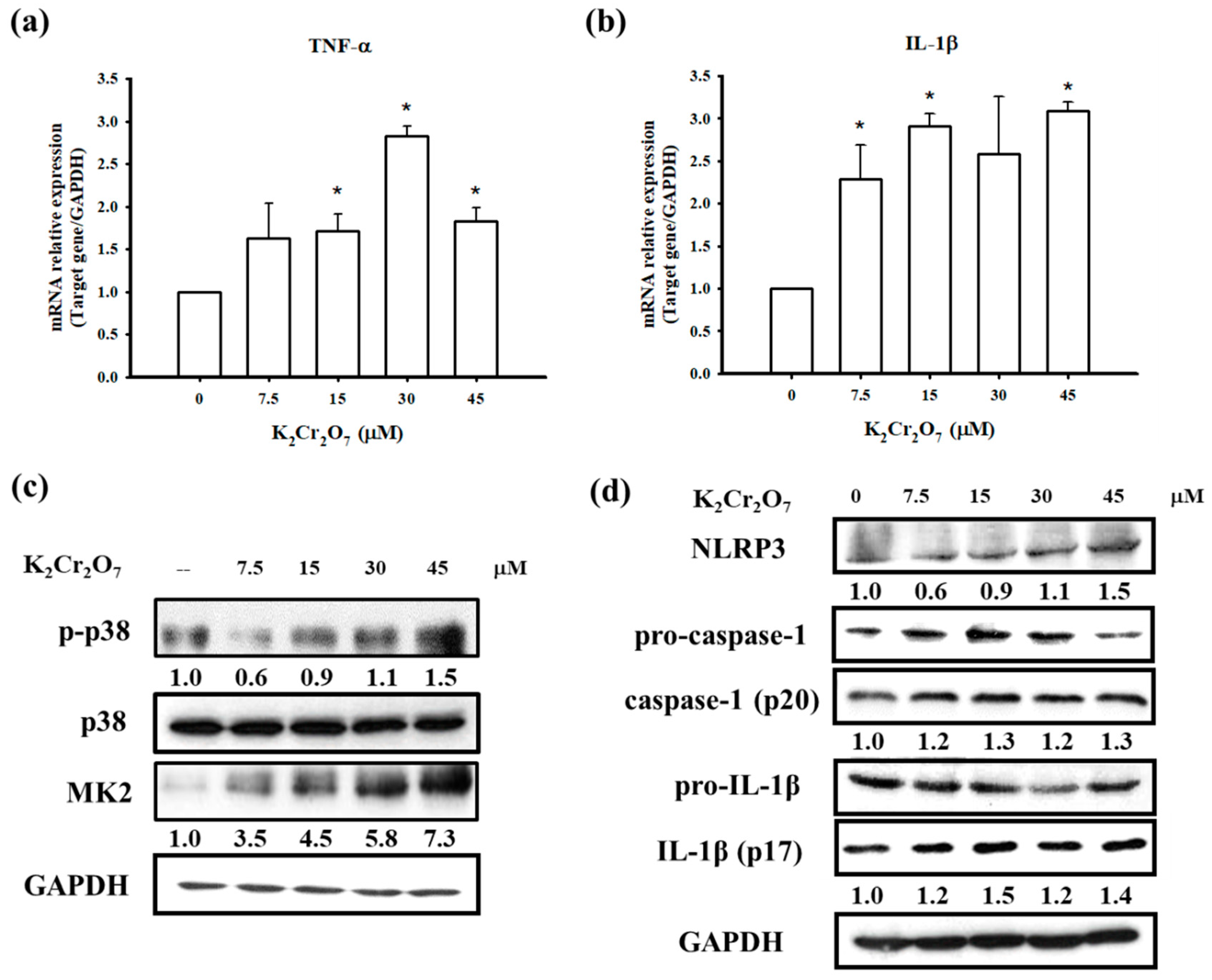
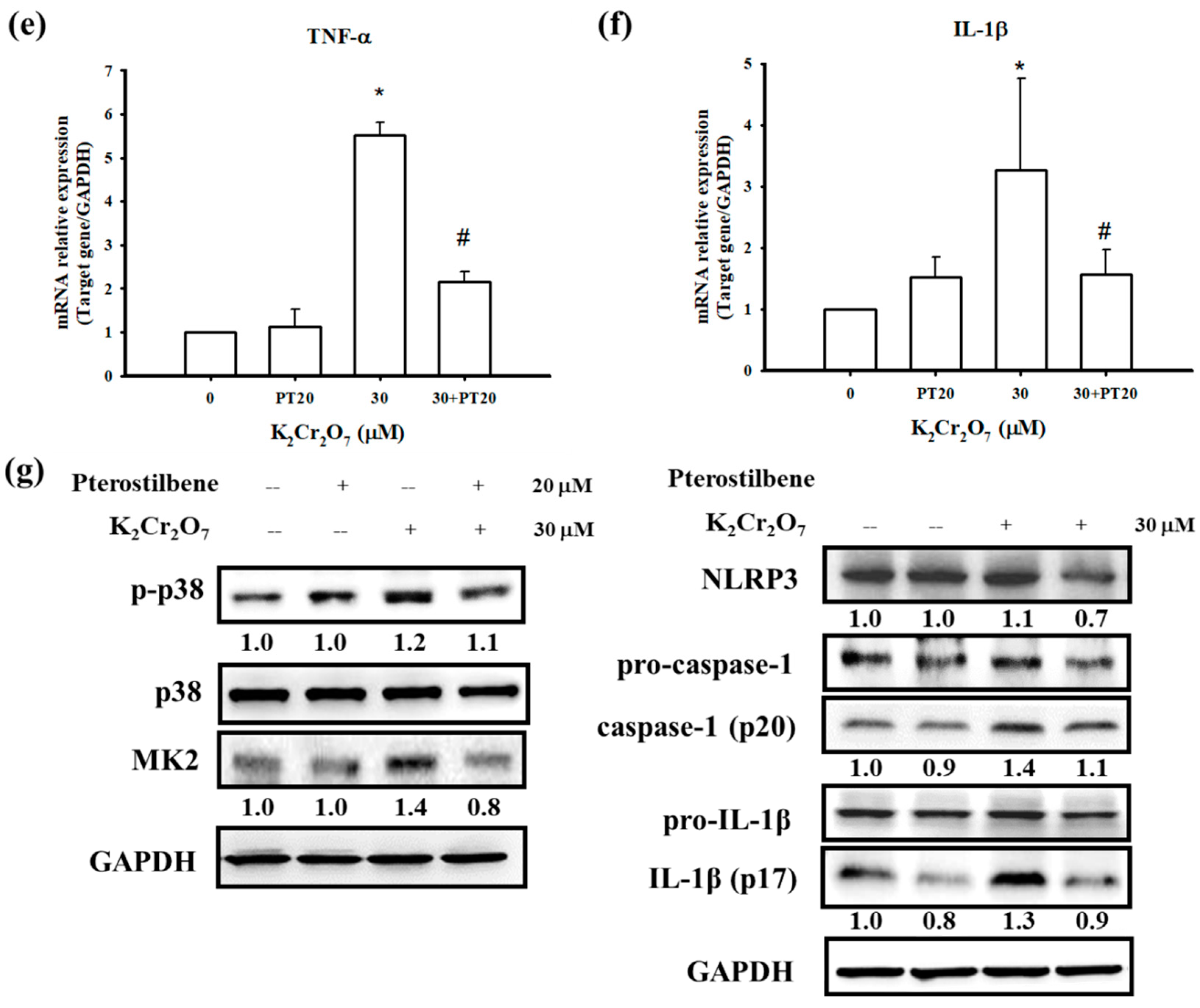
3.4. PT Inhibited Pro-Inflammatory Cytokines Production by Regulating p38 MAPK/ MK2 Pathway and the NLRP3-Inflammasome
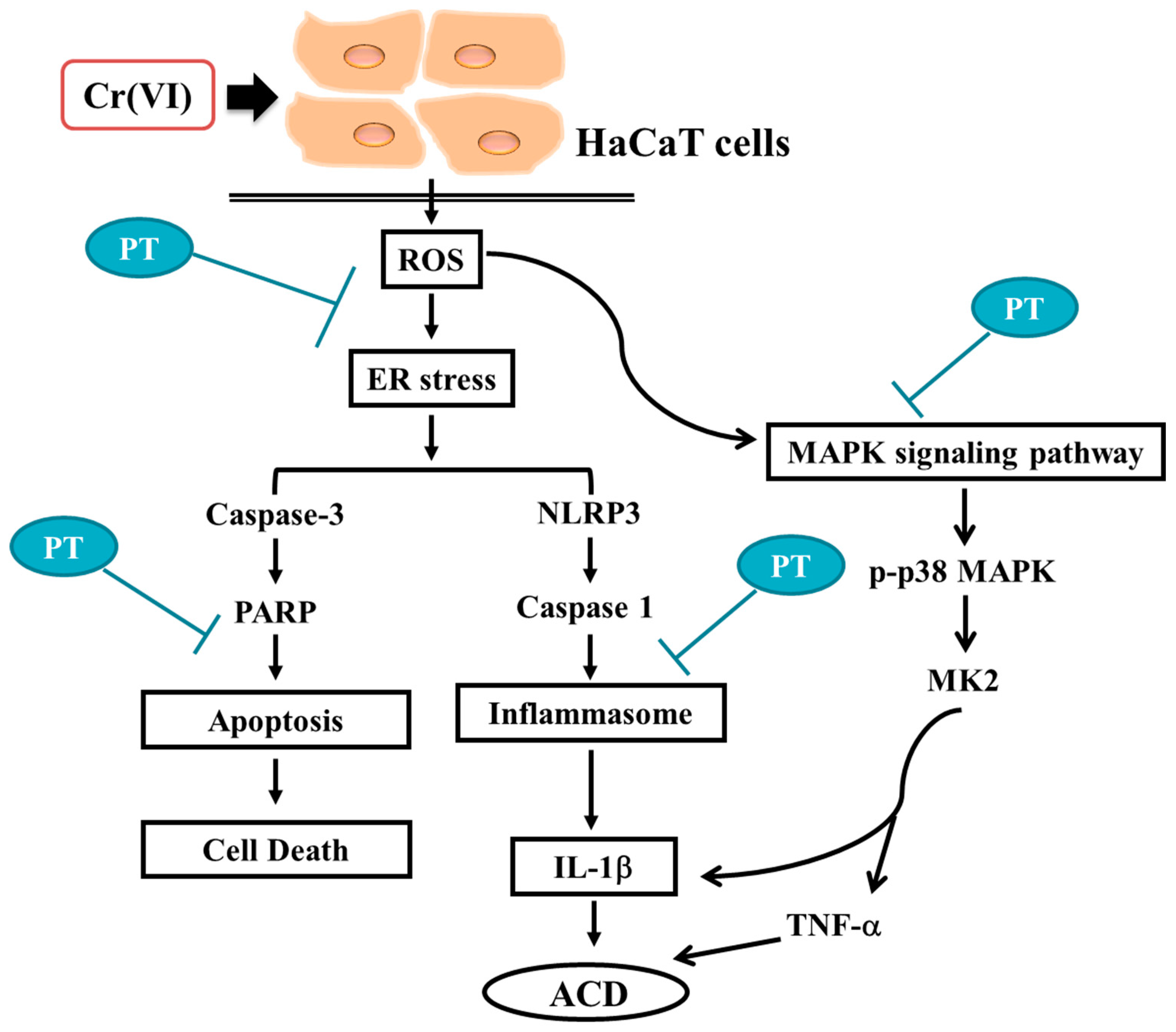
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shelnutt, S.R.; Goad, P.; Belsito, D.V. Dermatological toxicity of hexavalent chromium. Crit. Rev. Toxicol. 2007, 37, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Goh, C.L.; Gan, S.L.; Ngui, S.J. Occupational dermatitis in a prefabrication construction factory. Contact Dermat. 1986, 15, 235–240. [Google Scholar] [CrossRef]
- Guo, Y.L.; Wang, B.J.; Yeh, K.C.; Wang, J.C.; Kao, H.H.; Wang, M.T.; Shih, H.C.; Chen, C.J. Dermatoses in cement workers in southern Taiwan. Contact Dermat. 1999, 40, 1–7. [Google Scholar] [CrossRef]
- Toncic, R.J.; Lipozencic, J.; Martinac, I.; Greguric, S. Immunology of allergic contact dermatitis. Acta Dermatovenerol. Croat. 2011, 19, 51–68. [Google Scholar] [PubMed]
- Fuchs, J.; Zollner, T.M.; Kaufmann, R.; Podda, M. Redox-modulated pathways in inflammatory skin diseases. Free Radic. Biol. Med. 2001, 30, 337–353. [Google Scholar] [CrossRef]
- Matos, T.J.; Duarte, C.B.; Goncalo, M.; Lopes, M.C. Role of oxidative stress in ERK and p38 MAPK activation induced by the chemical sensitizer DNFB in a fetal skin dendritic cell line. Immunol. Cell Biol. 2005, 83, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Chao, W.; Deng, J.-S.; Huang, S.-S.; Li, P.-Y.; Liang, Y.-C.; Huang, G.-J. 3,4-dihydroxybenzalacetone attenuates lipopolysaccharide-induced inflammation in acute lung injury via down-regulation of MMP-2 and MMP-9 activities through suppressing ROS-mediated MAPK and PI3K/AKT signaling pathways. Int. Immunopharmacol. 2017, 50, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Najmi, A.K.; Dastidar, S.G. Biological functions and role of mitogen-activated protein kinase activated protein kinase 2 (MK2) in inflammatory diseases. Pharmacol. Rep. 2017, 69, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.J.; Lee, Y.H.; Yeh, Y.L.; Wang, Y.J.; Wang, B.J. The Roles of Autophagy and the Inflammasome during Environmental Stress-Triggered Skin Inflammation. Int. J. Mol. Sci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Corsini, E.; Galbiati, V.; Nikitovic, D.; Tsatsakis, A.M. Role of oxidative stress in chemical allergens induced skin cells activation. Food Chem. Toxicol. 2013, 61, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.J.; Sheu, H.M.; Guo, Y.L.; Lee, Y.H.; Lai, C.S.; Pan, M.H.; Wang, Y.J. Hexavalent chromium induced ROS formation, Akt, NF-kappaB, and MAPK activation, and TNF-alpha and IL-1alpha production in keratinocytes. Toxicol. Lett. 2010, 198, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Su, S.B.; Huang, C.C.; Sheu, H.M.; Tsai, J.C.; Lin, C.H.; Wang, Y.J.; Wang, B.J. N-acetylcysteine attenuates hexavalent chromium-induced hypersensitivity through inhibition of cell death, ROS-related signaling and cytokine expression. PLoS ONE 2014, 9, e108317. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xiao, F.; Liu, X.; Liu, K.; Zhou, X.; Zhong, C. Cr(VI) induces cytotoxicity in vitro through activation of ROS-mediated endoplasmic reticulum stress and mitochondrial dysfunction via the PI3K/Akt signaling pathway. Toxicol. In Vitro 2017, 41, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Kaufman, R.J. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008, 454, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.E.; Takagi, Y.; Nishizaka, T.; Baek, J.H.; Kim, H.J.; Lee, S.H. Subclinical cutaneous inflammation remained after permeability barrier disruption enhances UV sensitivity by altering ER stress responses and topical pseudoceramide prevents them. Arch. Dermatol. Res. 2017, 309, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed]
- Raj, D.; Brash, D.E.; Grossman, D. Keratinocyte apoptosis in epidermal development and disease. J. Investig. Dermatol. 2006, 126, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Osorio, F.; Lambrecht, B.N.; Janssens, S. Antigen presentation unfolded: Identifying convergence points between the UPR and antigen presentation pathways. Curr. Opin. Immunol. 2018, 52, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Kepp, O.; Kroemer, G. Decoding cell death signals in inflammation and immunity. Cell 2010, 140, 798–804. [Google Scholar] [CrossRef] [PubMed]
- McCormack, D.; McFadden, D. A review of pterostilbene antioxidant activity and disease modification. Oxid. Med. Cell Longev. 2013, 2013, 575482. [Google Scholar] [CrossRef] [PubMed]
- Perecko, T.; Jancinova, V.; Drabikova, K.; Nosal, R.; Harmatha, J. Structure-efficiency relationship in derivatives of stilbene. Comparison of resveratrol, pinosylvin and pterostilbene. Neuro. Endocrinol. Lett. 2008, 29, 802–805. [Google Scholar] [PubMed]
- Dvorakova, M.; Landa, P. Anti-inflammatory activity of natural stilbenoids: A review. Pharmacol. Res. 2017, 124, 126–145. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.J.; Lee, Y.H.; Yeh, Y.L.; Wu, W.S.; Ho, C.T.; Li, C.Y.; Wang, B.J.; Wang, Y.J. Autophagy-inducing effect of pterostilbene: A prospective therapeutic/preventive option for skin diseases. J. Food Drug Anal. 2017, 25, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Grealish, M.P.; Jung, M.K.; Hamel, E.; Pettit, R.K.; Chapuis, J.C.; Schmidt, J.M. Antineoplastic agents. 465. Structural modification of resveratrol: Sodium resverastatin phosphate. J. Med. Chem. 2002, 45, 2534–2542. [Google Scholar] [CrossRef] [PubMed]
- Yeo, S.C.; Ho, P.C.; Lin, H.S. Pharmacokinetics of pterostilbene in Sprague-Dawley rats: The impacts of aqueous solubility, fasting, dose escalation, and dosing route on bioavailability. Mol. Nutr. Food Res. 2013, 57, 1015–1025. [Google Scholar] [CrossRef] [PubMed]
- van Hoogstraten, I.M.; de Groot, J.; Boden, D.; von Blomberg, B.M.; Kraal, G.; Scheper, R.J. Development of a concomitant nickel and chromium sensitization model in the guinea pig. Int. Arch Allergy Immunol. 1992, 97, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Abdelrahim, M.; Newman, K.; Vanderlaag, K.; Samudio, I.; Safe, S. 3,3′-Diindolylmethane (DIM) and its derivatives induce apoptosis in pancreatic cancer cells through endoplasmic reticulum stress-dependent upregulation of DR5. Carcinogenesis 2006, 27, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Avnstorp, C. Cement eczema. An epidemiological intervention study. Acta. Derm. Venereol. Suppl. (Stockh) 1992, 179, 1–22. [Google Scholar] [PubMed]
- Crowley, L.C.; Chojnowski, G.; Waterhouse, N.J. Measuring the DNA content of cells in apoptosis and at different cell-cycle stages by propidium iodide staining and flow cytometry. Cold Spring Harb. Protoc. 2016. [Google Scholar] [CrossRef] [PubMed]
- Chaitanya, G.V.; Steven, A.J.; Babu, P.P. PARP-1 cleavage fragments: Signatures of cell-death proteases in neurodegeneration. Cell Commun. Signal. 2010, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, S.H.; Desnoyers, S.; Ottaviano, Y.; Davidson, N.E.; Poirier, G.G. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: An early marker of chemotherapy-induced apoptosis. Cancer Res. 1993, 53, 3976–3985. [Google Scholar] [PubMed]
- Husain, N.; Mahmood, R. Hexavalent chromium induces reactive oxygen species and impairs the antioxidant power of human erythrocytes and lymphocytes: Decreased metal reducing and free radical quenching ability of the cells. Toxicol. Ind. Health 2017, 33, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.J.; Guo, Y.L.; Chang, H.Y.; Sheu, H.M.; Pan, M.H.; Lee, Y.H.; Wang, Y.J. N-acetylcysteine inhibits chromium hypersensitivity in coadjuvant chromium-sensitized albino guinea pigs by suppressing the effects of reactive oxygen species. Exp. Dermatol. 2010, 19, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Valko, M. Advances in metal-induced oxidative stress and human disease. Toxicology 2011, 283, 65–87. [Google Scholar] [CrossRef] [PubMed]
- Schleicher, S.M.; Moretti, L.; Varki, V.; Lu, B. Progress in the unraveling of the endoplasmic reticulum stress/autophagy pathway and cancer: Implications for future therapeutic approaches. Drug Resist. Updates 2010, 13, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Menon, M.B.; Tiedje, C.; Lafera, J.; Ronkina, N.; Konen, T.; Kotlyarov, A.; Gaestel, M. Endoplasmic reticulum-associated ubiquitin-conjugating enzyme Ube2j1 is a novel substrate of MK2 (MAPKAP kinase-2) involved in MK2-mediated TNFalpha production. Biochem. J. 2013, 456, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Funding, A.T.; Johansen, C.; Gaestel, M.; Bibby, B.M.; Lilleholt, L.L.; Kragballe, K.; Iversen, L. Reduced oxazolone-induced skin inflammation in MAPKAP kinase 2 knockout mice. J. Investig. Dermatol. 2009, 129, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Menu, P.; Vince, J.E. The NLRP3 inflammasome in health and disease: The good, the bad and the ugly. Clin. Exp. Immunol. 2011, 166, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Adam, C.; Wohlfarth, J.; Haussmann, M.; Sennefelder, H.; Rodin, A.; Maler, M.; Martin, S.F.; Goebeler, M.; Schmidt, M. Allergy-Inducing Chromium Compounds Trigger Potent Innate Immune Stimulation Via ROS-Dependent Inflammasome Activation. J. Investig. Dermatol. 2017, 137, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Stocks, S.J.; McNamee, R.; Turner, S.; Carder, M.; Agius, R.M. Has European Union legislation to reduce exposure to chromate in cement been effective in reducing the incidence of allergic contact dermatitis attributed to chromate in the UK? Occup. Environ. Med. 2012, 69, 150–152. [Google Scholar] [CrossRef] [PubMed]
- Bregnbak, D.; Johansen, J.D.; Jellesen, M.S.; Zachariae, C.; Menne, T.; Thyssen, J.P. Chromium allergy and dermatitis: Prevalence and main findings. Contact Dermat. 2015, 73, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Kotlyarov, A.; Neininger, A.; Schubert, C.; Eckert, R.; Birchmeier, C.; Volk, H.D.; Gaestel, M. MAPKAP kinase 2 is essential for LPS-induced TNF-alpha biosynthesis. Nat. Cell Biol. 1999, 1, 94–97. [Google Scholar] [PubMed]
- Buters, J.; Biedermann, T. Chromium(VI) Contact Dermatitis: Getting Closer to Understanding the Underlying Mechanisms of Toxicity and Sensitization! J. Investig. Dermatol. 2017, 137, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Gross, O.; Thomas, C.J.; Guarda, G.; Tschopp, J. The inflammasome: An integrated view. Immunol. Rev. 2011, 243, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Gehrke, S.; Contassot, E.; Roques, S.; Tschopp, J.; Friedmann, P.S.; French, L.E.; Gaide, O. Danger signaling through the inflammasome acts as a master switch between tolerance and sensitization. J. Immunol. 2008, 180, 5826–5832. [Google Scholar] [CrossRef] [PubMed]
- Brauer, S.L.; Hneihen, A.S.; McBride, J.S.; Wetterhahn, K.E. Chromium(VI) Forms Thiolate Complexes with gamma-Glutamylcysteine, N-Acetylcysteine, Cysteine, and the Methyl Ester of N-Acetylcysteine. Inorg. Chem. 1996, 35, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Riche, D.M.; McEwen, C.L.; Riche, K.D.; Sherman, J.J.; Wofford, M.R.; Deschamp, D.; Griswold, M. Analysis of safety from a human clinical trial with pterostilbene. J. Toxicol. 2013, 2013, 463595. [Google Scholar] [PubMed]
- Ruiz, M.J.; Fernandez, M.; Pico, Y.; Manes, J.; Asensi, M.; Carda, C.; Asensio, G.; Estrela, J.M. Dietary administration of high doses of pterostilbene and quercetin to mice is not toxic. J. Agric. Food Chem. 2009, 57, 3180–3186. [Google Scholar] [CrossRef] [PubMed]
- Kapetanovic, I.M.; Muzzio, M.; Huang, Z.; Thompson, T.N.; McCormick, D.L. Pharmacokinetics, oral bioavailability, and metabolic profile of resveratrol and its dimethylether analog, pterostilbene, in rats. Cancer Chemother. Pharmacol. 2011, 68, 593–601. [Google Scholar] [PubMed]
- Sun, Y.; Wu, X.; Cai, X.; Song, M.; Zheng, J.; Pan, C.; Qiu, P.; Zhang, L.; Zhou, S.; Tang, Z.; et al. Identification of pinostilbene as a major colonic metabolite of pterostilbene and its inhibitory effects on colon cancer cells. Mol. Nutr. Food Res. 2016, 60, 1924–1932. [Google Scholar] [CrossRef] [PubMed]
- Xue, E.X.; Lin, J.P.; Zhang, Y.; Sheng, S.R.; Liu, H.X.; Zhou, Y.L.; Xu, H. Pterostilbene inhibits inflammation and ROS production in chondrocytes by activating Nrf2 pathway. Oncotarget 2017, 8, 41988–42000. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhao, L.; Yue, L.; Wang, B.; Li, X.; Guo, H.; Ma, Y.; Yao, C.; Gao, L.; Deng, J.; et al. Pterostilbene attenuates early brain injury following subarachnoid hemorrhage via inhibition of the NLRP3 inflammasome and Nox2-related oxidative stress. Mol. Neurobiol. 2017, 54, 5928–5940. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Xiao, F.; Luo, L.; Zhong, C. Activation of autophagy protects against ROS-mediated mitochondria-dependent apoptosis in L-02 hepatocytes induced by Cr(VI). Cell Physiol. Biochem. 2014, 33, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Kosuru, R.; Cai, Y.; Kandula, V.; Yan, D.; Wang, C.; Zheng, H.; Li, Y.; Irwin, M.G.; Singh, S.; Xia, Z. AMPK contributes to cardioprotective effects of pterostilbene against myocardial ischemia-reperfusion injury in diabetic rats by suppressing cardiac oxidative stress and apoptosis. Cell Physiol. Biochem. 2018, 46, 1381–1397. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Cai, N.; Jia, H. Pterostilbene attenuates myocardial ischemia-reperfusion injury via the phosphatidylinositol 3′-kinase-protein kinase B signaling pathway. Exp. Ther. Med. 2017, 14, 5509–5514. [Google Scholar] [PubMed]
- Schroder, M. Endoplasmic reticulum stress responses. Cell Mol. Life Sci. 2008, 65, 862–894. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, T.; Rubio, N.; Garg, A.D.; Bultynck, G.; Rizzuto, R.; Decuypere, J.P.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A.; et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012, 19, 1880–1891. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Fan, C.; Yu, L.; Yang, Y.; Jiang, S.; Ma, Z.; Hu, W.; Li, T.; Yang, Z.; Tian, T.; et al. Pterostilbene exerts an anti-inflammatory effect via regulating endoplasmic reticulum stress in endothelial cells. Cytokine 2016, 77, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Sirerol, J.A.; Feddi, F.; Mena, S.; Rodriguez, M.L.; Sirera, P.; Aupi, M.; Perez, S.; Asensi, M.; Ortega, A.; Estrela, J.M. Topical treatment with pterostilbene, a natural phytoalexin, effectively protects hairless mice against UVB radiation-induced skin damage and carcinogenesis. Free Radic. Biol. Med. 2015, 85, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sample, A.; He, Y.Y. Autophagy in UV damage response. Photochem. Photobiol. 2017, 93, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Strozyk, E.; Kulms, D. The role of akt/mtor pathway in stress response to UV-irradiation: Implication in skin carcinogenesis by regulation of apoptosis, autophagy and senescence. Int. J. Mol. Sci. 2013, 14, 15260–15285. [Google Scholar] [CrossRef] [PubMed]
- Nagar, R. Autophagy: A brief overview in perspective of dermatology. Indian J. Dermatol. Venereol. Leprol. 2017, 83, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.J.; Tsai, S.J.; Ho, C.T.; Pan, M.H.; Ho, Y.S.; Wu, C.H.; Wang, Y.J. Chemopreventive effects of pterostilbene on urethane-induced lung carcinogenesis in mice via the inhibition of EGFR-mediated pathways and the induction of apoptosis and autophagy. J. Agric. Food Chem. 2012, 60, 11533–11541. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Feeding Regimen and Antioxidants Administration | Application Procedure | Epicutaneous Test |
|---|---|---|---|
| A (Control) | Normal diet | Injected with saline | 21 days after injection treatment |
| B (K2Cr2O7) | Normal diet and continually gave 5% β-Cyclodextrin (200 μL/day) by gavage until the end of the experiment | Injected with K2Cr2O7 in FCA emulsion | 21 days after injection treatment |
| C (NAC) | NAC 1200 mg/kg/day for 2 weeks, and then fed with the same dosage for a further 3 weeks after injection treatment | Injected with K2Cr2O7 in FCA emulsion | 21 days after injection treatment |
| D (PT) | PT 500 mg/kg/day for 2 weeks, and then fed with the same dosage for a further 3 weeks after injection treatment | Injected with K2Cr2O7 in FCA emulsion | 21 days after injection treatment |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, B.-J.; Chiu, H.-W.; Lee, Y.-L.; Li, C.-Y.; Wang, Y.-J.; Lee, Y.-H. Pterostilbene Attenuates Hexavalent Chromium-Induced Allergic Contact Dermatitis by Preventing Cell Apoptosis and Inhibiting IL-1β-Related NLRP3 Inflammasome Activation. J. Clin. Med. 2018, 7, 489. https://doi.org/10.3390/jcm7120489
Wang B-J, Chiu H-W, Lee Y-L, Li C-Y, Wang Y-J, Lee Y-H. Pterostilbene Attenuates Hexavalent Chromium-Induced Allergic Contact Dermatitis by Preventing Cell Apoptosis and Inhibiting IL-1β-Related NLRP3 Inflammasome Activation. Journal of Clinical Medicine. 2018; 7(12):489. https://doi.org/10.3390/jcm7120489
Chicago/Turabian StyleWang, Bour-Jr, Hui-Wen Chiu, Yong-Lin Lee, Chia-Yi Li, Ying-Jan Wang, and Yu-Hsuan Lee. 2018. "Pterostilbene Attenuates Hexavalent Chromium-Induced Allergic Contact Dermatitis by Preventing Cell Apoptosis and Inhibiting IL-1β-Related NLRP3 Inflammasome Activation" Journal of Clinical Medicine 7, no. 12: 489. https://doi.org/10.3390/jcm7120489
APA StyleWang, B.-J., Chiu, H.-W., Lee, Y.-L., Li, C.-Y., Wang, Y.-J., & Lee, Y.-H. (2018). Pterostilbene Attenuates Hexavalent Chromium-Induced Allergic Contact Dermatitis by Preventing Cell Apoptosis and Inhibiting IL-1β-Related NLRP3 Inflammasome Activation. Journal of Clinical Medicine, 7(12), 489. https://doi.org/10.3390/jcm7120489