PG-Priming Enhances Doxorubicin Influx to Trigger Necrotic and Autophagic Cell Death in Oral Squamous Cell Carcinoma



Abstract
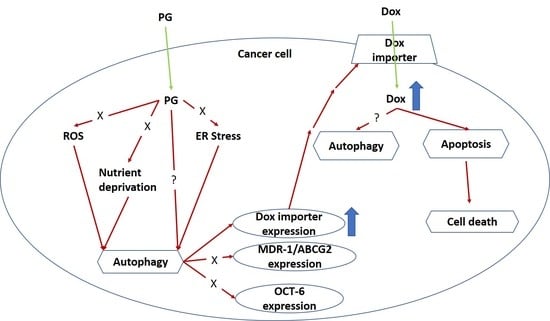
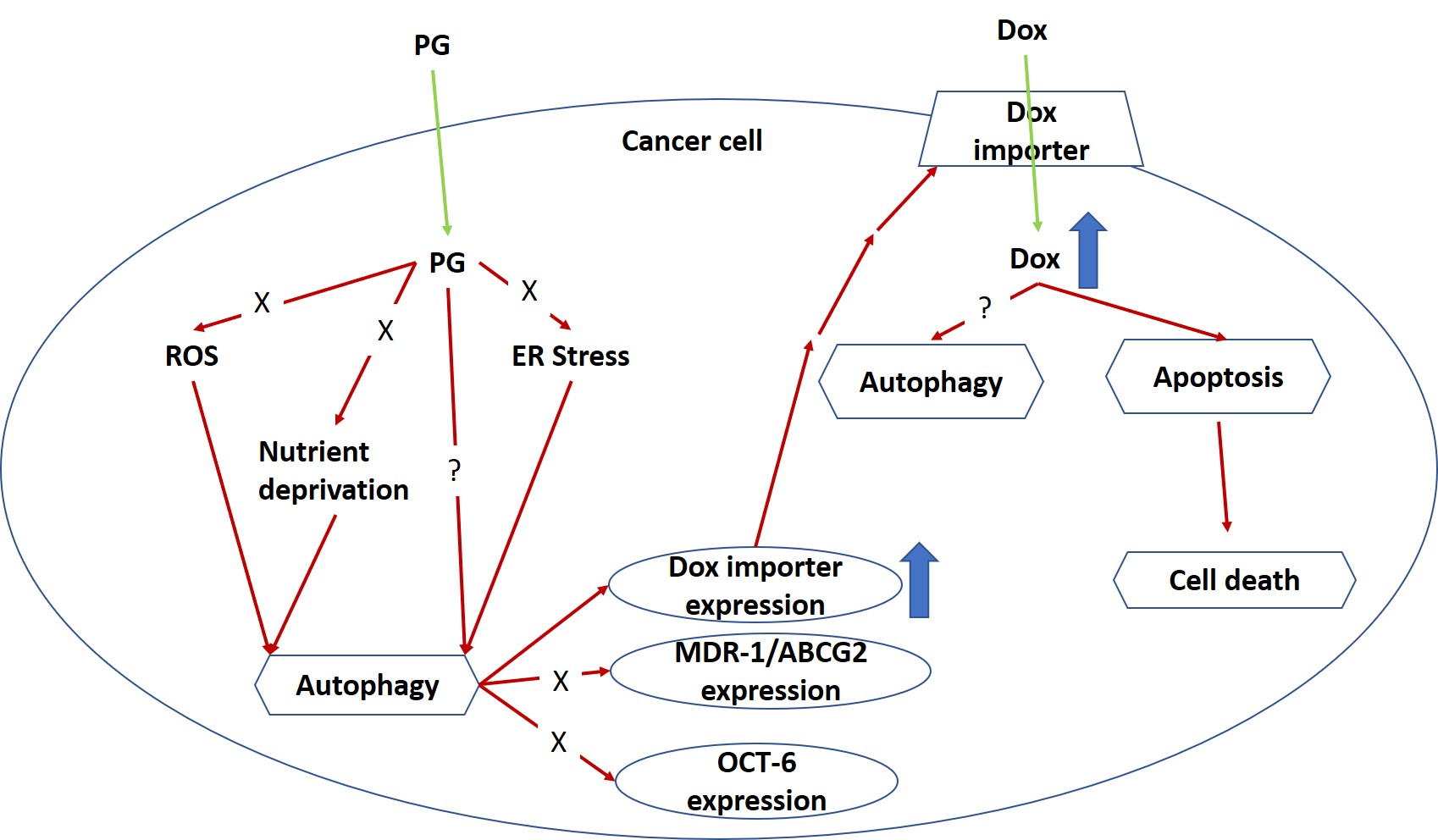
1. Introduction
2. Experimental Section
2.1. Research Aims
2.2. Reagents
2.3. Cell Culture
2.4. Cytotoxicity Assay
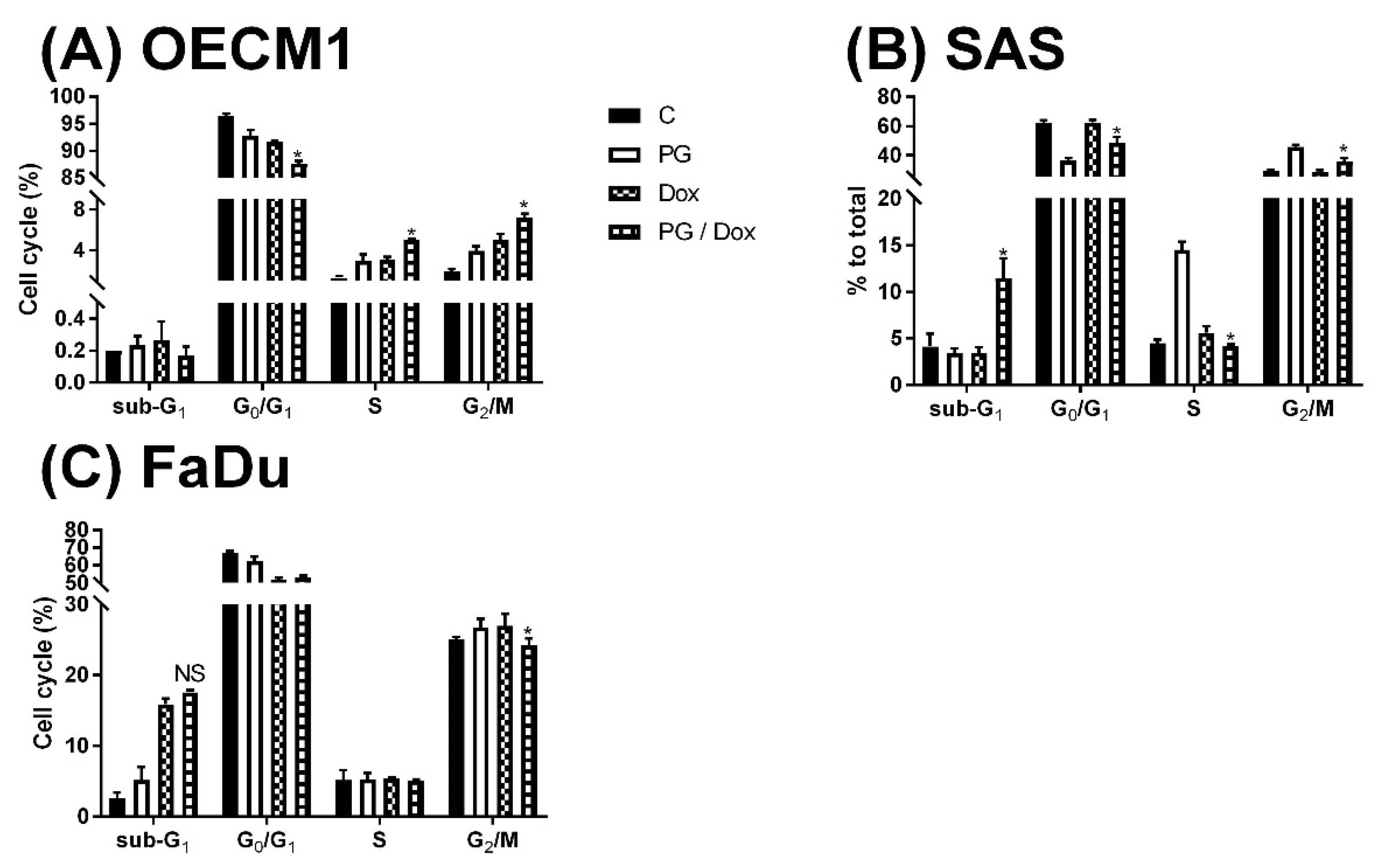
2.5. Cell Cycle Analysis
2.6. Doxorubicin Flux Analysis
2.7. Western Blotting
2.8. Statistical Analysis
3. Results
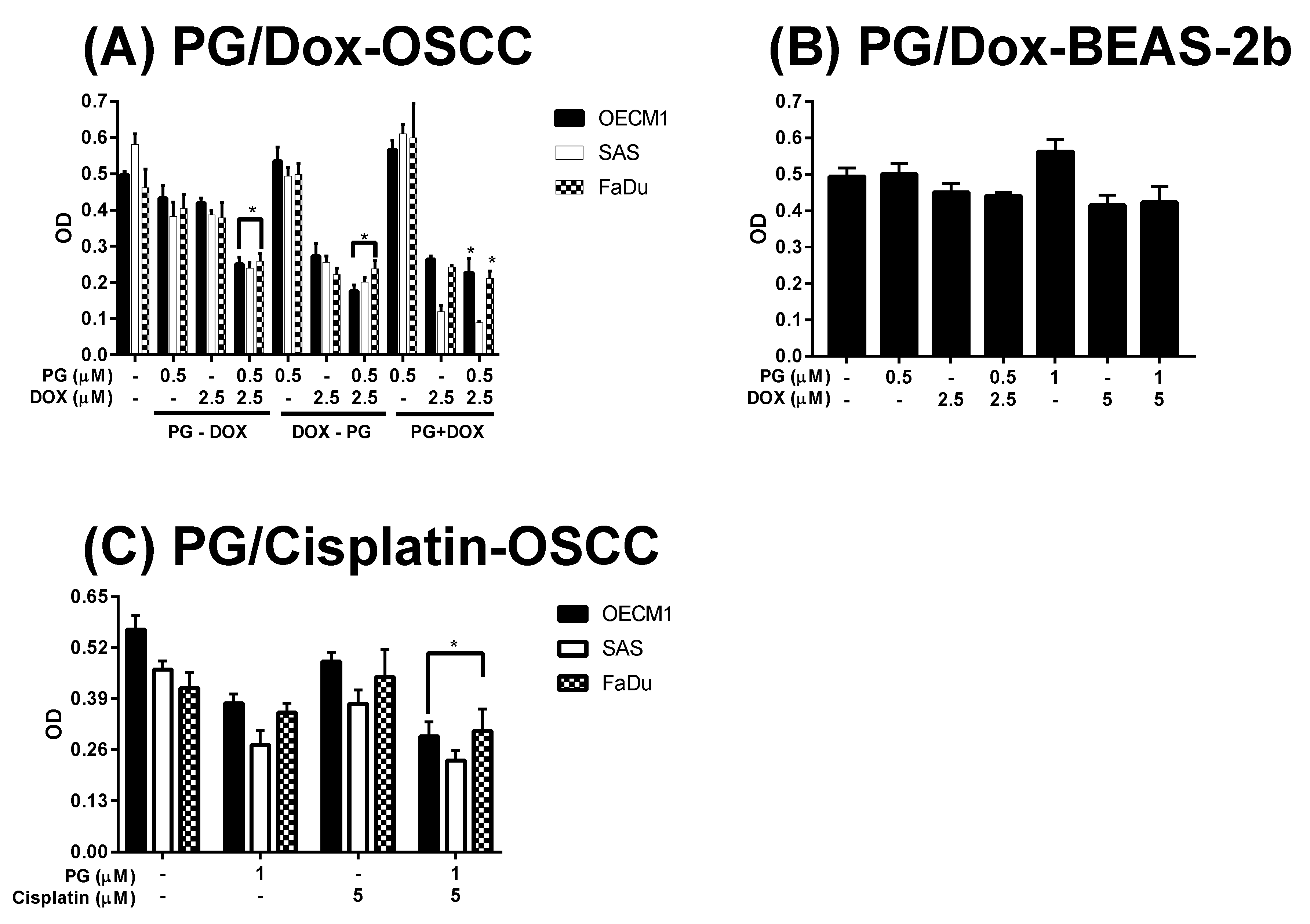
3.1. Cytotoxicity Change of PG/Dox Treated Strategies
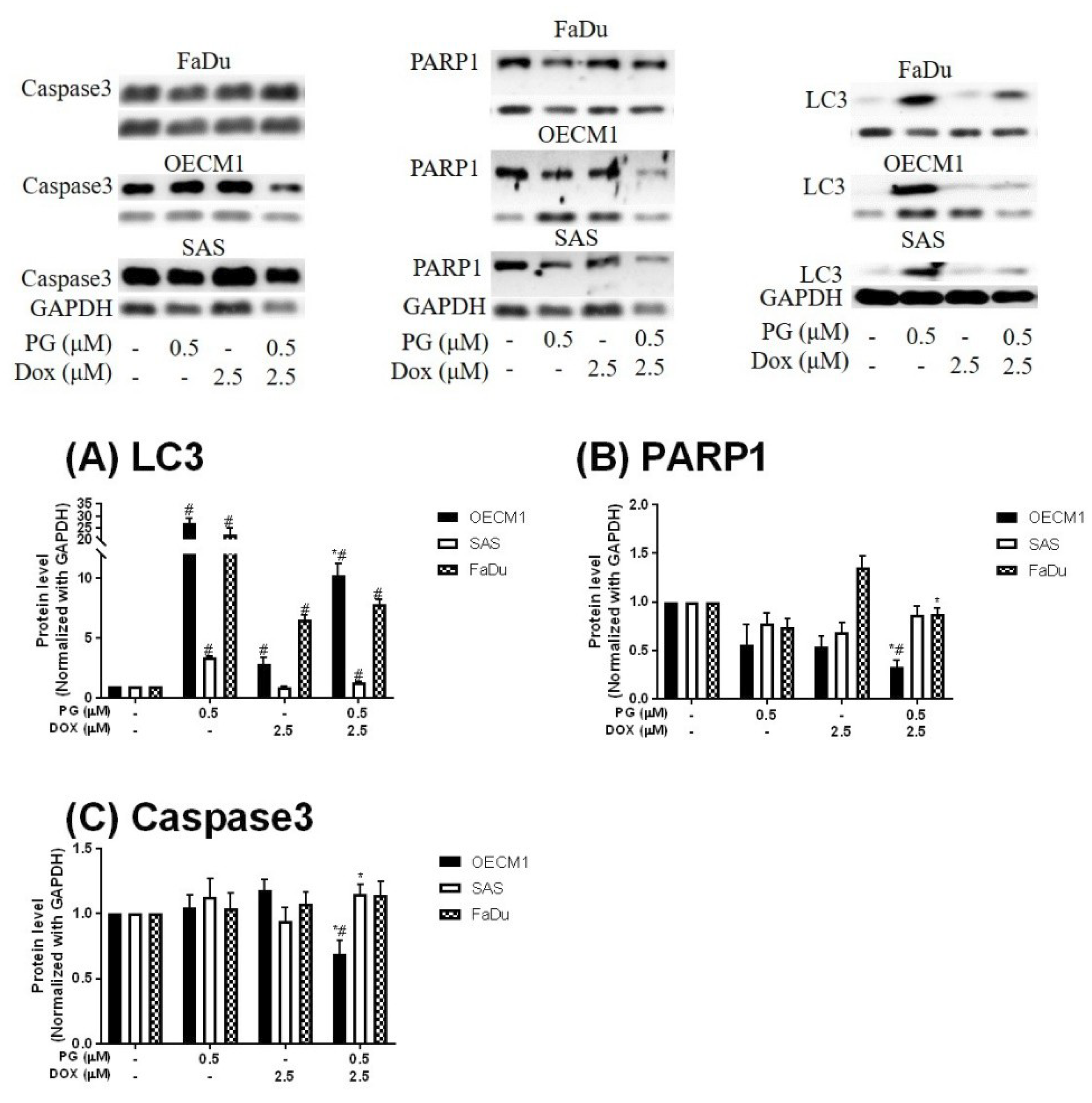
3.2. Identification of Cell Death Characteristics
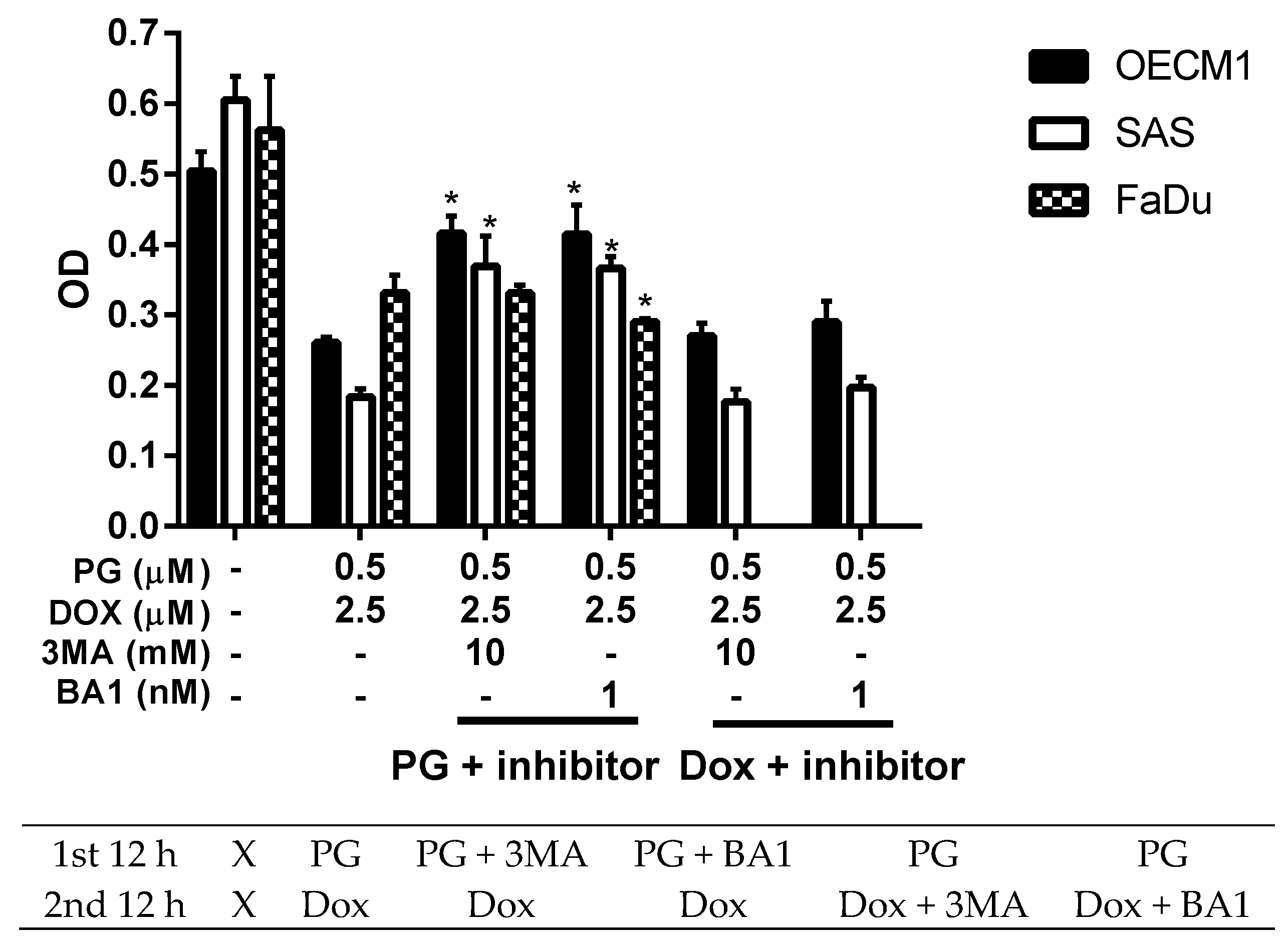
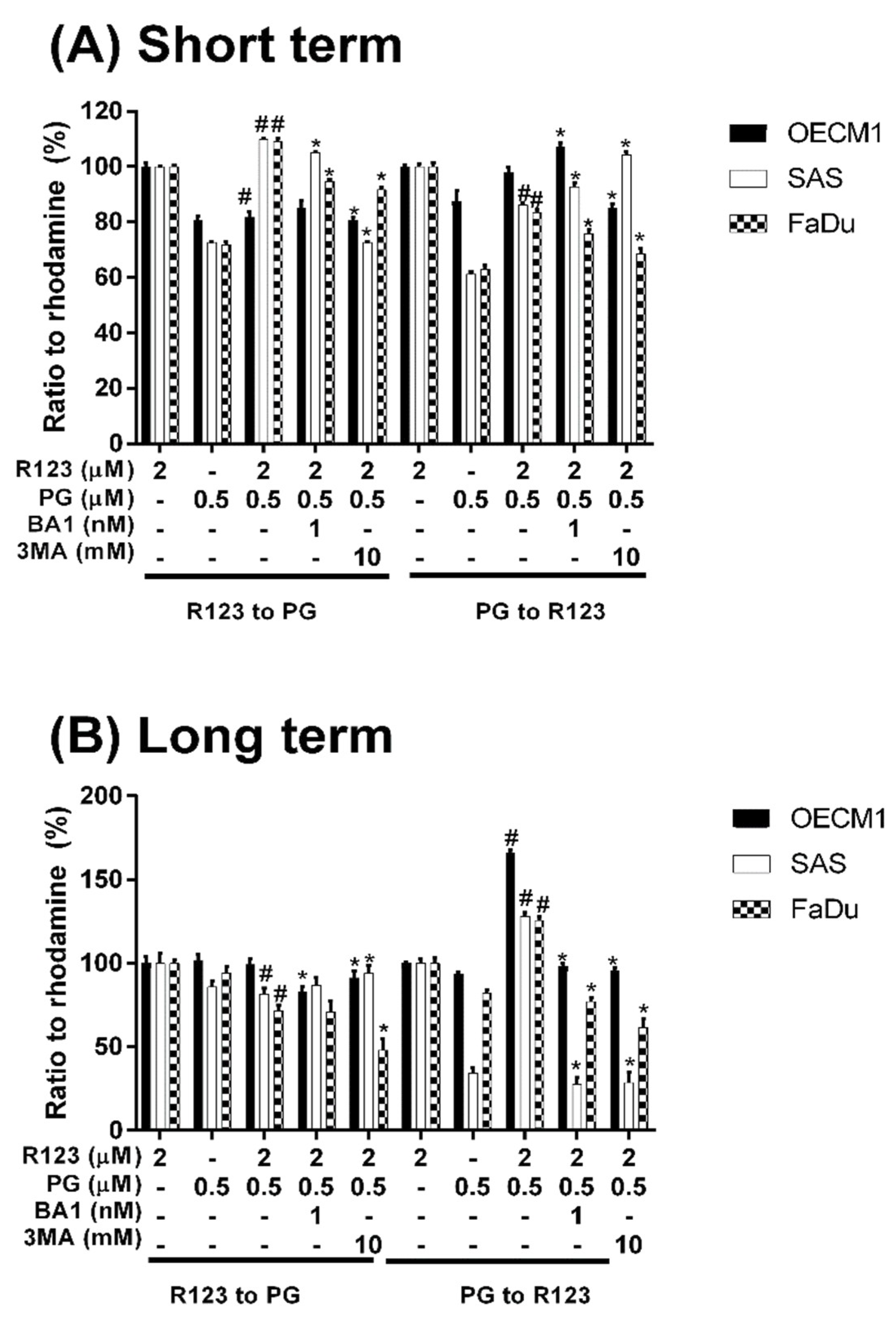
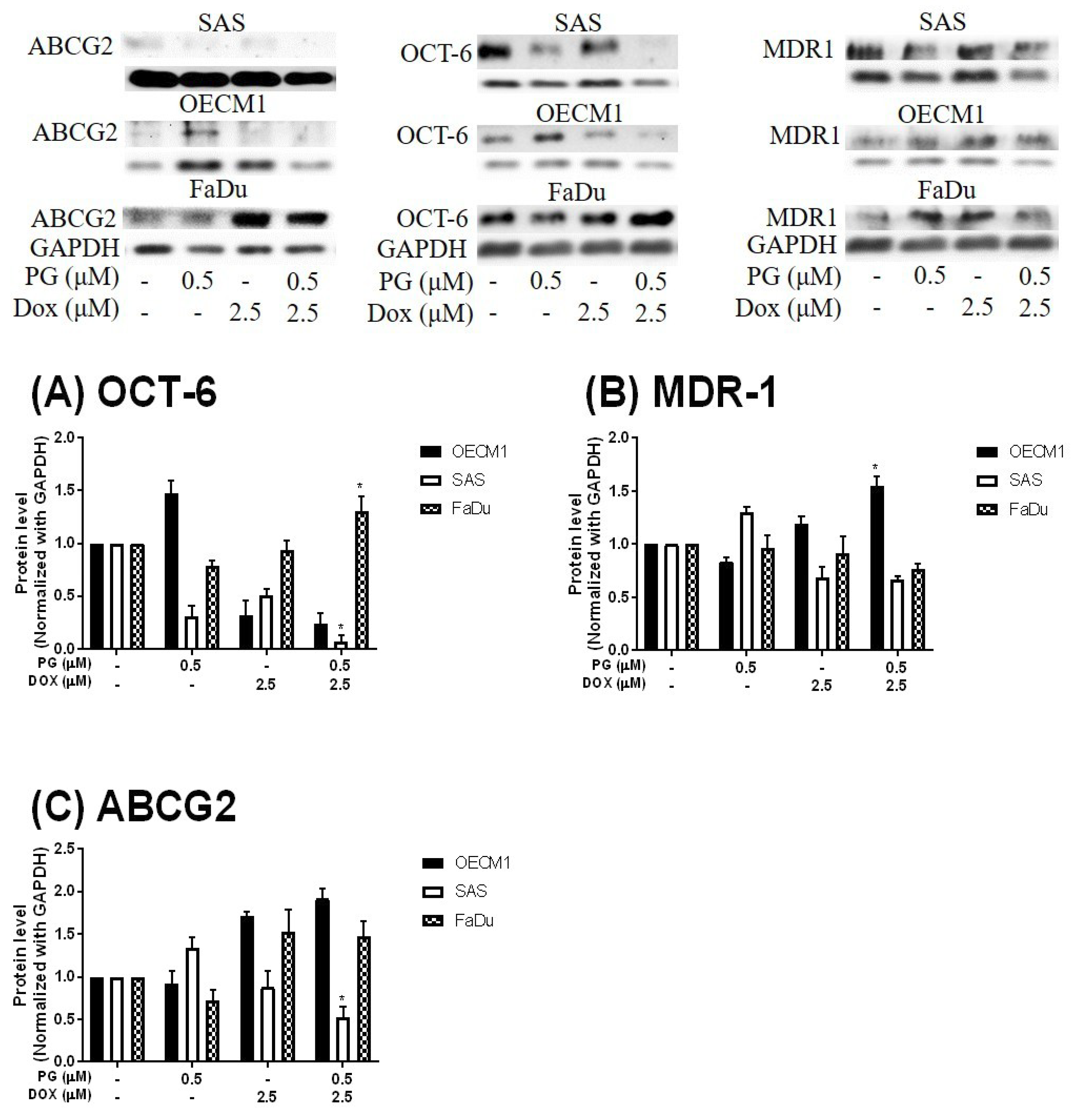
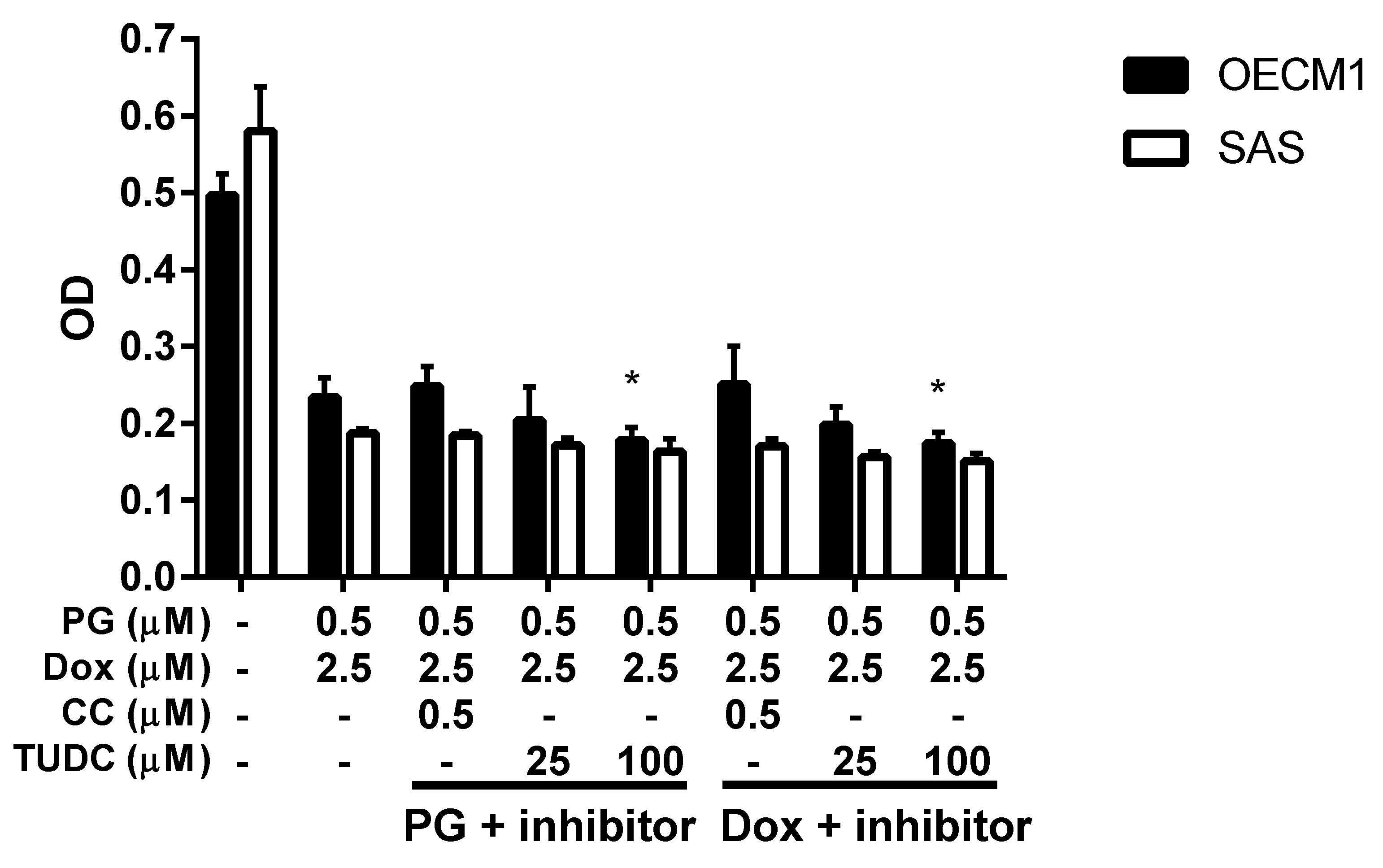
3.3. Doxorubicin Flux Affected by PG-Induced Autophagy
3.4. ER Stress and Energy Deprivation Analysis in PG-Priming OSCC Cells
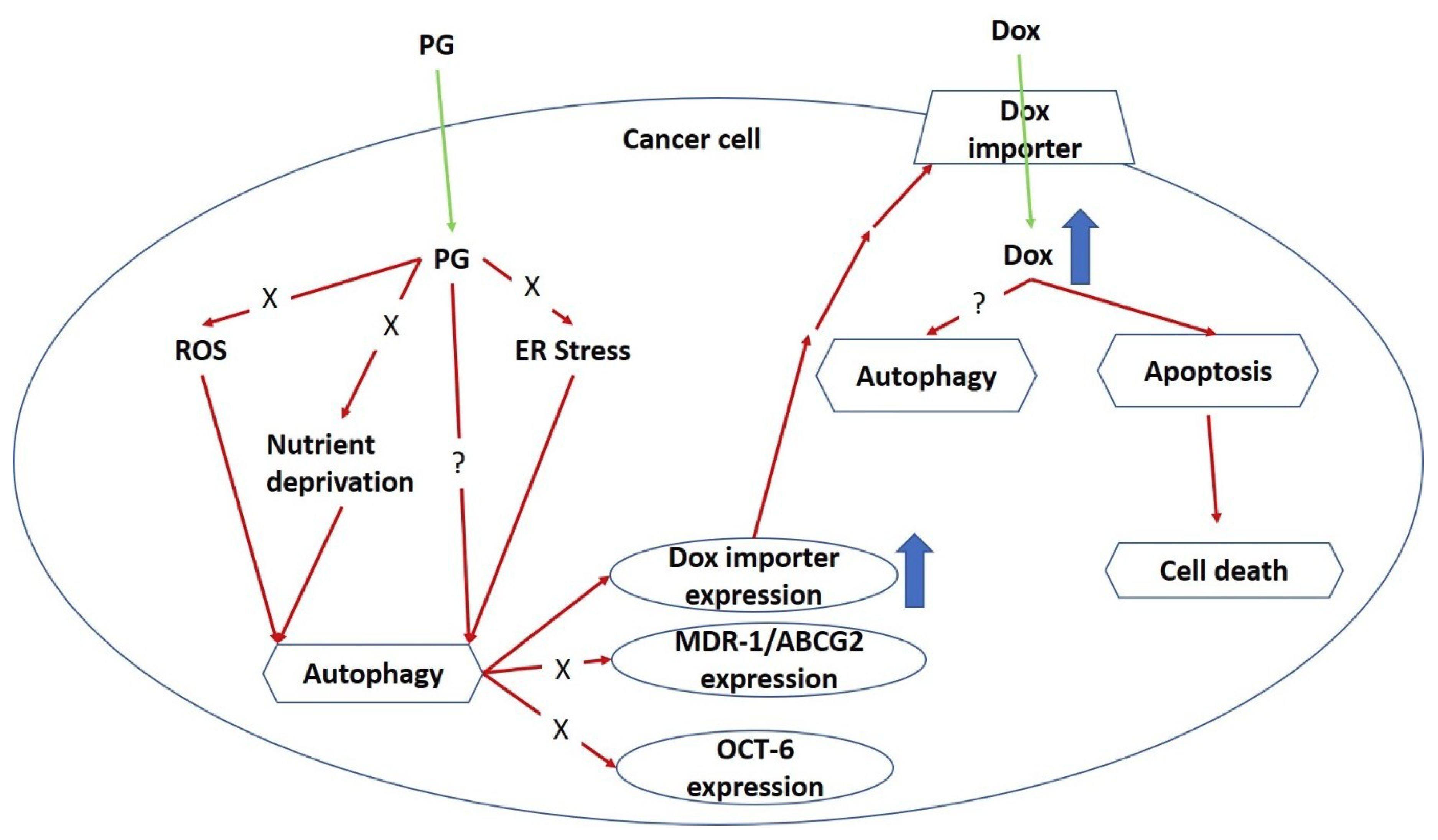
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McGowan, J.V.; Chung, R.; Maulik, A.; Piotrowska, I.; Walker, J.M.; Yellon, D.M. Anthracycline Chemotherapy and Cardiotoxicity. Cardiovasc. Drugs Ther. 2017, 31, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Damiani, R.M.; Moura, D.J.; Viau, C.M.; Caceres, R.A.; Henriques, J.A.; Saffi, J. Pathways of cardiac toxicity: Comparison between chemotherapeutic drugs doxorubicin and mitoxantrone. Arch. Toxicol. 2016, 90, 2063–2076. [Google Scholar] [CrossRef] [PubMed]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharmacogenet. Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Johnson-Arbor, K.; Dubey, R. Doxorubicin. In StatPearls; StatPearls Publisher: Treasure Island, FL, USA, 2018. [Google Scholar]
- Renu, K.; Abilash, V.G.; Pirupathi Pichiah, P.B.; Arunachalam, S. Molecular mechanism of doxorubicin-induced cardiomyopathy—An update. Eur. J. Pharmacol. 2018, 818, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Shafei, A.; El-Bakly, W.; Sobhy, A.; Wagdy, O.; Reda, A.; Aboelenin, O.; Marzouk, A.; El Habak, K.; Mostafa, R.; Ali, M.A.; et al. A review on the efficacy and toxicity of different doxorubicin nanoparticles for targeted therapy in metastatic breast cancer. Biomed. Pharmacother. 2017, 95, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- Asensio-Lopez, M.C.; Soler, F.; Pascual-Figal, D.; Fernandez-Belda, F.; Lax, A. Doxorubicin-induced oxidative stress: The protective effect of nicorandil on HL-1 cardiomyocytes. PLoS ONE 2017, 12, e0172803. [Google Scholar] [CrossRef] [PubMed]
- Kwatra, M.; Kumar, V.; Jangra, A.; Mishra, M.; Ahmed, S.; Ghosh, P.; Vohora, D.; Khanam, R. Ameliorative effect of naringin against doxorubicin-induced acute cardiac toxicity in rats. Pharm. Biol. 2016, 54, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Meredith, A.M.; Dass, C.R. Increasing role of the cancer chemotherapeutic doxorubicin in cellular metabolism. J. Pharm. Pharmacol. 2016, 68, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; He, Q.; Zhao, W.; Luo, J.; Gao, W. Tumor-homing, pH- and ultrasound-responsive polypeptide-doxorubicin nanoconjugates overcome doxorubicin resistance in cancer therapy. J. Control. Release 2017, 264, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Zhang, L.; Gao, H.; Liu, Y.; Zhang, Q.; Ran, R.; Zhang, Z.; He, Q. Co-delivery of doxorubicin and P-gp inhibitor by a reduction-sensitive liposome to overcome multidrug resistance, enhance anti-tumor efficiency and reduce toxicity. Drug Deliv. 2016, 23, 1130–1143. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Yu, Z.; Liu, Y.; Yuan, W.; Yang, T.; You, J.; He, X.; Lee, R.J.; Li, L.; Xu, C. Delivery of miR-375 and doxorubicin hydrochloride by lipid-coated hollow mesoporous silica nanoparticles to overcome multiple drug resistance in hepatocellular carcinoma. Int. J. Nanomed. 2017, 12, 5271–5287. [Google Scholar] [CrossRef] [PubMed]
- Gupta, B.; Ramasamy, T.; Poudel, B.K.; Pathak, S.; Regmi, S.; Choi, J.Y.; Son, Y.; Thapa, R.K.; Jeong, J.H.; Kim, J.R.; et al. Development of Bioactive PEGylated Nanostructured Platforms for Sequential Delivery of Doxorubicin and Imatinib to Overcome Drug Resistance in Metastatic Tumors. ACS Appl. Mater. Interfaces 2017, 9, 9280–9290. [Google Scholar] [CrossRef] [PubMed]
- Perillo, E.; Porto, S.; Falanga, A.; Zappavigna, S.; Stiuso, P.; Tirino, V.; Desiderio, V.; Papaccio, G.; Galdiero, M.; Giordano, A.; et al. Liposome armed with herpes virus-derived gH625 peptide to overcome doxorubicin resistance in lung adenocarcinoma cell lines. Oncotarget 2016, 7, 4077–4092. [Google Scholar] [CrossRef] [PubMed]
- Kaminskas, L.M.; McLeod, V.M.; Kelly, B.D.; Sberna, G.; Boyd, B.J.; Williamson, M.; Owen, D.J.; Porter, C.J. A comparison of changes to doxorubicin pharmacokinetics, antitumor activity, and toxicity mediated by PEGylated dendrimer and PEGylated liposome drug delivery systems. Nanomedicine 2012, 8, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Broxterman, H.J.; Gotink, K.J.; Verheul, H.M. Understanding the causes of multidrug resistance in cancer: A comparison of doxorubicin and sunitinib. Drug Resist. Updat. 2009, 12, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [PubMed]
- Bradley, G.; Juranka, P.F.; Ling, V. Mechanism of multidrug resistance. Biochim. Biophys. Acta 1988, 948, 87–128. [Google Scholar] [CrossRef]
- Liu, T.; Li, Z.; Zhang, Q.; De Amorim Bernstein, K.; Lozano-Calderon, S.; Choy, E.; Hornicek, F.J.; Duan, Z. Targeting ABCB1 (MDR1) in multi-drug resistant osteosarcoma cells using the CRISPR-Cas9 system to reverse drug resistance. Oncotarget 2016, 7, 83502–83513. [Google Scholar] [CrossRef] [PubMed]
- Komori, Y.; Arisawa, S.; Takai, M.; Yokoyama, K.; Honda, M.; Hayashi, K.; Ishigami, M.; Katano, Y.; Goto, H.; Ueyama, J.; et al. Ursodeoxycholic acid inhibits overexpression of P-glycoprotein induced by doxorubicin in HepG2 cells. Eur. J. Pharmacol. 2014, 724, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.M.; Haniadka, R.; Chacko, P.P.; Palatty, P.L.; Baliga, M.S. Zingiber officinale Roscoe (ginger) as an adjuvant in cancer treatment: A review. J. BUON 2011, 16, 414–424. [Google Scholar] [PubMed]
- Laatsch, H.; Kellner, M.; Weyland, H. Butyl-meta-cycloheptylprodiginine—A revision of the structure of the former ortho-isomer. J. Antibiot. 1991, 44, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Soliev, A.B.; Hosokawa, K.; Enomoto, K. Bioactive pigments from marine bacteria: Applications and physiological roles. Evid. Based Complement. Alternat. Med. 2011, 2011, 670349. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chen, W.C.; Ho, T.F.; Wu, H.S.; Wei, Y.H. Development of natural anti-tumor drugs by microorganisms. J. Biosci. Bioeng. 2011, 111, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Marchal, E.; Smithen, D.A.; Uddin, M.I.; Robertson, A.W.; Jakeman, D.L.; Mollard, V.; Goodman, C.D.; MacDougall, K.S.; McFarland, S.A.; McFadden, G.I.; et al. Synthesis and antimalarial activity of prodigiosenes. Org. Biomol. Chem. 2014, 12, 4132–4142. [Google Scholar] [CrossRef] [PubMed]
- Lapenda, J.C.; Silva, P.A.; Vicalvi, M.C.; Sena, K.X.; Nascimento, S.C. Antimicrobial activity of prodigiosin isolated from Serratia marcescens UFPEDA 398. World J. Microbiol. Biotechnol. 2015, 31, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Nakajima, A.; Hosokawa, K.; Soliev, A.B.; Osaka, I.; Arakawa, R.; Enomoto, K. Cytotoxic prodigiosin family pigments from Pseudoalteromonas sp. 1020R isolated from the Pacific coast of Japan. Biosci. Biotechnol. Biochem. 2012, 76, 1229–1232. [Google Scholar] [CrossRef] [PubMed]
- Kimyon, O.; Das, T.; Ibugo, A.I.; Kutty, S.K.; Ho, K.K.; Tebben, J.; Kumar, N.; Manefield, M. Serratia Secondary Metabolite Prodigiosin Inhibits Pseudomonas aeruginosa Biofilm Development by Producing Reactive Oxygen Species that Damage Biological Molecules. Front. Microbiol. 2016, 7, 972. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Liu, G.; Li, J.; Huang, H.; Zhang, X.; Zhang, H.; Ju, J. Cytotoxic and antibacterial angucycline- and prodigiosin-analogues from the deep-sea derived Streptomyces sp. SCSIO 11594. Mar. Drugs 2015, 13, 1304–1316. [Google Scholar] [CrossRef] [PubMed]
- Kancharla, P.; Lu, W.; Salem, S.M.; Kelly, J.X.; Reynolds, K.A. Stereospecific synthesis of 23-hydroxyundecylprodiginines and analogues and conversion to antimalarial premarineosins via a Rieske oxygenase catalyzed bicyclization. J. Org. Chem. 2014, 79, 11674–11689. [Google Scholar] [CrossRef] [PubMed]
- Perez-Tomas, R.; Vinas, M. New insights on the antitumoral properties of prodiginines. Curr. Med. Chem. 2010, 17, 2222–2231. [Google Scholar] [CrossRef] [PubMed]
- Sam, S.; Sam, M.R.; Esmaeillou, M.; Safaralizadeh, R. Effective Targeting Survivin, Caspase-3 and MicroRNA-16-1 Expression by Methyl-3-pentyl-6-methoxyprodigiosene Triggers Apoptosis in Colorectal Cancer Stem-Like Cells. Pathol. Oncol. Res. 2016, 22, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.J.; Ou, J.H.; Wang, M.L.; Jialielihan, N.; Liu, Y.H. Elevated survivin mediated multidrug resistance and reduced apoptosis in breast cancer stem cells. J. BUON 2015, 20, 1287–1294. [Google Scholar] [PubMed]
- Chiu, W.-J.; Lin, S.-R.; Chen, Y.-H.; Tsai, M.-J.; Leong, M.; Weng, C.-F. Prodigiosin-Emerged PI3K/Beclin-1-Independent Pathway Elicits Autophagic Cell Death in Doxorubicin-Sensitive and -Resistant Lung Cancer. J. Clin. Med. 2018, 7, 321. [Google Scholar] [CrossRef] [PubMed]
- Llagostera, E.; Soto-Cerrato, V.; Montaner, B.; Perez-Tomas, R. Prodigiosin induces apoptosis by acting on mitochondria in human lung cancer cells. Ann. N.Y. Acad. Sci. 2003, 1010, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Llagostera, E.; Soto-Cerrato, V.; Joshi, R.; Montaner, B.; Gimenez-Bonafe, P.; Perez-Tomas, R. High cytotoxic sensitivity of the human small cell lung doxorubicin-resistant carcinoma (GLC4/ADR) cell line to prodigiosin through apoptosis activation. Anticancer Drugs 2005, 16, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Jin, Z.X.; Wan, Y.J. Apoptosis of human lung adenocarcinoma A549 cells induced by prodigiosin analogue obtained from an entomopathogenic bacterium Serratia marcescens. Appl. Microbiol. Biotechnol. 2010, 88, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Soto-Cerrato, V.; Llagostera, E.; Montaner, B.; Scheffer, G.L.; Perez-Tomas, R. Mitochondria-mediated apoptosis operating irrespective of multidrug resistance in breast cancer cells by the anticancer agent prodigiosin. Biochem. Pharmacol. 2004, 68, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Monge, M.; Vilaseca, M.; Soto-Cerrato, V.; Montaner, B.; Giralt, E.; Perez-Tomas, R. Proteomic analysis of prodigiosin-induced apoptosis in a breast cancer mitoxantrone-resistant (MCF-7 MR) cell line. Investig. New Drugs 2007, 25, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Montaner, B.; Perez-Tomas, R. Prodigiosin-induced apoptosis in human colon cancer cells. Life Sci. 2001, 68, 2025–2036. [Google Scholar] [CrossRef]
- Dalili, D.; Fouladdel, S.; Rastkari, N.; Samadi, N.; Ahmadkhaniha, R.; Ardavan, A.; Azizi, E. Prodigiosin, the red pigment of Serratia marcescens, shows cytotoxic effects and apoptosis induction in HT-29 and T47D cancer cell lines. Nat. Prod. Res. 2012, 26, 2078–2083. [Google Scholar] [PubMed]
- Hassankhani, R.; Sam, M.R.; Esmaeilou, M.; Ahangar, P. Prodigiosin isolated from cell wall of Serratia marcescens alters expression of apoptosis-related genes and increases apoptosis in colorectal cancer cells. Med. Oncol. 2015, 32, 366. [Google Scholar] [CrossRef] [PubMed]
- Sam, M.R.; Pourpak, R.S. Regulation of p53 and survivin by prodigiosin compound derived from Serratia marcescens contribute to caspase-3-dependent apoptosis in acute lymphoblastic leukemia cells. Hum. Exp. Toxicol. 2018, 37, 608–617. [Google Scholar] [CrossRef] [PubMed]
- Campas, C.; Dalmau, M.; Montaner, B.; Barragan, M.; Bellosillo, B.; Colomer, D.; Pons, G.; Perez-Tomas, R.; Gil, J. Prodigiosin induces apoptosis of B and T cells from B-cell chronic lymphocytic leukemia. Leukemia 2003, 17, 746–750. [Google Scholar] [CrossRef] [PubMed]
- Yenkejeh, R.A.; Sam, M.R.; Esmaeillou, M. Targeting survivin with prodigiosin isolated from cell wall of Serratia marcescens induces apoptosis in hepatocellular carcinoma cells. Hum. Exp. Toxicol. 2017, 36, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Wang, Y.H.; Chern, C.M.; Liou, K.T.; Hou, Y.C.; Peng, Y.T.; Shen, Y.C. Prodigiosin inhibits gp91(phox) and iNOS expression to protect mice against the oxidative/nitrosative brain injury induced by hypoxia-ischemia. Toxicol. Appl. Pharmacol. 2011, 257, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.R.; Fu, Y.S.; Tsai, M.J.; Cheng, H.; Weng, C.F. Natural Compounds from Herbs that can Potentially Execute as Autophagy Inducers for Cancer Therapy. Int. J. Mol. Sci. 2017, 18, e1412. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.F.; Lin, C.S.; Chen, Y.H.; Sung, P.J.; Lin, S.R.; Tong, Y.W.; Weng, C.F. Inhibitory Growth of Oral Squamous Cell Carcinoma Cancer via Bacterial Prodigiosin. Mar. Drugs 2017, 15, e224. [Google Scholar] [CrossRef] [PubMed]
- McGrail, D.J.; Khambhati, N.N.; Qi, M.X.; Patel, K.S.; Ravikumar, N.; Brandenburg, C.P.; Dawson, M.R. Alterations in ovarian cancer cell adhesion drive taxol resistance by increasing microtubule dynamics in a FAK-dependent manner. Sci. Rep. 2015, 5, 9529. [Google Scholar] [CrossRef] [PubMed]
- Nabekura, T.; Hiroi, T.; Kawasaki, T.; Uwai, Y. Effects of natural nuclear factor-kappa B inhibitors on anticancer drug efflux transporter human P-glycoprotein. Biomed. Pharmacother. 2015, 70, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Thiyagarajan, V.; Lin, S.X.; Lee, C.H.; Weng, C.F. A focal adhesion kinase inhibitor 16-hydroxy-cleroda-3,13-dien-16,15-olide incorporated into enteric-coated nanoparticles for controlled anti-glioma drug delivery. Colloids Surf. B Biointerfaces 2016, 141, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Kessel, D. Exploring multidrug resistance using rhodamine 123. Cancer Commun. 1989, 1, 145–149. [Google Scholar] [PubMed]
- Wang, J.; Yuan, Z. Gambogic acid sensitizes ovarian cancer cells to doxorubicin through ROS-mediated apoptosis. Cell Biochem. Biophys. 2013, 67, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Zhang, N.; Wang, X.; Cai, C.; Cun, J.; Li, Y.; Lv, S.; Yang, Q. Nitidine chloride induces apoptosis, cell cycle arrest, and synergistic cytotoxicity with doxorubicin in breast cancer cells. Tumour Biol. 2014, 35, 10201–10212. [Google Scholar] [CrossRef] [PubMed]
- Al-Abbasi, F.A.; Alghamdi, E.A.; Baghdadi, M.A.; Alamoudi, A.J.; El-Halawany, A.M.; El-Bassossy, H.M.; Aseeri, A.H.; Al-Abd, A.M. Gingerol Synergizes the Cytotoxic Effects of Doxorubicin against Liver Cancer Cells and Protects from Its Vascular Toxicity. Molecules 2016, 21, 886. [Google Scholar] [CrossRef] [PubMed]
- Guerriero, E.; Sorice, A.; Capone, F.; Storti, G.; Colonna, G.; Ciliberto, G.; Costantini, S. Combining doxorubicin with a phenolic extract from flaxseed oil: Evaluation of the effect on two breast cancer cell lines. Int. J. Oncol. 2017, 50, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, L.; Shi, Z.; Zhong, Z.; Chen, M.; Wang, Y. Evodiamine synergizes with doxorubicin in the treatment of chemoresistant human breast cancer without inhibiting P-glycoprotein. PLoS ONE 2014, 9, e97512. [Google Scholar] [CrossRef] [PubMed]
- Poornima, P.; Kumar, V.B.; Weng, C.F.; Padma, V.V. Doxorubicin induced apoptosis was potentiated by neferine in human lung adenocarcima, A549 cells. Food Chem. Toxicol. 2014, 68, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Sun, Y.; Zheng, J.M.; Yan, X.L.; Chen, H.M.; Chen, J.K.; Huang, H.Q. Formononetin sensitizes glioma cells to doxorubicin through preventing EMT via inhibition of histone deacetylase 5. Int. J. Clin. Exp. Pathol. 2015, 8, 6434–6441. [Google Scholar] [PubMed]
- Yu, L.L.; Wu, J.G.; Dai, N.; Yu, H.G.; Si, J.M. Curcumin reverses chemoresistance of human gastric cancer cells by downregulating the NF-kappaB transcription factor. Oncol. Rep. 2011, 26, 1197–1203. [Google Scholar] [PubMed]
- Mansingh, D.P.; O, J.S.; Sali, V.K.; Vasanthi, H.R. [6]-Gingerol-induced cell cycle arrest, reactive oxygen species generation, and disruption of mitochondrial membrane potential are associated with apoptosis in human gastric cancer (AGS) cells. J. Biochem. Mol. Toxicol. 2018, 10, e22206. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Yang, L.; Feng, J. Nitidine chloride inhibits proliferation and induces apoptosis in ovarian cancer cells by activating the Fas signalling pathway. J. Pharm. Pharmacol. 2018, 70, 778–786. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Wang, M.; Jiang, Y.; Wu, H.; Lu, G.; Shi, W.; Cong, D.; Song, S.; Liu, K.; Wang, H. Gambogic Acid Induces Apoptosis of Non-Small Cell Lung Cancer (NSCLC) Cells by Suppressing Notch Signaling. Med. Sci. Monit. 2018, 24, 7146–7151. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Deng, D.; Shao, N.; Xu, Y.; Xue, L.; Peng, Y.; Liu, Y.; Zhi, F. Evodiamine activates cellular apoptosis through suppressing PI3K/AKT and activating MAPK in glioma. Onco Targets Ther. 2018, 11, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Bazer, F.W.; Lim, W.; Song, G. The O-methylated isoflavone, formononetin, inhibits human ovarian cancer cell proliferation by sub G0/G1 cell phase arrest through PI3K/AKT and ERK1/2 inactivation. J. Cell. Biochem. 2018, 119, 7377–7387. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Li, D.; Luo, X.; Li, L.; Gu, S.; Yu, L.; Ma, Y. Curcumin may serve an anticancer role in human osteosarcoma cell line U-2 OS by targeting ITPR1. Oncol. Lett. 2018, 15, 5593–5601. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.F.; Peng, Y.T.; Chuang, S.M.; Lin, S.C.; Feng, B.L.; Lu, C.H.; Yu, W.J.; Chang, J.S.; Chang, C.C. Prodigiosin down-regulates survivin to facilitate paclitaxel sensitization in human breast carcinoma cell lines. Toxicol. Appl. Pharmacol. 2009, 235, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Sah, N.K.; Khan, Z.; Khan, G.J.; Bisen, P.S. Structural, functional and therapeutic biology of survivin. Cancer Lett. 2006, 244, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Jabbour-Leung, N.A.; Chen, X.; Bui, T.; Jiang, Y.; Yang, D.; Vijayaraghavan, S.; McArthur, M.J.; Hunt, K.K.; Keyomarsi, K. Sequential Combination Therapy of CDK Inhibition and Doxorubicin Is Synthetically Lethal in p53-Mutant Triple-Negative Breast Cancer. Mol. Cancer Ther. 2016, 15, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Geretti, E.; Leonard, S.C.; Dumont, N.; Lee, H.; Zheng, J.; De Souza, R.; Gaddy, D.F.; Espelin, C.W.; Jaffray, D.A.; Moyo, V.; et al. Cyclophosphamide-Mediated Tumor Priming for Enhanced Delivery and Antitumor Activity of HER2-Targeted Liposomal Doxorubicin (MM-302). Mol. Cancer Ther. 2015, 14, 2060–2071. [Google Scholar] [CrossRef] [PubMed]
- Ihnat, M.A.; Nervi, A.M.; Anthony, S.P.; Kaltreider, R.C.; Warren, A.J.; Pesce, C.A.; Davis, S.A.; Lariviere, J.P.; Hamilton, J.W. Effects of mitomycin C and carboplatin pretreatment on multidrug resistance-associated P-glycoprotein expression and on subsequent suppression of tumor growth by doxorubicin and paclitaxel in human metastatic breast cancer xenografted nude mice. Oncol. Res. 1999, 11, 303–310. [Google Scholar] [PubMed]
- Tenconi, E.; Guichard, P.; Motte, P.; Matagne, A.; Rigali, S. Use of red autofluorescence for monitoring prodiginine biosynthesis. J. Microbiol. Methods 2013, 93, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Elahian, F.; Moghimi, B.; Dinmohammadi, F.; Ghamghami, M.; Hamidi, M.; Mirzaei, S.A. The Anticancer Agent Prodigiosin Is Not a Multidrug Resistance Protein Substrate. DNA Cell Biol. 2013, 32, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Okabe, M.; Unno, M.; Harigae, H.; Kaku, M.; Okitsu, Y.; Sasaki, T.; Mizoi, T.; Shiiba, K.; Takanaga, H.; Terasaki, T.; et al. Characterization of the organic cation transporter SLC22A16: A doxorubicin importer. Biochem. Biophys. Res. Commun. 2005, 333, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Darshan, N.; Manonmani, H.K. Prodigiosin and its potential applications. J. Food Sci. Technol. 2015, 52, 5393–5407. [Google Scholar] [CrossRef] [PubMed]
- Anding, A.L.; Baehrecke, E.H. Autophagy in Cell Life and Cell Death. Apoptosis Dev. 2015, 114, 67–91. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.S.; Yoo, W.H.; Chae, H.J. ER Stress and Autophagy. Curr. Mol. Med. 2015, 15, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol. 2013, 65, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Dirks-Naylor, A.J. The role of autophagy in doxorubicin-induced cardiotoxicity. Life Sci. 2013, 93, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Yuan, H.; Yi, F.; Meng, C.; Zhu, Q. Autophagy prevents doxorubicininduced apoptosis in osteosarcoma. Mol. Med. Rep. 2014, 9, 1975–1981. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.R.; Vessoni, A.T.; Menck, C.F. Three-dimensional microenvironment confers enhanced sensitivity to doxorubicin by reducing p53-dependent induction of autophagy. Oncogene 2015, 34, 5329–5340. [Google Scholar] [CrossRef] [PubMed]
- Zilinyi, R.; Czompa, A.; Czegledi, A.; Gajtko, A.; Pituk, D.; Lekli, I.; Tosaki, A. The Cardioprotective Effect of Metformin in Doxorubicin-Induced Cardiotoxicity: The Role of Autophagy. Molecules 2018, 23, E1184. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Host | Source | RRID | MW (kDa) | Dilution |
|---|---|---|---|---|---|
| MDR-1 | Human | Mouse | AB_2565004 | 180 | 1:200 |
| PARP1 | Human | Mouse | AB_1127036 | 116 | 1:200 |
| ABCG2 | Human | Mouse | AB_629007 | 80 | 1:200 |
| OCT-6 | Human | Mouse | AB_10989254 | 46 | 1:200 |
| GAPDH | Human | Mouse | AB_1124759 | 37 | 1:1000 |
| Caspase3 | Human | Mouse | AB_1119997 | 32 | 1:200 |
| LC3 I/II | Human | Mouse | AB_2137722 | 15/18 | 1:200 |
| HRP-conjugated 2nd Ab | Mouse | Goat | AB_92635 | 1:5000 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, S.-R.; Weng, C.-F. PG-Priming Enhances Doxorubicin Influx to Trigger Necrotic and Autophagic Cell Death in Oral Squamous Cell Carcinoma. J. Clin. Med. 2018, 7, 375. https://doi.org/10.3390/jcm7100375
Lin S-R, Weng C-F. PG-Priming Enhances Doxorubicin Influx to Trigger Necrotic and Autophagic Cell Death in Oral Squamous Cell Carcinoma. Journal of Clinical Medicine. 2018; 7(10):375. https://doi.org/10.3390/jcm7100375
Chicago/Turabian StyleLin, Shian-Ren, and Ching-Feng Weng. 2018. "PG-Priming Enhances Doxorubicin Influx to Trigger Necrotic and Autophagic Cell Death in Oral Squamous Cell Carcinoma" Journal of Clinical Medicine 7, no. 10: 375. https://doi.org/10.3390/jcm7100375
APA StyleLin, S.-R., & Weng, C.-F. (2018). PG-Priming Enhances Doxorubicin Influx to Trigger Necrotic and Autophagic Cell Death in Oral Squamous Cell Carcinoma. Journal of Clinical Medicine, 7(10), 375. https://doi.org/10.3390/jcm7100375