
Use of FGF-21 as a Biomarker of Mitochondrial Disease in Clinical Practice

 , ,
, ,
Abstract
1. Introduction
2. Methods
2.1. Individual Populations and Investigations
2.2. Laboratory Analyses
2.3. Statistics
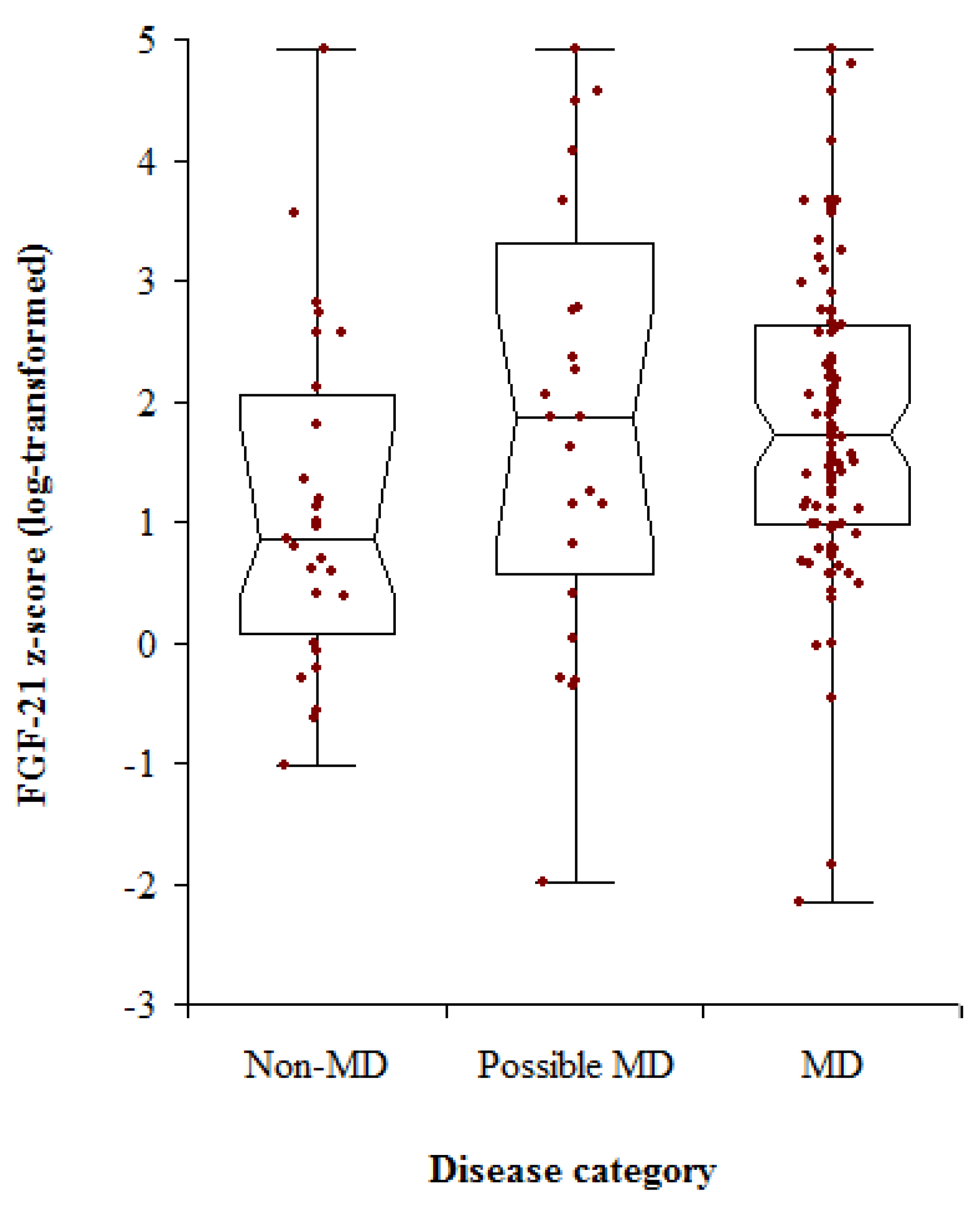
3. Results
3.1. FGF-21 Reference Values
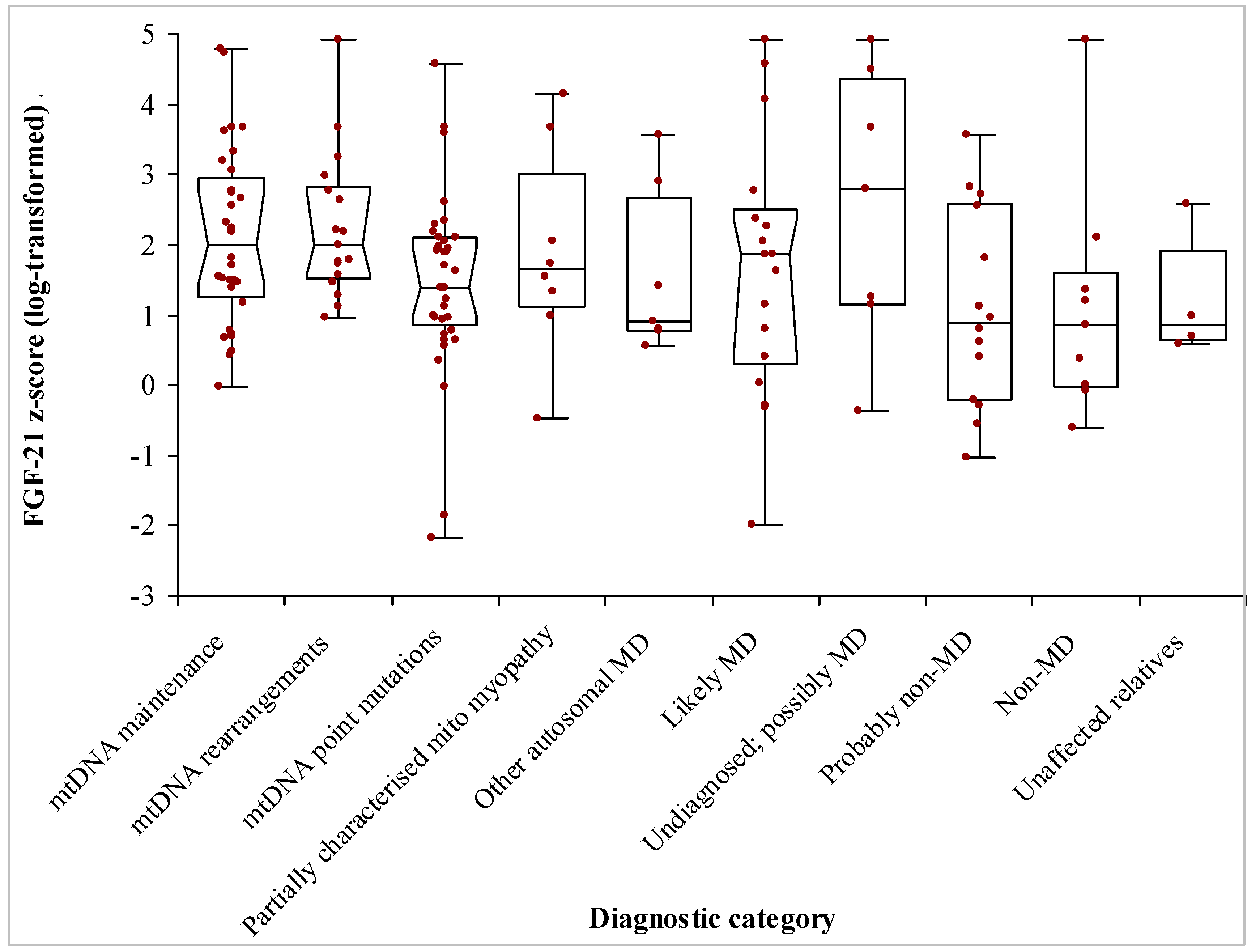
3.2. Patients’ Demographics and Diagnoses
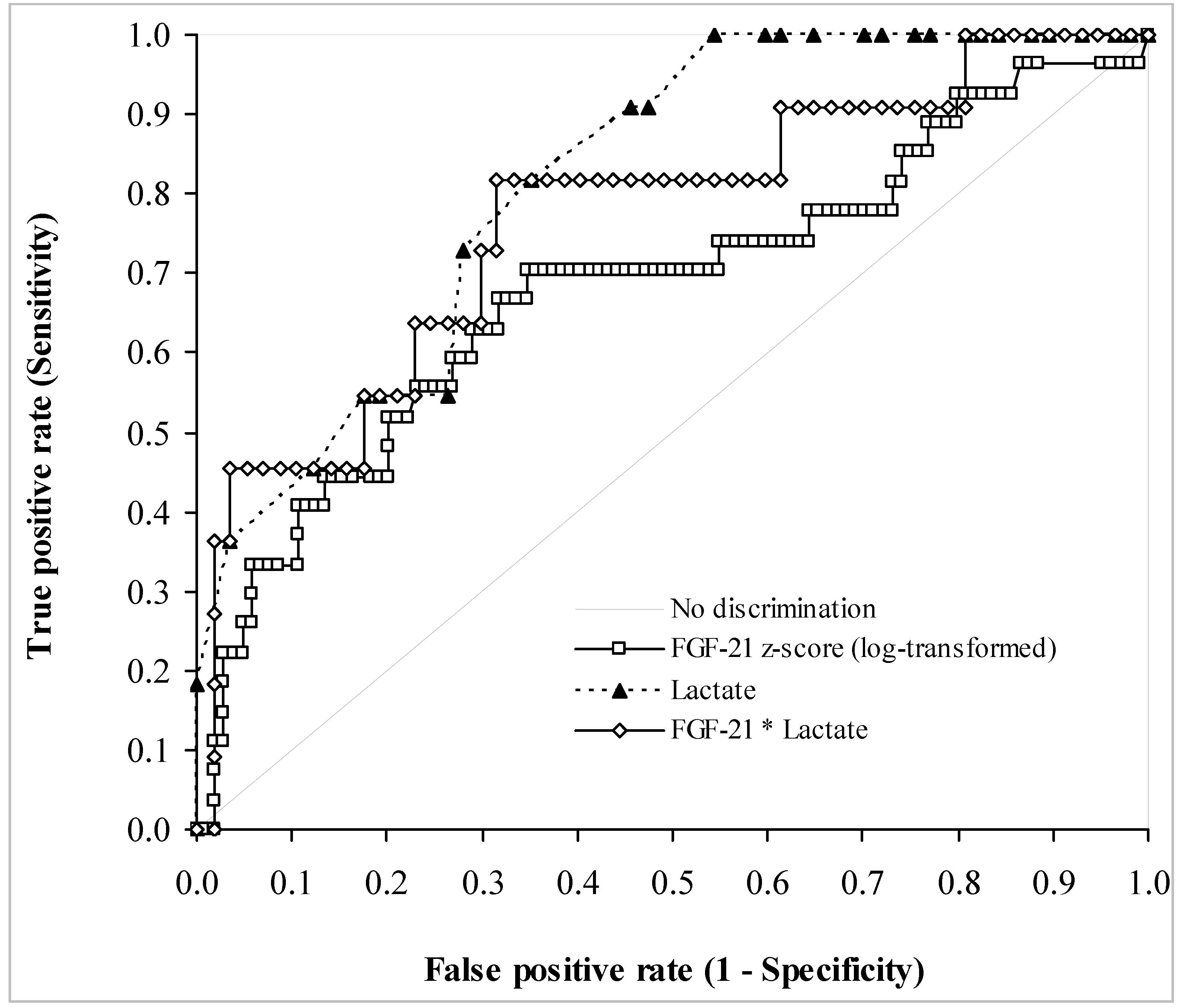
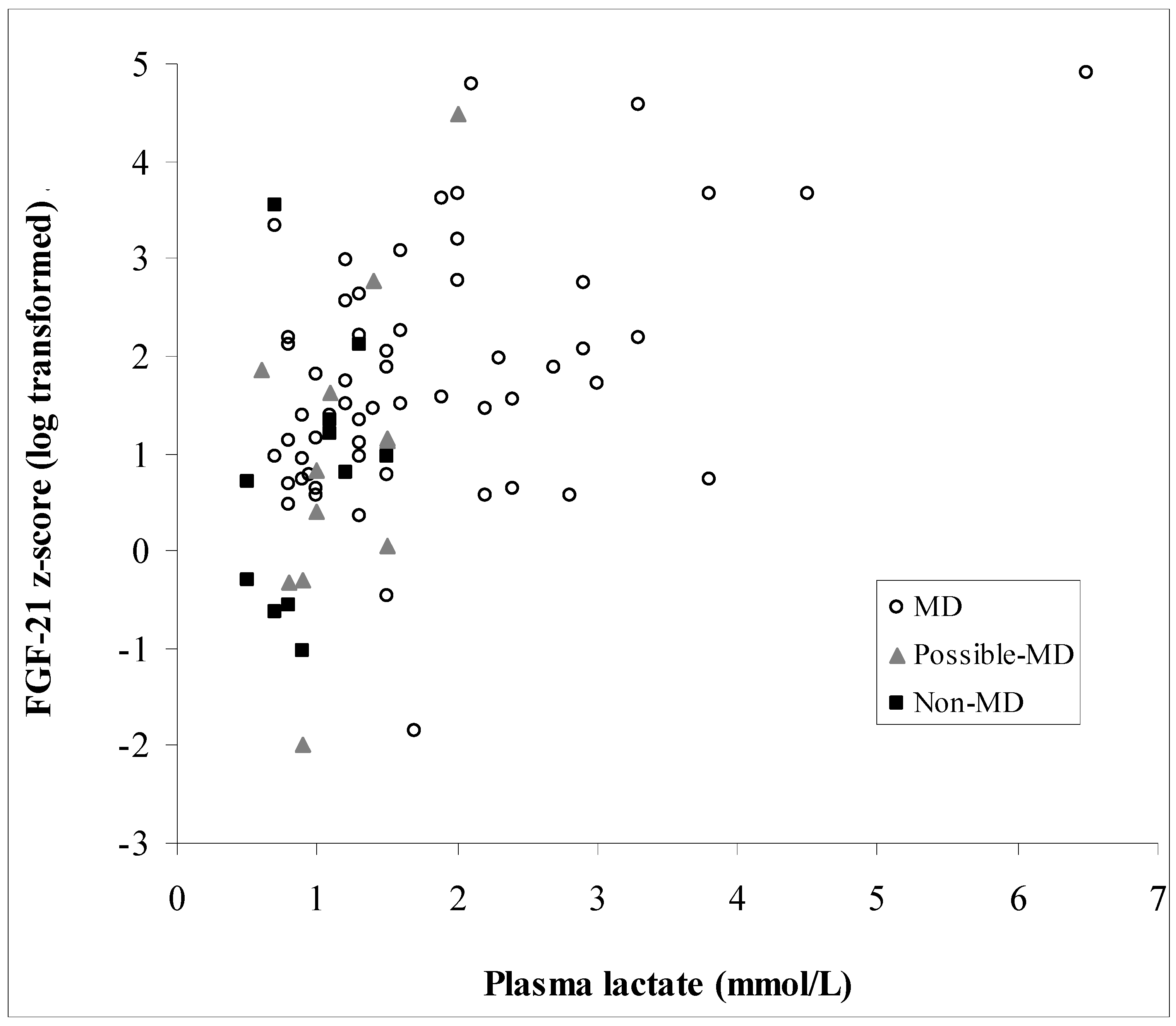
3.3. Biochemical Data
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Manwaring, N.; Jones, M.M.; Wang, J.J.; Rochtchina, E.; Howard, C.; Mitchell, P.; Sue, C.M. Population prevalence of the MELAS A3243G mutation. Mitochondrion 2007, 7, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Skladal, D.; Halliday, J.; Thorburn, D.R. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain 2003, 126, 1905–1912. [Google Scholar] [CrossRef] [PubMed]
- Gorman, G.S.; Schaefer, A.M.; Ng, Y.; Gomez, N.; Blakely, E.L.; Alston, C.L.; Feeney, C.; Horvath, R.; Yu-Wai-Man, P.; Chinnery, P.F.; et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 2015, 77, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Oglesbee, D.; Freedenberg, D.; Kramer, K.A.; Anderson, B.D.; Hahn, S.H. Normal muscle respiratory chain enzymes can complicate mitochondrial disease diagnosis. Pediatr. Neurol. 2006, 35, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Suomalainen, A.; Elo, J.M.; Pietilainen, K.H.; Hakonen, A.H.; Sevastianova, K.; Korpela, M.; Isohanni, P.; Marjavaara, S.K.; Tyni, T.; Kiuru-Enari, S.; et al. FGF-21 as a biomarker for muscle-manifesting mitochondrial respiratory chain deficiencies: A diagnostic study. Lancet Neurol. 2011, 10, 806–818. [Google Scholar] [CrossRef]
- Suomalainen, A. Fibroblast growth factor 21: A novel biomarker for human muscle-manifesting mitochondrial disorders. Expert Opin. Med. Diagn. 2013, 7, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.L.; Liang, C.; Edema-Hildebrand, F.; Riley, C.; Needham, M.; Sue, C.M. Fibroblast growth factor 21 is a sensitive biomarker of mitochondrial disease. Neurology 2013, 81, 1819–1826. [Google Scholar] [CrossRef] [PubMed]
- Woo, Y.C.; Xu, A.; Wang, Y.; Lam, K.S. Fibroblast growth factor 21 as an emerging metabolic regulator: Clinical perspectives. Clin. Endocrinol. 2013, 78, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Christodoulides, C.; Dyson, P.; Sprecher, D.; Tsintzas, K.; Karpe, F. Circulating fibroblast growth factor 21 is induced by peroxisome proliferator-activated receptor agonists but not ketosis in man. J. Clin. Endocrinol. Metab. 2009, 94, 3594–3601. [Google Scholar] [CrossRef] [PubMed]
- Lundåsen, T.; Hunt, M.C.; Nilsson, L.M.; Sanyal, S.; Angelin, B.; Alexson, S.E.; Rudling, M. PPARalpha is a key regulator of hepatic FGF21. Biochem. Biophys. Res. Commun. 2007, 360, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Nakatake, Y.; Konishi, M.; Itoh, N. Identification of a novel FGF, FGF-21, preferentially expressed in the liver. Biochim. Biophys. Acta 2000, 1492, 203–206. [Google Scholar] [CrossRef]
- Fisher, F.M.; Maratos-Flier, E. Understanding the physiology of FGF21. Annu. Rev. Physiol. 2016, 78, 223–241. [Google Scholar] [CrossRef] [PubMed]
- Hojman, P.; Pedersen, M.; Nielsen, A.R.; Krogh-Madsen, R.; Yfanti, C.; Akerstrom, T.; Nielsen, S.; Pedersen, B.K. Fibroblast growth factor-21 is induced in human skeletal muscles by hyperinsulinemia. Diabetes 2009, 58, 2797–2801. [Google Scholar] [CrossRef] [PubMed]
- Planavila, A.; Redondo-Angulo, I.; Ribas, F.; Garrabou, G.; Casademont, J.; Giralt, M.; Villarroya, F. Fibroblast growth factor 21 protects the heart from oxidative stress. Cardiovasc. Res. 2015, 106, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Thiessen, S.E.; Vanhorebeek, I.; Derese, I.; Gunst, J.; Van den Berghe, G. FGF21 Response to critical illness: Effect of blood glucose control and relation with cellular stress and survival. J. Clin. Endocrinol. Metab. 2015, 100, E1319–E1327. [Google Scholar] [CrossRef] [PubMed]
- Ost, M.; Coleman, V.; Voigt, A.; van Schothorst, E.M.; Keipert, S.; van der Stelt, I.; Ringel, S.; Graja, A.; Ambrosi, T.; Kipp, A.P.; et al. Muscle mitochondrial stress adaptation operates independently of endogenous FGF21 action. Mol. Metab. 2015, 5, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Ji, K.; Zheng, J.; Lv, J.; Xu, J.; Ji, X.; Luo, Y.B.; Li, W.; Zhao, Y.; Yan, C. Skeletal muscle increases FGF21 expression in mitochondrial disorders to compensate for energy metabolic insufficiency by activating the mTOR-YY1-PGC1α pathway. Free Radic. Biol. Med. 2015, 84, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Mashili, F.L.; Austin, R.L.; Deshmukh, A.S.; Fritz, T.; Caidahl, K.; Bergdahl, K.; Zierath, J.R.; Chibalin, A.V.; Moller, D.E.; Kharitonenkov, A.; et al. Direct effects of FGF21 on glucose uptake in human skeletal muscle: Implications for type 2 diabetes and obesity. Diabetes Metab. Res. Rev. 2011, 27, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.V.; Li, D.; Yan, Q.; Zhu, Y.; Goodwin, B.; Calle, R.; Brenner, M.B.; Talukdar, S. Fibroblast growth factor 21 improves insulin sensitivity and synergizes with insulin in human adipose stem cell-derived (hASC) adipocytes. PLoS ONE 2014, 9, e111767. [Google Scholar] [CrossRef] [PubMed]
- Crooks, D.R.; Natarajan, T.G.; Jeong, S.Y.; Chen, C.; Park, S.Y.; Huang, H.; Ghosh, M.C.; Tong, W.H.; Haller, R.G.; Wu, C.; et al. Elevated FGF21 secretion, PGC-1α and ketogenic enzyme expression are hallmarks of iron-sulfur cluster depletion in human skeletal muscle. Hum. Mol. Genet. 2014, 23, 24–39. [Google Scholar] [CrossRef] [PubMed]
- Ribas, F.; Villarroya, J.; Hondares, E.; Giralt, M.; Villarroya, F. FGF21 expression and release in muscle cells: Involvement of MyoD and regulation by mitochondria-driven signalling. Biochem. J. 2014, 463, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Lehtonen, J.M.; Forsström, S.; Bottani, E.; Viscomi, C.; Baris, O.R.; Isoniemi, H.; Höckerstedt, K.; Österlund, P.; Hurme, M.; Jylhävä, J.; et al. FGF21 is a biomarker for mitochondrial translation and mtDNA maintenance disorders. Neurology 2016, 87, 2290–2299. [Google Scholar] [CrossRef] [PubMed]
- Koene, S.; de Laat, P.; van Tienoven, D.H.; Vriens, D.; Brandt, A.M.; Sweep, F.C.; Rodenburg, R.J.; Donders, A.R.; Janssen, M.C.; Smeitink, J.A. Serum FGF21 levels in adult m.3243A>G carriers: Clinical implications. Neurology 2014, 83, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Dong, K.; Fang, Q.; Hou, X.; Zhou, M.; Bao, Y.; Xiang, K.; Xu, A.; Jia, W. High serum level of fibroblast growth factor 21 is an independent predictor of non-alcoholic fatty liver disease: A 3-year prospective study in China. J. Hepatol. 2013, 58, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Dushay, J.; Chui, P.C.; Gopalakrishnan, G.S.; Varela-Rey, M.; Crawley, M.; Fisher, F.M.; Badman, M.K.; Martinez-Chantar, M.L.; Maratos-Flier, E. Increased fibroblast growth factor 21 in obesity and nonalcoholic fatty liver disease. Gastroenterology 2010, 139, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Tyynismaa, H.; Raivio, T.; Hakkarainen, A.; Ortega-Alonso, A.; Lundbom, N.; Kaprio, J.; Rissanen, A.; Suomalainen, A.; Pietiläinen, K.H. Liver fat but not other adiposity measures influence circulating FGF21 levels in healthy young adult twins. J. Clin. Endocrinol. Metab. 2011, 96, E351–E355. [Google Scholar] [CrossRef] [PubMed]
- Badman, M.K.; Pissios, P.; Kennedy, A.R.; Koukos, G.; Flier, J.S.; Maratos-Flier, E. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007, 5, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Zhou, Z.; Liu, Y.; Gong, Q.; Yan, X.; Xiao, J.; Wang, X.; Lin, S.; Feng, W.; Li, X. Circulating FGF21 levels are progressively increased from the early to end stages of chronic kidney diseases and are associated with renal function in Chinese. PLoS ONE 2011, 6, e18398. [Google Scholar] [CrossRef] [PubMed]
- Dushay, J.R.; Toschi, E.; Mitten, E.K.; Fisher, F.M.; Herman, M.A.; Maratos-Flier, E. Fructose ingestion acutely stimulates circulating FGF21 levels in humans. Mol. Metab. 2014, 4, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.L.; Liang, C.; Sue, C.M. A comparison of current serum biomarkers as diagnostic indicators of mitochondrial diseases. Neurology 2016, 86, 2010–2015. [Google Scholar] [CrossRef] [PubMed]
- Planavila, A.; Redondo-Angulo, I.; Villarroya, F. FGF21 and Cardiac Physiopathology. Front. Endocrinol. 2015, 6, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Dogan, S.A.; Pujol, C.; Maiti, P.; Kukat, A.; Wang, S.; Hermans, S.; Senft, K.; Wibom, R.; Rugarli, E.I.; Trifunovic, A. Tissue-specific loss of DARS2 activates stress responses independently of respiratory chain deficiency in the heart. Cell Metab. 2014, 19, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Tyynismaa, H.; Carroll, C.J.; Raimundo, N.; Ahola-Erkkilä, S.; Wenz, T.; Ruhanen, H.; Guse, K.; Hemminki, A.; Peltola-Mjøsund, K.E.; Tulkki, V.; et al. Mitochondrial myopathy induces a starvation-like response. Hum. Mol. Genet. 2010, 19, 3948–3958. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; McKeehan, W.L. Stressed liver and muscle call on adipocytes with FGF21. Front. Endocrinol. 2013, 4, 194. [Google Scholar] [CrossRef] [PubMed]
- Jeanson, Y.; Ribas, F.; Galinier, A.; Arnaud, E.; Ducos, M.; André, M.; Chenouard, V.; Villarroya, F.; Casteilla, L.; Carrière, A. Lactate induces FGF21 expression in adipocytes through a p38-MAPK pathway. Biochem. J. 2016, 473, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Jeong, Y.T.; Oh, H.; Kim, S.H.; Cho, J.M.; Kim, Y.N.; Kim, S.S.; Kim, D.H.; Hur, K.Y.; Kim, H.K.; et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing FGF21 as a mitokine. Nat. Med. 2013, 19, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Rath, E.; Berger, E.; Messlik, A.; Nunes, T.; Liu, B.; Kim, S.C.; Hoogenraad, N.; Sans, M.; Sartor, R.B.; Haller, D. Induction of dsRNA-activated protein kinase links mitochondrial unfolded protein response to the pathogenesis of intestinal inflammation. Gut 2012, 61, 1269–1278. [Google Scholar] [CrossRef] [PubMed]
- Michel, S.; Canonne, M.; Arnould, T.; Renard, P. Inhibition of mitochondrial genome expression triggers the activation of CHOP-10 by a cell signaling dependent on the integrated stress response but not the mitochondrial unfolded protein response. Mitochondrion 2015, 21, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Ambrosi, J.; Gallego-Escuredo, J.M.; Catalán, V.; Rodríguez, A.; Domingo, P.; Moncada, R.; Valentí, V.; Salvador, J.; Giralt, M.; Villarroya, F.; et al. FGF19 and FGF21 serum concentrations in human obesity and type 2 diabetes behave differently after diet- or surgically-induced weight loss. Clin. Nutr. 2017, 36, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Wolny, S.; McFarland, R.; Chinnery, P.; Cheetham, T. Abnormal growth in mitochondrial disease. Acta Paediatr. 2009, 98, 553–554. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, J.; Chen, M.Z.; Baruch, A. FGF21-receptor agonists: An emerging therapeutic class for obesity-related diseases. Horm. Mol. Biol. Clin. Investig. 2017. [CrossRef] [PubMed]
- Gaich, G.; Chien, J.Y.; Fu, H.; Glass, L.C.; Deeg, M.A.; Holland, W.L.; Kharitonenkov, A.; Bumol, T.; Schilske, H.K.; Moller, D.E. The effects of LY2405319, an FGF21 analog, in obese human subjects with type 2 diabetes. Cell Metab. 2013, 18, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Phadke, R. Myopathology of adult and paediatric mitochondrial diseases. J. Clin. Med. 2017, 6, 64. [Google Scholar] [CrossRef] [PubMed]
- Kalko, S.G.; Paco, S.; Jou, C.; Rodriguez, M.A.; Meznaric, M.; Rogac, M.; Jekovec-Vrhovsek, M.; Sciacco, M.; Moggio, M.; Fagiolari, G.; et al. Transcriptomic profiling of TK2 deficient human skeletal muscle suggests a role for the p53 signalling pathway and identifies growth and differentiation factor-15 as a potential novel biomarker for mitochondrial myopathies. BMC Genomics 2014, 15, 91. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diagnostic Category | n | Median Age (Range) | Median FGF-21 z-Score (95% C.I.) | FGF-21 z-Score IQR |
|---|---|---|---|---|
| Non-MD | 27 | 45 (0.5–78) | 0.86 (−0.39–1.80) | 2.00 |
| Possible MD | 24 | 11 (0–63) | 1.87 (1.81–2.78) | 2.72 |
| MD | 104 | 39.5 (2–87) | 1.72 (1.46–1.99) | 1.65 |
| -mtDMA maintenance defects | 32 | 50 (7–87) | 1.99 (1.46–2.73) | 1.69 |
| -mtDNA rearrangements | 17 | 35 (2–82) | 1.99 (1.56–2.76) | 1.30 |
| -mt DNA point mutations | 40 | 40 (14–72) | 1.40 (0.99–1.94) | 1.26 |
| -Partially characterised mitochondrial myopathy | 8 | 22.5 (2–71) | 1.64 (−0.47–4.15) | 1.88 |
| -Other autosomal MD | 7 | 36 (3–56) | 0.90 (0.56–3.56) | 1.88 |
| Patients without (cardio)myopathy | 117 | 37 (0–84) | 1.13 (1.30–1.94) | 1.85 |
| Patients with (cardio)myopathy | 39 | 40 (3–87) | 1.74 (1.52–2.11) | 1.35 |
| Patients without ophthalmoplegia | 93 | 35 | 1.33 (0.99–1.88) | 1.88 |
| Patients with ophthalmoplegia | 63 | 43 | 1.74 (1.49–2.11) | 1.51 |
| FGF-21 z-Score | Sensitivity | Specificity | Positive PV | Negative PV |
|---|---|---|---|---|
| 1.35 | 0.65 (0.55–0.74) | 0.70 (0.50–0.86) | 0.90 | 0.35 |
| 2.82 | 0.20 (0.13–0.29) | 0.93 (0.76–0.99) | 0.91 | 0.23 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morovat, A.; Weerasinghe, G.; Nesbitt, V.; Hofer, M.; Agnew, T.; Quaghebeur, G.; Sergeant, K.; Fratter, C.; Guha, N.; Mirzazadeh, M.; et al. Use of FGF-21 as a Biomarker of Mitochondrial Disease in Clinical Practice. J. Clin. Med. 2017, 6, 80. https://doi.org/10.3390/jcm6080080
Morovat A, Weerasinghe G, Nesbitt V, Hofer M, Agnew T, Quaghebeur G, Sergeant K, Fratter C, Guha N, Mirzazadeh M, et al. Use of FGF-21 as a Biomarker of Mitochondrial Disease in Clinical Practice. Journal of Clinical Medicine. 2017; 6(8):80. https://doi.org/10.3390/jcm6080080
Chicago/Turabian StyleMorovat, Alireza, Gayani Weerasinghe, Victoria Nesbitt, Monika Hofer, Thomas Agnew, Geralrine Quaghebeur, Kate Sergeant, Carl Fratter, Nishan Guha, Mehdi Mirzazadeh, and et al. 2017. "Use of FGF-21 as a Biomarker of Mitochondrial Disease in Clinical Practice" Journal of Clinical Medicine 6, no. 8: 80. https://doi.org/10.3390/jcm6080080
APA StyleMorovat, A., Weerasinghe, G., Nesbitt, V., Hofer, M., Agnew, T., Quaghebeur, G., Sergeant, K., Fratter, C., Guha, N., Mirzazadeh, M., & Poulton, J. (2017). Use of FGF-21 as a Biomarker of Mitochondrial Disease in Clinical Practice. Journal of Clinical Medicine, 6(8), 80. https://doi.org/10.3390/jcm6080080