Hypertrophic Cardiomyopathy—Past, Present and Future

Abstract
:1. Introduction
2. Early Descriptions of HCM
3. Left Ventricular Outflow Tract Obstruction (LVOTO)
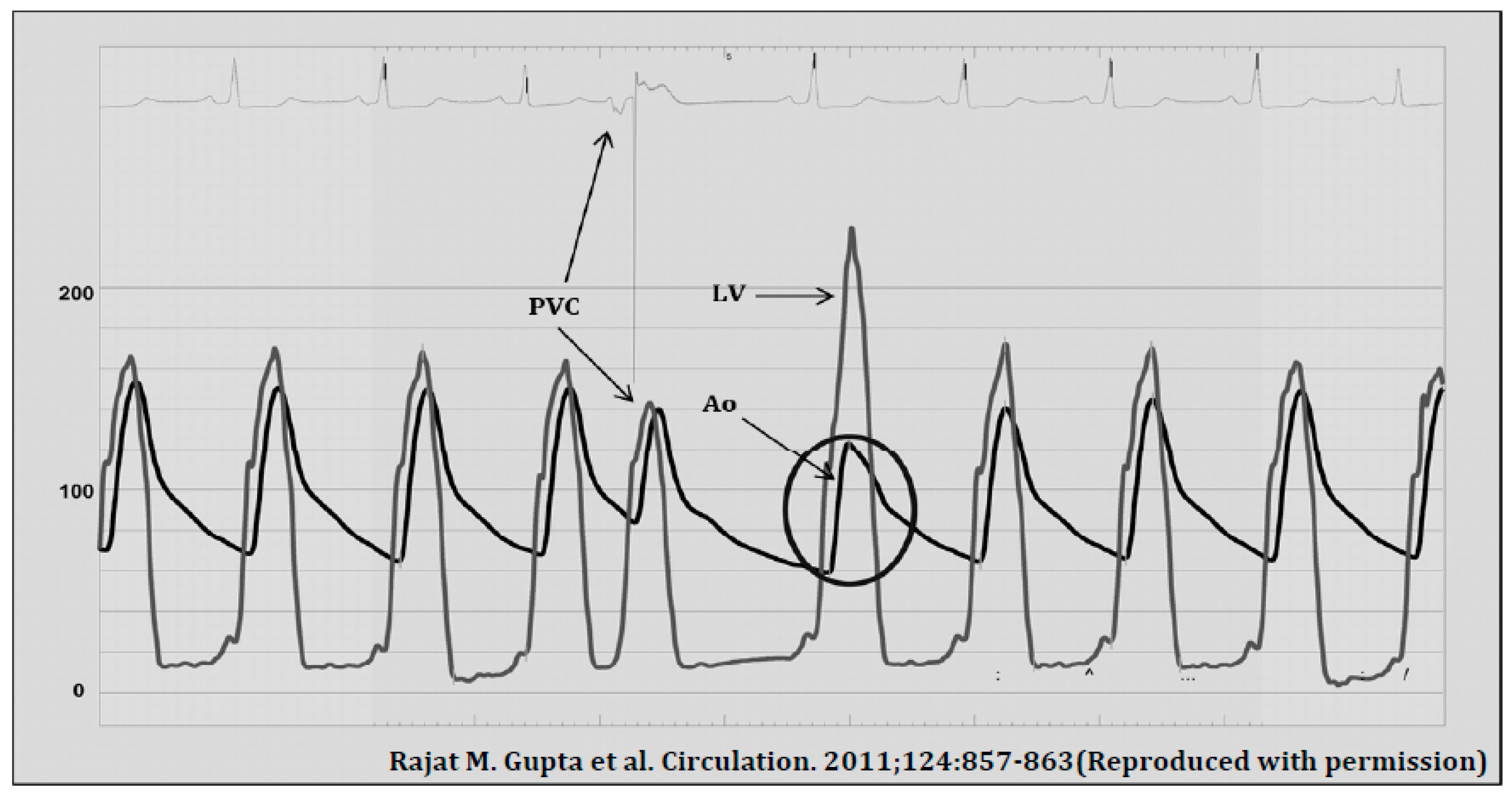
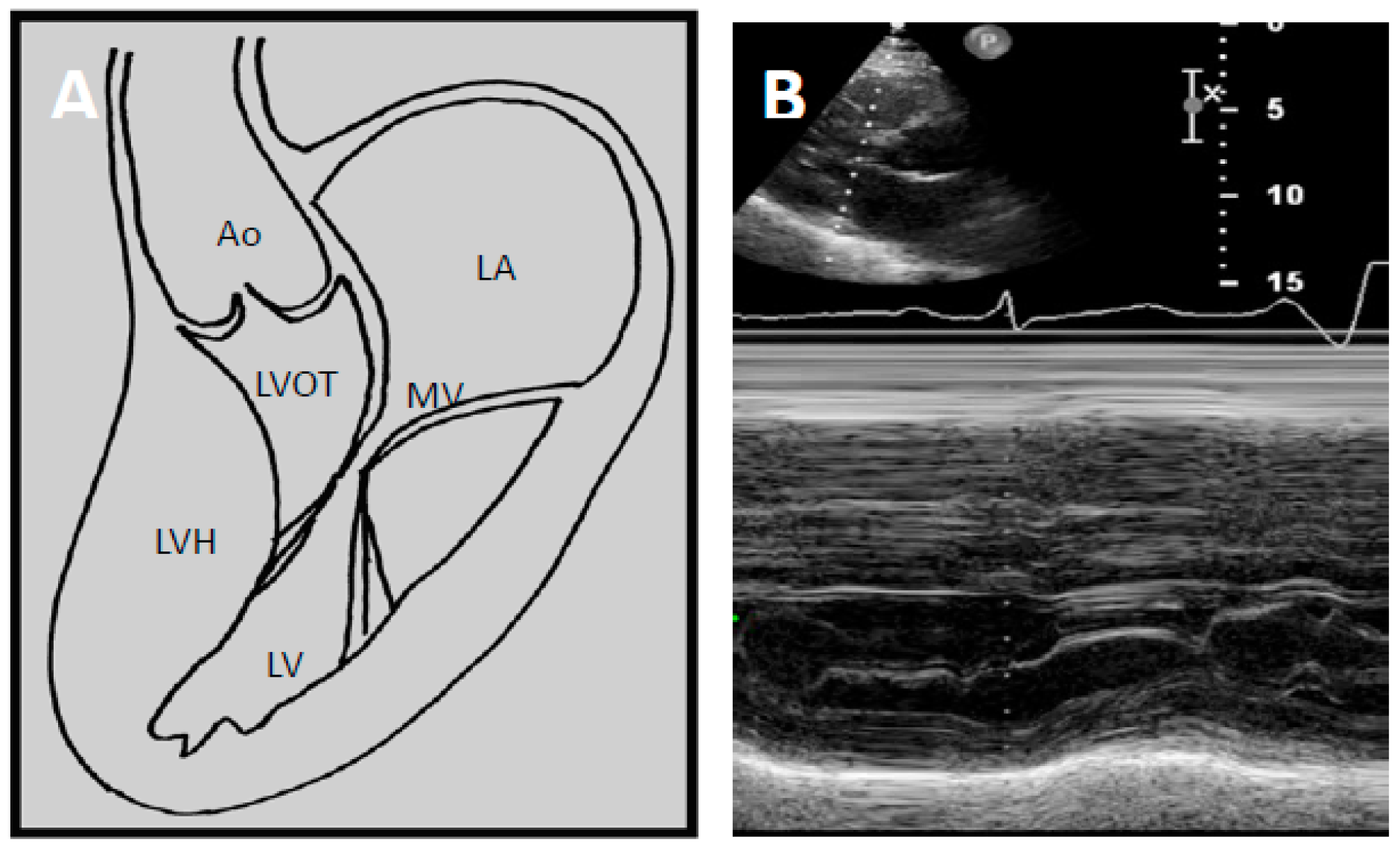
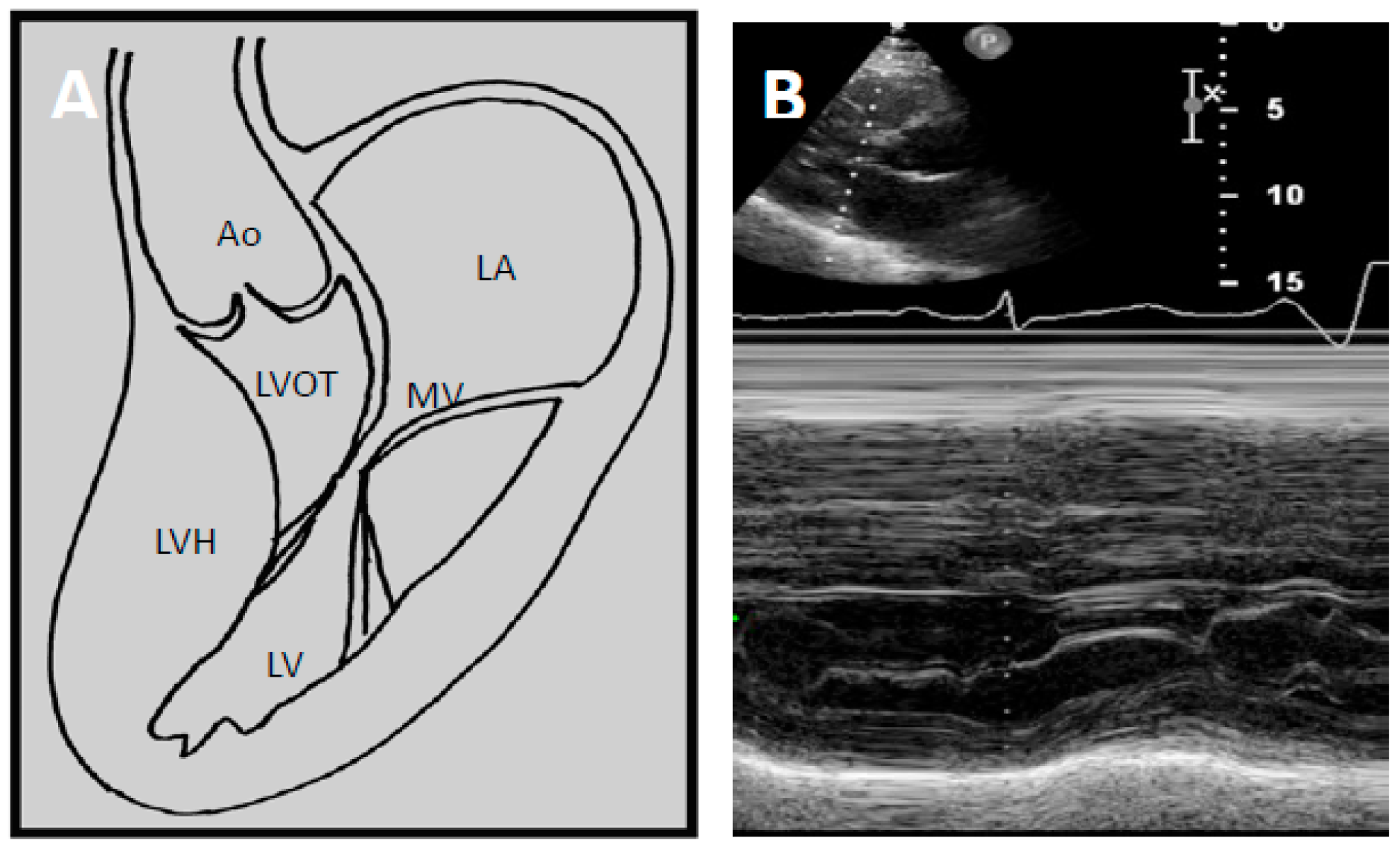
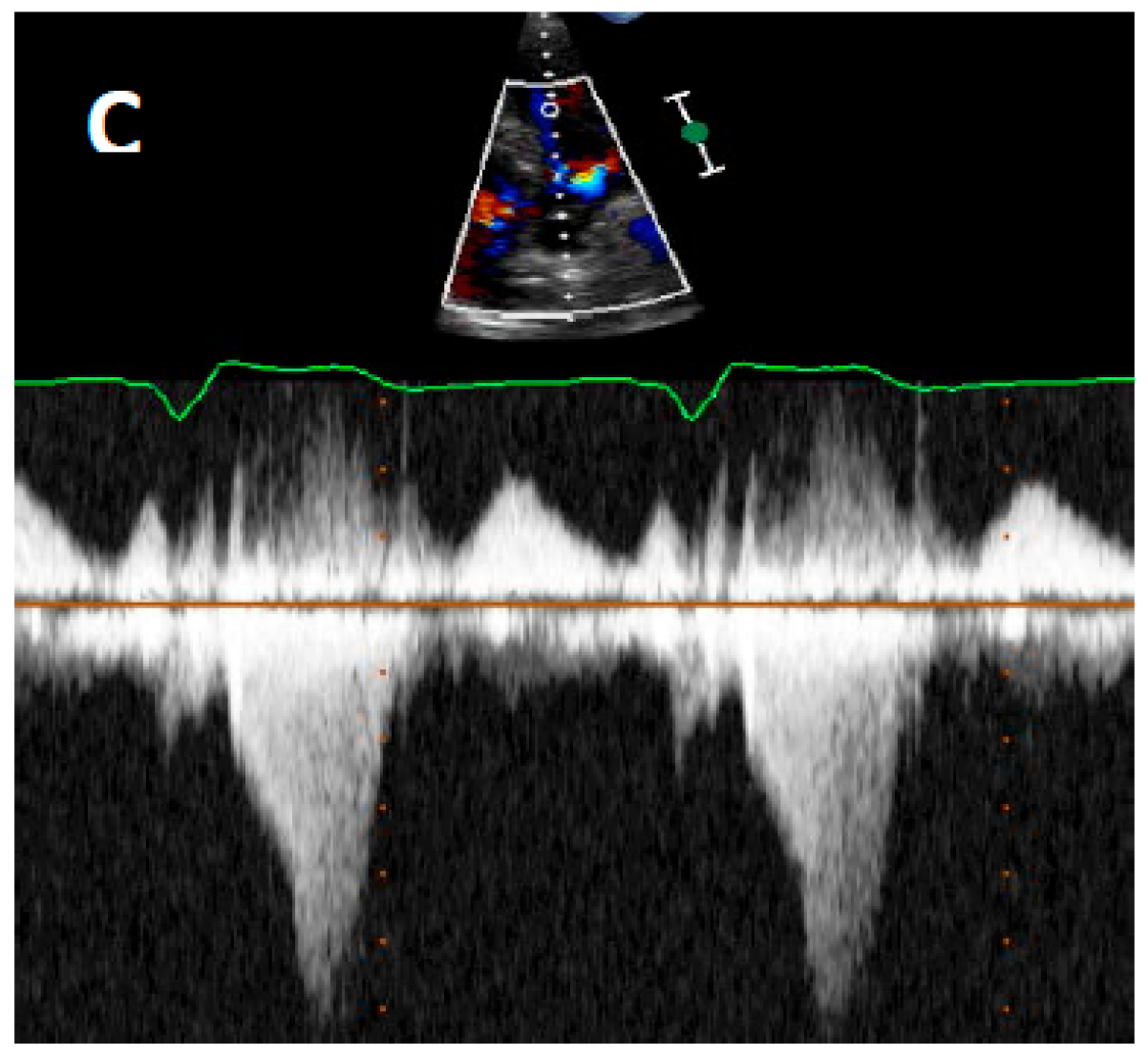
Pathophysiology of LVOTO
4. Diagnosis
4.1. Cardiac Catheterisation
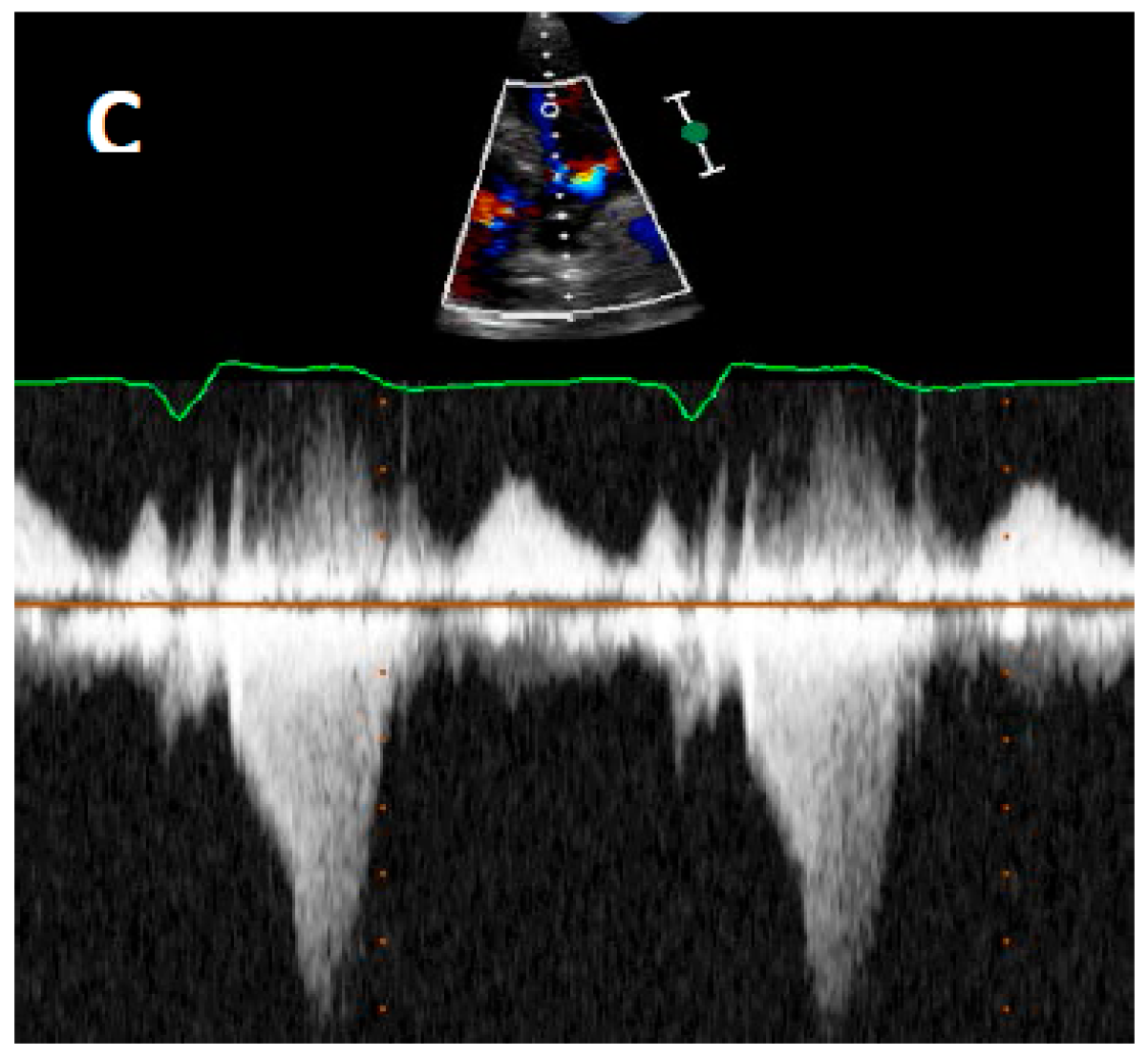
4.2. Echocardiography
5. Current Diagnostic Techniques
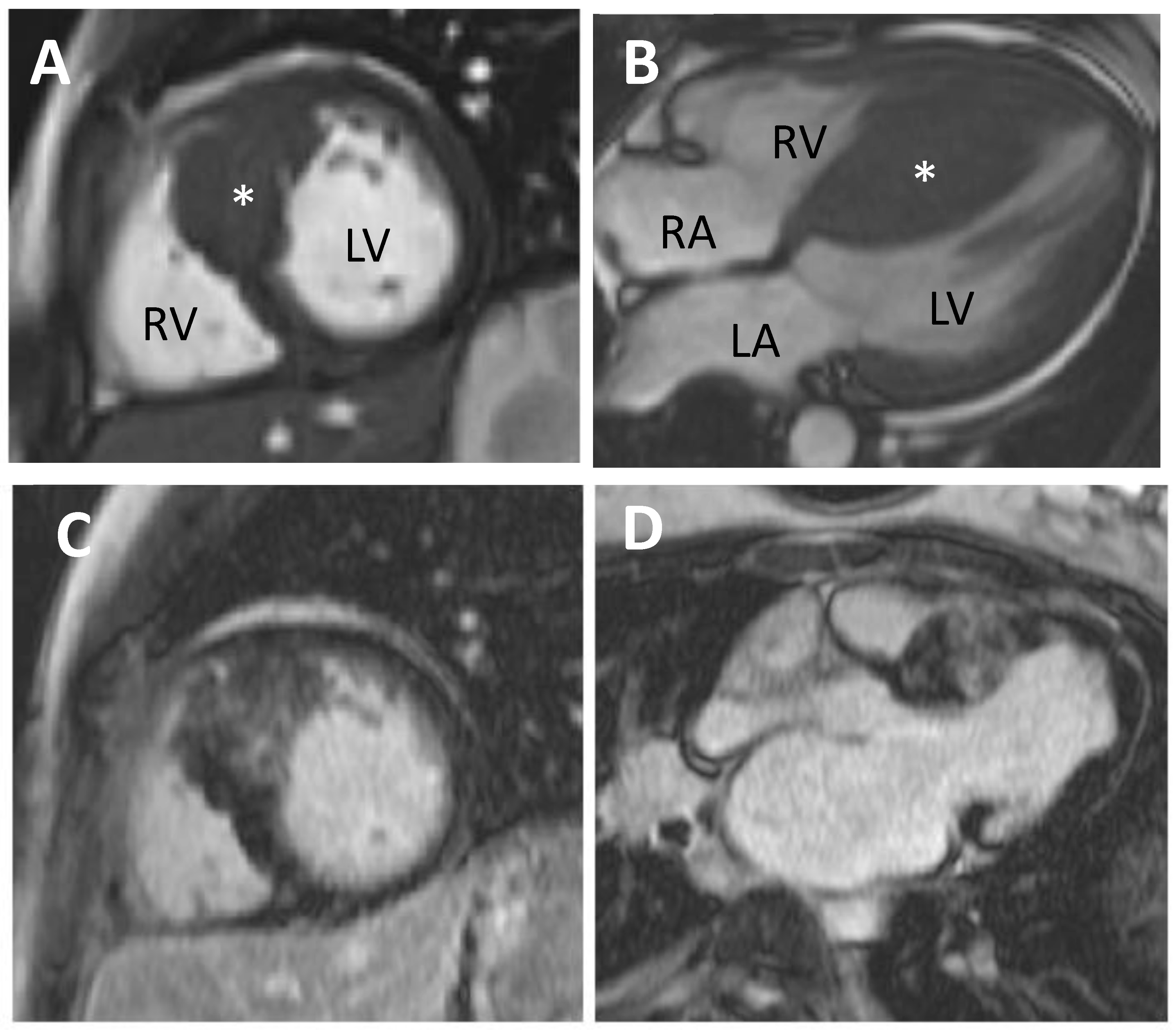
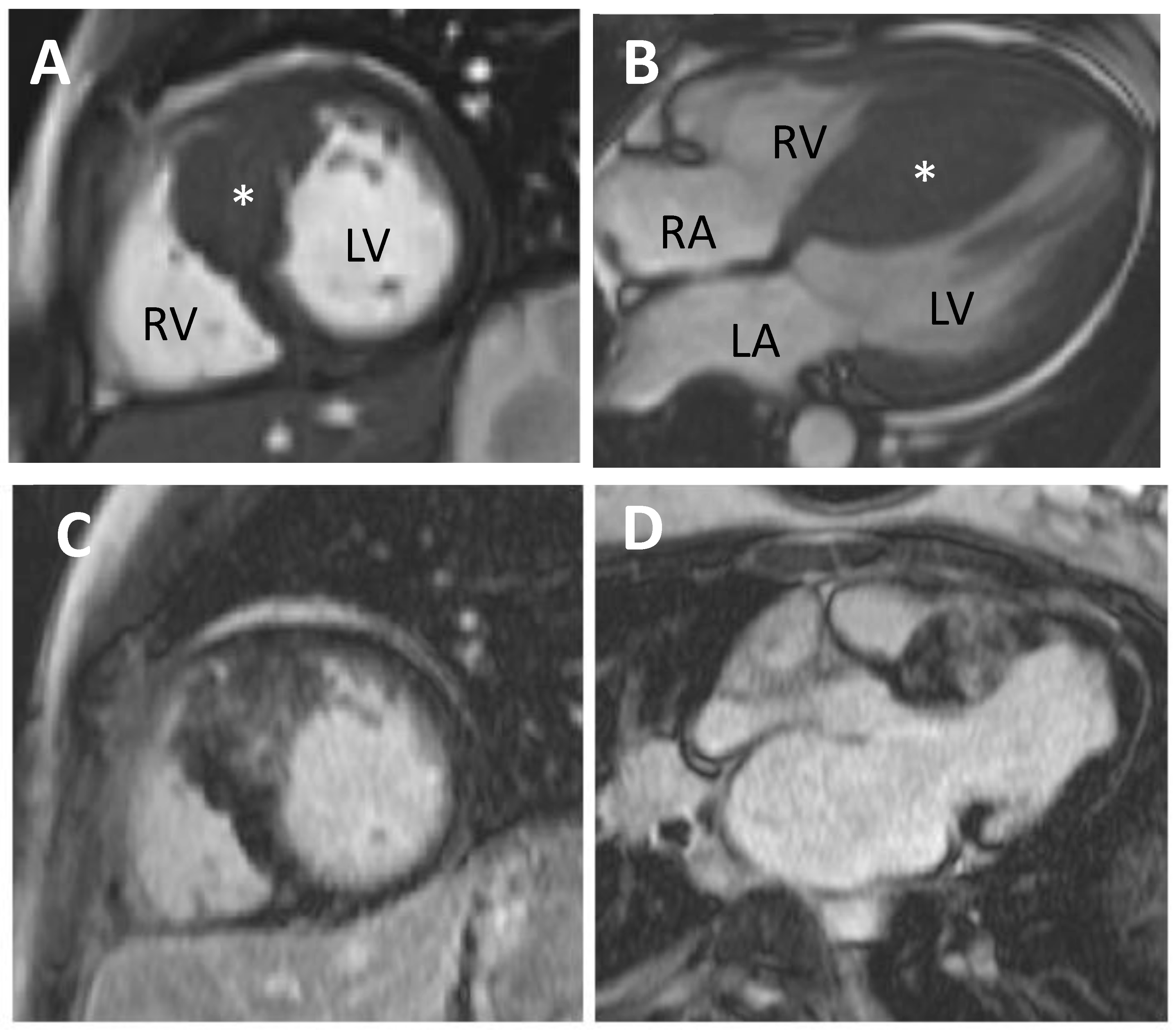
5.1. Cardiovascular Magnetic Resonance (CMR)
5.2. Positron Emission Tomography (PET)
5.3. Genetics
6. Management of HCM
7. Sudden Cardiac Death-Risk Stratification and ICD Therapies
- Maximal LV wall thickness,
- Left atrial diameter,
- Maximal LVOT gradient (at rest or by Valsalva),
- Family history of SCD,
- Unexplained syncope,
- Non-sustained ventricular tachycardia (NSVT), and
- Age.
- NSVT,
- family history of SCD,
- maximal LV wall thickness ≥30 mm,
- unexplained syncope, and
- abnormal blood pressure response during exercise (defined as a decrease in blood pressure or inability to increase blood pressure by ≥20 mmHg during exercise).
8. Management of LVOTO
8.1. Non-Invasive Therapy
8.2. Invasive Therapy
9. Atrial Fibrillation
9.1. Non-Invasive
9.2. Invasive
10. Myocardial Ischaemia in HCM: Why Does It Happen and How Should We Manage It?
11. Future Directions in HCM
11.1. Diagnosis
11.1.1. CMR
11.1.2. Biomarkers
- (1)
- Fibrotic development occurs when the equilibrium between collagen synthesis and degradation is disrupted
- (2)
- Sarcomeric mutations may directly induce the development of fibrosis, irrespective of the presence of ischaemia.
11.1.3. Genetics
11.1.4. Medical Therapy
11.1.5. Interventional
11.1.6. Molecular Therapy
12. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ACC | American College of Cardiology |
| ACCF | American College of Cardiology Foundation |
| AF | Atrial fibrillation |
| AHA | American Heart Association |
| ARB | Angiotensin receptor blocker |
| ASA | Alcohol septal ablation |
| CMR | Cardiovascular magnetic resonance |
| DOAC | Direct oral anticoagulant |
| DT-CMR | Diffusion tensor cardiovascular magnetic resonance |
| ERA | Endocardial radiofrequency ablation |
| ESC | European Society of Cardiology |
| HCM | Hypertrophic cardiomyopathy |
| ICD | Implantable cardioverter defibrillator |
| LA | Left atrium |
| LGE | Late gadolinium enhancement |
| LV | Left ventricle |
| LVEF | Left ventricular ejection fraction |
| LVH | Left ventricular hypertrophy |
| LVOT | Left ventricular outflow tract |
| LVOTO | Left ventricular outflow tract obstruction |
| MV | Mitral valve |
| NASPE | North American Society of Pacing and Electrophysiology |
| NYHA | New York Heart Association |
| PET | Positron Emission Tomography |
| PICP | C-terminal propeptide of type I procollagen |
| PVC | Premature ventricular contraction |
| SAM | Systolic anterior motion |
| SCD | Sudden cardiac death |
| VF | Ventricular fibrillation |
| VT | Ventricular tachycardia |
References
- Teare, D. Asymmetrical hypertrophy of the heart in young adults. Br. Heart J. 1958, 20, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Gardin, J.M.; Flack, J.M.; Gidding, S.S.; Kurosaki, T.T.; Bild, D.E. Prevalence of Hypertrophic Cardiomyopathy in a General Population of Young Adults. Circulation 1995, 92, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Spirito, P.; Autore, C.; Formisano, F.; Assenza, G.E.; Biagini, E.; Haas, T.S.; Bongioanni, S.; Semsarian, C.; Devoto, E.; Musumeci, B.; et al. Risk of sudden death and outcome in patients with hypertrophic cardiomyopathy with benign presentation and without risk factors. Am. J. Cardiol. 2014, 113, 1550–1555. [Google Scholar] [CrossRef] [PubMed]
- Veselka, J.; Zemanek, D.; Jahnlova, D.; Krejci, J.; Januska, J.; Dabrowski, M.; Bartel, T.; Tomasov, P. Risk and Causes of Death in Patients after Alcohol Septal Ablation for Hypertrophic Obstructive Cardiomyopathy. Can. J. Cardiol. 2015, 31, 1245–1251. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Rowin, E.J.; Casey, S.A.; Link, M.S.; Lesser, J.R.; Chan, R.H.M.; Garberich, R.F.; Udelson, J.E.; Maron, M.S. Hypertrophic Cardiomyopathy in Adulthood Associated with Low Cardiovascular Mortality with Contemporary Management Strategies. J. Am. Coll. Cardiol. 2015, 65, 1915–1928. [Google Scholar] [CrossRef] [PubMed]
- Vulpian, A. Contribution à l’étude des rétrécissements de l’orifice ventriculo-aortique. Arch. Physiol. 1868, 3, 456–457. [Google Scholar]
- Liouville, H. Rétrécissement cardiaque sous aortique. Gaz. Med. Paris 1869, 24, 3. [Google Scholar]
- Hallopeau, M. Rétrécissement ventriculo-aortique. Gaz. Med. Paris 1869, 24, 4. [Google Scholar]
- Brock, R. Functional obstruction of the left ventricle; acquired aortic subvalvar stenosis. Guys Hosp. Rep. 1957, 106, 221–238. [Google Scholar] [PubMed]
- Bercu, B.A.; Diettert, G.A.; Danforth, W.H.; Pund, E.E.; Ahlvin, R.C.; Belliveau, R.R. Pseudoaortic stenosis produced by ventricular hypertrophy. Am. J. Med. 1958, 25, 814–818. [Google Scholar] [CrossRef]
- Brockenbrough, E.C.; Braunwald, E.; Morrow, A.G. A Hemodynamic Technic for the Detection of Hypertrophic Subaortic Stenosis. Circulation 1961, 23, 189–194. [Google Scholar] [CrossRef]
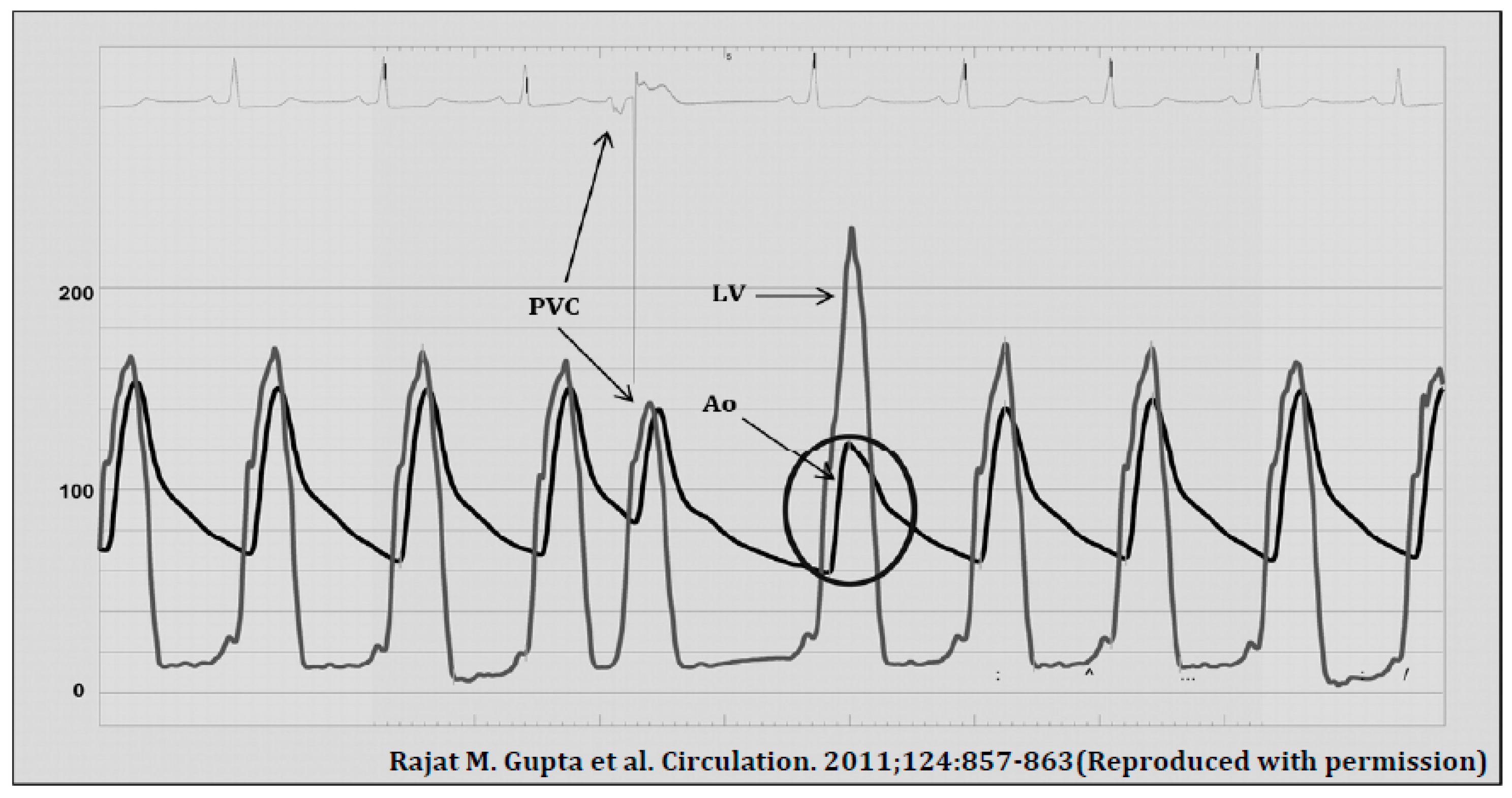
- Gupta, R.M.; Weiner, R.B.; Baggish, A.L.; Fifer, M.A. Still a Kid at Heart. Circulation 2011, 124, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Braunwald, E.; Aygen, M.M. Idiopathic myocardial hypertrophy without congestive heart failure or obstruction to blood flow: Clinical, hemodynamic and angiocardiographic studies in fourteen patients. Am. J. Med. 1963, 35, 7–19. [Google Scholar] [CrossRef]
- Pridie, R.B.; Oakley, C. Mitral valve movement in hypertrophic obstructive cardiomyopathy. Br. Heart J. 1969, 31, 390. [Google Scholar] [PubMed]
- Abbasi, A.S.; MacAlpin, R.N.; Eber, L.M.; Pearce, M.L. Left ventricular hypertrophy diagnosed by echocardiography. N. Engl. J. Med. 1973, 289, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Henry, W.L.; Clark, C.E.; Epstein, S.E. Asymmetric septal hypertrophy. Echocardiographic identification of the pathognomonic anatomic abnormality of IHSS. Circulation 1973, 47, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.M.; Gramiak, R.; Adelman, A.G.; Wigle, E.D. Role of echocardiography in diagnostic and hemodynamic assessment of hypertrophic subaortic stenosis. Circulation 1971, 44, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, A.S.; Macalpin, R.N.; Eber, L.M.; Pearce, M.L. Echocardiographic Diagnosis of Idiopathic Hypertrophic Cardiomyopathy without Outflow Obstruction. Circulation 1972, 46, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Morrow, A.G.; Lambrew, C.; Braunwald, E. Idiopathic Hypertrophic Subaortic Stenosis: II. Operative Treatment and the Results of Pre- and Postoperative Hemodynamic Evaluations. Circulation 1964, 29, 120–151. [Google Scholar] [CrossRef]
- Wehrmacher, W. Cardiomyopathies: Ciba foundation symposium. Arch. Intern. Med. 1965, 116, 642–643. [Google Scholar] [CrossRef]
- Moreyra, E.; Klein, J.J.; Shimada, H.; Segal, B.L. Idiopathic hypertrophic subaortic stenosis diagnosed by reflected ultrasound. Am. J. Cardiol. 1969, 23, 32–37. [Google Scholar] [CrossRef]
- Shah, P.M.; Gramiak, R.; Kramer, D.H. Ultrasound localization of left ventricular outflow obstruction in hypertrophic obstructive cardiomyopathy. Circulation 1969, 40, 3–11. [Google Scholar] [CrossRef] [PubMed]
- King, D. A stop-action technique for imaging intra-cardiac anatomy. Radiology 1972, 103, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Mirowski, M.; Reid, P.R.; Mower, M.M.; Watkins, L.; Gott, V.L.; Schauble, J.F.; Langer, A.; Heilman, M.S.; Kolenik, S.A.; Fischell, R.E.; et al. Termination of Malignant Ventricular Arrhythmias with an Implanted Automatic Defibrillator in Human Beings. N. Engl. J. Med. 1980, 303, 322–324. [Google Scholar] [CrossRef] [PubMed]
- Geisterfer-Lowrance, A.A.; Kass, S.; Tanigawa, G.; Vosberg, H.P.; McKenna, W.; Seidman, C.E.; Seidman, J.G. A molecular basis for familial hypertrophic cardiomyopathy: A beta cardiac myosin heavy chain gene missense mutation. Cell 1990, 62, 999–1006. [Google Scholar] [CrossRef]
- Sigwart, U. Non-surgical myocardial reduction for hypertrophic obstructive cardiomyopathy. Lancet 1995, 346, 211–214. [Google Scholar] [CrossRef]
- Maron, B.J.; Shen, W.K.; Link, M.S.; Epstein, A.E.; Almquist, A.K.; Daubert, J.P.; Bardy, G.H.; Favale, S.; Rea, R.F.; Boriani, G.; et al. Efficacy of implantable cardioverter-defibrillators for the prevention of sudden death in patients with hypertrophic cardiomyopathy. N. Engl. J. Med. 2000, 342, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Gregoratos, G.; Abrams, J.; Epstein, A.E.; Freedman, R.A.; Hayes, D.L.; Hlatky, M.A.; Kerber, R.E.; Naccarelli, G.V.; Schoenfeld, M.H.; Silka, M.J.; et al. ACC/AHA/NASPE 2002 guideline update for implantation of cardiac pacemakers and antiarrhythmia devices: Summary article: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (ACC/AHA/NASPE Committee to. Circulation 2002, 106, 2145–2161. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Casey, S.A.; Poliac, L.C.; Gohman, T.E.; Almquist, A.K.; Aeppli, D.M. Clinical course of hypertrophic cardiomyopathy in a regional United States cohort. JAMA 1999, 281, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Spirito, P.; Bellone, P.; Harris, K.M.; Bernabo, P.; Bruzzi, P.; Maron, B.J. Magnitude of left ventricular hypertrophy and risk of sudden death in hypertrophic cardiomyopathy. N. Engl. J. Med. 2000, 342, 1778–1785. [Google Scholar] [CrossRef] [PubMed]
- Maki, S.; Ikeda, H.; Muro, A.; Yoshida, N.; Shibata, A.; Koga, Y.; Imaizumi, T. Predictors of sudden cardiac death in hypertrophic cardiomyopathy. Am. J. Cardiol. 1998, 82, 774–778. [Google Scholar] [CrossRef]
- Elliott, P.M.; Gimeno, J.R.; Tome, M.T.; Shah, J.; Ward, D.; Thaman, R.; Mogensen, J.; McKenna, W.J. Left ventricular outflow tract obstruction and sudden death risk in patients with hypertrophic cardiomyopathy. Eur. Heart J. 2006, 27, 1933–1941. [Google Scholar] [CrossRef] [PubMed]
- Maron, M.S.; Olivotto, I.; Betocchi, S.; Casey, S.A.; Lesser, J.R.; Losi, M.A.; Cecchi, F.; Maron, B.J. Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy. N. Engl. J. Med. 2003, 348, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Sherrid, M.V.; Chu, C.K.; Delia, E.; Mograder, A.; Dwyer, E.M. An echocardiographic study of the fluid machanics of obstruction in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 1993, 22, 816–825. [Google Scholar] [CrossRef]
- Levine, R.A.; Schwammenthal, E.; Song, J.-K. Diastolic Leading to Systolic Anterior Motion. J. Am. Coll. Cardiol. 2014, 64, 1996–1999. [Google Scholar] [CrossRef] [PubMed]
- Raphael, C.E.; Cooper, R.; Parker, K.H.; Collinson, J.; Vassiliou, V.; Pennell, D.J.; de Silva, R.; Hsu, L.Y.; Greve, A.M.; Nijjer, S.; et al. Mechanisms of Myocardial Ischemia in Hypertrophic Cardiomyopathy: Insights From Wave Intensity Analysis and Magnetic Resonance. J. Am. Coll. Cardiol. 2016, 68, 1651–1660. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.; Braunwald, E.; Gault, J.H.; Mason, D.T.; Morrow, A.G. The Mechanism of the Intraventricular Pressure Gradient in Idiopathic Hypertrophic Subaortic Stenosis. Circulation 1966, 34, 558–578. [Google Scholar] [CrossRef] [PubMed]
- Adelman, A.G.; McLoughlin, M.J.; Marquis, Y.; Auger, P.; Wigle, E.D. Left ventricular cineangiographic observations in muscular subaortic stenosis. Am. J. Cardiol. 1969, 24, 689–697. [Google Scholar] [CrossRef]
- Simon, A.L.; Ross, J.; Gault, J.H. Angiographic Anatomy of the Left Ventricle and Mitral Valve in Idiopathic Hypertrophic Subaortic Stenosis. Circulation 1967, 36, 852–867. [Google Scholar] [CrossRef] [PubMed]
- Stokke, T.M.; Hasselberg, N.E.; Smedsrud, M.K.; Sarvari, S.I.; Haugaa, K.H.; Smiseth, O.A.; Edvardsen, T.; Remme, E.W. Geometry as a Confounder When Assessing Ventricular Systolic Function: Comparison Between Ejection Fraction and Strain. J. Am. Coll. Cardiol. 2017, 70, 942–954. [Google Scholar] [CrossRef] [PubMed]
- Tower-Rader, A.; Betancor, J.; Popovic, Z.B.; Sato, K.; Thamilarasan, M.; Smedira, N.G.; Lever, H.M.; Desai, M.Y. Incremental Prognostic Utility of Left Ventricular Global Longitudinal Strain in Hypertrophic Obstructive Cardiomyopathy Patients and Preserved Left Ventricular Ejection Fraction. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Di Salvo, G.; Pacileo, G.; Limongelli, G.; Baldini, L.; Rea, A.; Verrengia, M.; D’Andrea, A.; Russo, M.G.; Calabro, R. Non sustained ventricular tachycardia in hypertrophic cardiomyopathy and new ultrasonic derived parameters. J. Am. Soc. Echocardiogr. 2010, 23, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Correia, E.; Rodrigues, B.; Santos, L.F.; Moreira, D.; Gama, P.; Cabral, C.; Santos, O. Longitudinal left ventricular strain in hypertrophic cardiomyopathy: Correlation with nonsustained ventricular tachycardia. Echocardiography 2011, 28, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; Mahrholdt, H.; et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: The task force for the diagnosis and management of hypertrophic cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar] [CrossRef] [PubMed]
- Gersh, B.J.; Maron, B.J.; Bonow, R.O.; Dearani, J.A.; Fifer, M.A.; Link, M.S.; Naidu, S.S.; Nishimura, R.A.; Ommen, S.R.; Rakowski, H.; et al. 2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy: Executive Summary: A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2011, 124, 2761–2796. [Google Scholar] [CrossRef] [PubMed]
- Matsumori, A. Hepatitis C Virus Infection and Cardiomyopathies. Circ. Res. 2005, 96, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Veselka, J.; Anavekar, N.S.; Charron, P. Hypertrophic obstructive cardiomyopathy. Lancet 2017, 389, 1253–1267. [Google Scholar] [CrossRef]
- Noureldin, R.A.; Liu, S.; Nacif, M.S.; Judge, D.P.; Halushka, M.K.; Abraham, T.P.; Ho, C.; Bluemke, D.A. The diagnosis of hypertrophic cardiomyopathy by cardiovascular magnetic resonance. J. Cardiovasc. Magn. Reson. 2012, 14. [Google Scholar] [CrossRef] [PubMed]
- Bruder, O.; Wagner, A.; Jensen, C.J.; Schneider, S.; Ong, P.; Kispert, E.M.; Nassenstein, K.; Schlosser, T.; Sabin, G.V.; Sechtem, U.; et al. Myocardial scar visualized by cardiovascular magnetic resonance imaging predicts major adverse events in patients with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2010, 56, 875–887. [Google Scholar] [CrossRef] [PubMed]
- O’Hanlon, R.; Grasso, A.; Roughton, M.; Moon, J.C.; Clark, S.; Wage, R.; Webb, J.; Kulkarni, M.; Dawson, D.; Sulaibeekh, L.; et al. Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2010, 56, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Briasoulis, A.; Mallikethi-Reddy, S.; Palla, M.; Alesh, I.; Afonso, L. Myocardial fibrosis on cardiac magnetic resonance and cardiac outcomes in hypertrophic cardiomyopathy: A meta-analysis. Heart 2015, 101, 1406–1411. [Google Scholar] [CrossRef] [PubMed]
- Weng, Z.; Yao, J.; Chan, R.H.; He, J.; Yang, X.; Zhou, Y.; He, Y. Prognostic Value of LGE-CMR in HCM: A Meta-Analysis. JACC Cardiovasc. Imaging 2016, 9, 1392–1402. [Google Scholar] [CrossRef] [PubMed]
- Poutanen, T.; Tikanoja, T.; Jaaskelainen, P.; Jokinen, E.; Silvast, A.; Laakso, M.; Kuusisto, J. Diastolic dysfunction without left ventricular hypertrophy is an early finding in children with hypertrophic cardiomyopathy-causing mutations in the beta-myosin heavy chain, alpha-tropomyosin, and myosin-binding protein C genes. Am. Heart J. 2006, 151. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.M.; Towbin, J.A.; Ackerman, M.J. Diagnostic, prognostic, and therapeutic implications of genetic testing for hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2009, 54, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Fokstuen, S.; Lyle, R.; Munoz, A.; Gehrig, C.; Lerch, R.; Perrot, A.; Osterziel, K.J.; Geier, C.; Beghetti, M.; Mach, F.; et al. A DNA resequencing array for pathogenic mutation detection in hypertrophic cardiomyopathy. Hum. Mutat. 2008, 29, 879–885. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Seidman, J.G.; Seidman, C.E. The genetic basis for cardiac remodeling. Annu. Rev. Genom. Hum. Genet. 2005, 6, 185–216. [Google Scholar] [CrossRef] [PubMed]
- Marian, A.J.; Braunwald, E. Hypertrophic Cardiomyopathy. Circ. Res. 2017, 121, 749–770. [Google Scholar] [CrossRef] [PubMed]
- Van Driest, S.L.; Ellsworth, E.G.; Ommen, S.R.; Tajik, A.J.; Gersh, B.J.; Ackerman, M.J. Prevalence and spectrum of thin filament mutations in an outpatient referral population with hypertrophic cardiomyopathy. Circulation 2003, 108, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Richard, P.; Charron, P.; Carrier, L.; Ledeuil, C.; Cheav, T.; Pichereau, C.; Benaiche, A.; Isnard, R.; Dubourg, O.; Burban, M.; et al. Hypertrophic cardiomyopathy: Distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 2003, 107, 2227–2232. [Google Scholar] [CrossRef] [PubMed]
- Jahnlova, D.; Tomasov, P.; Zemanek, D.; Veselka, J. Transatlantic differences in assessment of risk of sudden cardiac death in patients with hypertrophic cardiomyopathy. Int. J. Cardiol. 2015, 186, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Casey, S.A.; Chan, R.H.; Garberich, R.F.; Rowin, E.J.; Maron, M.S. Independent Assessment of the European Society of Cardiology Sudden Death Risk Model for Hypertrophic Cardiomyopathy. Am. J. Cardiol. 2015, 116, 757–764. [Google Scholar] [CrossRef] [PubMed]
- O’Mahony, C.; Lambiase, P.D.; Quarta, G.; Cardona, M.; Calcagnino, M.; Tsovolas, K.; Al-Shaikh, S.; Rahman, S.M.; Arnous, S.; Jones, S.; et al. The long-term survival and the risks and benefits of implantable cardioverter defibrillators in patients with hypertrophic cardiomyopathy. Heart 2012, 98, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Axelsson, A.; Iversen, K.; Vejlstrup, N.; Ho, C.; Norsk, J.; Langhoff, L.; Ahtarovski, K.; Corell, P.; Havndrup, O.; Jensen, M.; et al. Efficacy and safety of the angiotensin II receptor blocker losartan for hypertrophic cardiomyopathy: The INHERIT randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2015, 3, 123–131. [Google Scholar] [CrossRef]
- Heric, B.; Lytle, B.W.; Miller, D.P.; Rosenkranz, E.R.; Lever, H.M.; Cosgrove, D.M. Surgical management of hypertrophic obstructive cardiomyopathy: Early and late results. J. Thorac. Cardiovasc. Surg. 1995, 110, 195–198. [Google Scholar] [CrossRef]
- Robbins, R.C.; Stinson, E.B. Long-term results of left ventricular myotomy and myectomy for obstructive hypertrophic cardiomyopathy. J. Thorac. Cardiovasc. Surg. 1996, 111, 586–594. [Google Scholar] [CrossRef]
- Schonbeck, M.H.; Brunner-La Rocca, H.P.; Vogt, P.R.; Lachat, M.L.; Jenni, R.; Hess, O.M.; Turina, M.I. Long-term follow-up in hypertrophic obstructive cardiomyopathy after septal myectomy. Ann. Thorac. Surg. 1998, 65, 1207–1214. [Google Scholar] [CrossRef]
- Schulte, H.D.; Borisov, K.; Gams, E.; Gramsch-Zabel, H.; Losse, B.; Schwartzkopff, B. Management of symptomatic hypertrophic obstructive cardiomyopathy—Long-term results after surgical therapy. Thorac. Cardiovasc. Surg. 1999, 47, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Ommen, S.R.; Maron, B.J.; Olivotto, I.; Maron, M.S.; Cecchi, F.; Betocchi, S.; Gersh, B.J.; Ackerman, M.J.; McCully, R.B.; Dearani, J.A.; et al. Long-term effects of surgical septal myectomy on survival in patients with obstructive hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2005, 46, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Bonow, R. 2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy: Executive Summary: A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J. Thorac. Cardiovasc. Surg. 2011, 142, 1303–1338. [Google Scholar] [CrossRef]
- Veselka, J.; Jensen, M.K.; Liebregts, M.; Januska, J.; Krejci, J.; Bartel, T.; Dabrowski, M.; Hansen, P.R.; Almaas, V.M.; Seggewiss, H.; et al. Long-term clinical outcome after alcohol septal ablation for obstructive hypertrophic cardiomyopathy: Results from the Euro-ASA registry. Eur. Heart J. 2016, 37, 1517–1523. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Tuzcu, E.M.; Desai, M.Y.; Smedira, N.; Lever, H.M.; Lytle, B.W.; Kapadia, S.R. Updated meta-analysis of septal alcohol ablation versus myectomy for hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2010, 55, 823–834. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.; Dokainish, H.; Lakkis, N.M. Hypertrophic obstructive cardiomyopathy-alcohol septal ablation vs. myectomy: A meta-analysis. Eur. Heart J. 2009, 30, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Wang, F.; Dou, X.; Zhang, S.; Pu, J. Comparison of percutaneous transluminal septal myocardial ablation versus septal myectomy for the treatment of patients with hypertrophic obstructive cardiomyopathy—A meta analysis. Int. J. Cardiol. 2006, 112, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, R.A.; Kransdorf, E.P.; Simel, D.L.; Wang, A. Meta-analyses of septal reduction therapies for obstructive hypertrophic cardiomyopathy: Comparative rates of overall mortality and sudden cardiac death after treatment. Circ. Cardiovasc. Interv. 2010, 3, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.K.; Prinz, C.; Horstkotte, D.; van Buuren, F.; Bitter, T.; Faber, L.; Bundgaard, H. Alcohol septal ablation in patients with hypertrophic obstructive cardiomyopathy: Low incidence of sudden cardiac death and reduced risk profile. Heart 2013, 99, 1012–1017. [Google Scholar] [CrossRef] [PubMed]
- McLeod, C.J.; Ommen, S.R.; Ackerman, M.J.; Weivoda, P.L.; Shen, W.K.; Dearani, J.A.; Schaff, H.V.; Tajik, A.J.; Gersh, B.J. Surgical septal myectomy decreases the risk for appropriate implantable cardioverter defibrillator discharge in obstructive hypertrophic cardiomyopathy. Eur. Heart J. 2007, 28, 2583–2588. [Google Scholar] [CrossRef] [PubMed]
- Dearani, J.A.; Ommen, S.R.; Gersh, B.J.; Schaff, H.V.; Danielson, G.K. Surgery insight: Septal myectomy for obstructive hypertrophic cardiomyopathy—The Mayo Clinic experience. Nat. Clin. Pract. Cardiovasc. Med. 2007, 4, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Schaff, H.V.; Dearani, J.A.; Ommen, S.R.; Sorajja, P.; Nishimura, R.A. Expanding the indications for septal myectomy in patients with hypertrophic cardiomyopathy: Results of operation in patients with latent obstruction. J. Thorac. Cardiovasc. Surg. 2012, 143, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Veselka, J.; Krejčí, J.; Tomašov, P.; Zemánek, D. Long-term survival after alcohol septal ablation for hypertrophic obstructive cardiomyopathy: A comparison with general population. Eur. Heart J. 2014, 35, 2040–2045. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, H.; Seggewiss, H.; Gietzen, F.H.; Boekstegers, P.; Neuhaus, L.; Seipel, L. Catheter-based therapy for hypertrophic obstructive cardiomyopathy. First in-hospital outcome analysis of the German TASH Registry. Z. Kardiol. 2004, 93, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Dearani, J.A.; Ommen, S.R.; Maron, M.S.; Schaff, H.V.; Nishimura, R.A.; Ralph-Edwards, A.; Rakowski, H.; Sherrid, M.V.; Swistel, D.G.; et al. Low Operative Mortality Achieved With Surgical Septal Myectomy: Role of Dedicated Hypertrophic Cardiomyopathy Centers in the Management of Dynamic Subaortic Obstruction. J. Am. Coll. Cardiol. 2015, 66, 1307–1308. [Google Scholar] [CrossRef] [PubMed]
- Guttmann, O.P.; Rahman, M.S.; O’Mahony, C.; Anastasakis, A.; Elliott, P.M. Atrial fibrillation and thromboembolism in patients with hypertrophic cardiomyopathy: Systematic review. Heart 2014, 100, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Merinopoulos, I.; Raphael, C.E.; Yardley, A.; Goonewardene, M.; Vassiliou, V.S. Device-identified atrial fibrillation at pacing clinics. Should it guide anticoagulation? Int. J. Cardiol. 2016, 207, 378–381. [Google Scholar] [CrossRef] [PubMed]
- Olivotto, I.; Cecchi, F.; Casey, S.A.; Dolara, A.; Traverse, J.H.; Maron, B.J. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation 2001, 104, 2517–2524. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Wang, Y.; Sun, K.; Wang, J.; Zou, Y.; Zhang, W.; Bao, J.; Zhu, L.; Shen, H.; Hui, R.; et al. Clinical profile and prognostic significance of atrial fibrillation in hypertrophic cardiomyopathy. Cardiology 2013, 126, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.; Frenneaux, M.P.; Stockins, B.; Karatasakis, G.; Poloniecki, J.D.; McKenna, W.J. Atrial fibrillation in hypertrophic cardiomyopathy: A longitudinal study. J. Am. Coll. Cardiol. 1990, 15, 1279–1285. [Google Scholar] [CrossRef]
- Providencia, R.; Elliott, P.; Patel, K.; McCready, J.; Babu, G.; Srinivasan, N.; Bronis, K.; Papageorgiou, N.; Chow, A.; Rowland, E.; et al. Catheter ablation for atrial fibrillation in hypertrophic cardiomyopathy: A systematic review and meta-analysis. Heart 2016, 102, 1533–1543. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.M.; Kaski, J.C.; Prasad, K.; Seo, H.; Slade, A.K.; Goldman, J.H.; McKenna, W.J. Chest pain during daily life in patients with hypertrophic cardiomyopathy: An ambulatory electrocardiographic study. Eur. Heart J. 1996, 17, 1056–1064. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J. Hypertrophic cardiomyopathy: A systematic review. JAMA 2002, 287, 1308–1320. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; McKenna, W.J.; Danielson, G.K.; Kappenberger, L.J.; Kuhn, H.J.; Seidman, C.E.; Shah, P.M.; Spencer, W.H., III; Spirito, P.; Ten Cate, F.J.; et al. American College of Cardiology/European Society of Cardiology Clinical Expert Consensus Document on Hypertrophic Cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. Eur. Heart J. 2003, 24, 1965–1991. [Google Scholar] [PubMed]
- Pasternac, A.; Noble, J.; Streulens, Y.; Elie, R.; Henschke, C.; Bourassa, M.G. Pathophysiology of chest pain in patients with cardiomyopathies and normal coronary arteries. Circulation 1982, 65, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Basso, C.; Thiene, G.; Corrado, D.; Buja, G.; Melacini, P.; Nava, A. Hypertrophic cardiomyopathy and sudden death in the young: Pathologic evidence of myocardial ischemia. Hum. Pathol. 2000, 31, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Gori, F.; Basso, C.; Thiene, G. Myocardial Infarction in a Patient with Hypertrophic Cardiomyopathy. N. Engl. J. Med. 2000, 342, 593–594. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Epstein, S.E.; Roberts, W.C. Hypertrophic cardiomyopathy and transmural myocardial infarction without significant atherosclerosis of the extramural coronary arteries. Am. J. Cardiol. 1979, 43, 1086–1102. [Google Scholar] [CrossRef]
- Maron, B.J.; Wolfson, J.K.; Epstein, S.E.; Roberts, W.C. Intramural (“small vessel”) coronary artery disease in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 1986, 8, 545–557. [Google Scholar] [CrossRef]
- O’Gorman, D.J.; Sheridan, D.J. Abnormalities of the coronary circulation associated with left ventricular hypertrophy. Clin. Sci. (Lond.) 1991, 81, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Varnava, A.M.; Elliott, P.M.; Sharma, S.; McKenna, W.J.; Davies, M.J. Hypertrophic cardiomyopathy: The interrelation of disarray, fibrosis, and small vessel disease. Heart 2000, 84, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Cecchi, F.; Olivotto, I.; Gistri, R.; Lorenzoni, R.; Chiriatti, G.; Camici, P.G. Coronary microvascular dysfunction and prognosis in hypertrophic cardiomyopathy. N. Engl. J. Med. 2003, 349, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- Haley, J.H.; Miller, T.D. Myocardial Ischemia on Thallium Scintigraphy in Hypertrophic Cardiomyopathy. Circulation 2001, 104. [Google Scholar] [CrossRef]
- Olivotto, I.; Cecchi, F.; Gistri, R.; Lorenzoni, R.; Chiriatti, G.; Girolami, F.; Torricelli, F.; Camici, P.G. Relevance of coronary microvascular flow impairment to long-term remodeling and systolic dysfunction in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2006, 47, 1043–1048. [Google Scholar] [CrossRef] [PubMed]
- Von Dohlen, T.W.; Prisant, L.M.; Frank, M.J. Significance of positive or negative thallium-201 scintigraphy in hypertrophic cardiomyopathy. Am. J. Cardiol. 1989, 64, 498–503. [Google Scholar] [CrossRef]
- Yamada, M.; Elliott, P.M.; Kaski, J.C.; Prasad, K.; Gane, J.N.; Lowe, C.M.; Doi, Y.; McKenna, W.J. Dipyridamole stress thallium-201 perfusion abnormalities in patients with hypertrophic cardiomyopathy. Relationship to clinical presentation and outcome. Eur. Heart J. 1998, 19, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Vassiliou, V.S.; Heng, E.L.; Gatehouse, P.D.; Donovan, J.; Raphael, C.E.; Giri, S.; Babu-Narayan, S.V.; Gatzoulis, M.A.; Pennell, D.J.; Prasad, S.K.; et al. Magnetic resonance imaging phantoms for quality-control of myocardial T1 and ECV mapping: Specific formulation, long-term stability and variation with heart rate and temperature. J. Cardiovasc. Magn. Reson. 2016, 18, 62. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Zhao, S.; Yin, G.; Jiang, S.; Zhao, T.; Chen, X.; Tian, L.; Zhang, Y.; Wei, Y.; Liu, Q.; et al. T1 mapping for detection of left ventricular myocardial fibrosis in hypertrophic cardiomyopathy: A preliminary study. Eur. J. Radiol. 2017, 82, e225–e231. [Google Scholar] [CrossRef] [PubMed]
- Puntmann, V.O.; Voigt, T.; Chen, Z.; Mayr, M.; Karim, R.; Rhode, K.; Pastor, A.; Carr-White, G.; Razavi, R.; Schaeffter, T.; et al. Native T1 mapping in differentiation of normal myocardium from diffuse disease in hypertrophic and dilated cardiomyopathy. JACC Cardiovasc. Imaging 2013, 6, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Nielles-Vallespin, S.; Khalique, Z.; Ferreira, P.F.; de Silva, R.; Scott, A.D.; Kilner, P.; McGill, L.-A.; Giannakidis, A.; Gatehouse, P.D.; Ennis, D.; et al. Assessment of Myocardial Microstructural Dynamics by In Vivo Diffusion Tensor Cardiac Magnetic Resonance. J. Am. Coll. Cardiol. 2017, 69, 661–676. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Lopez, B.; Coelho-Filho, O.R.; Lakdawala, N.K.; Cirino, A.L.; Jarolim, P.; Kwong, R.; Gonzalez, A.; Colan, S.D.; Seidman, J.G. Myocardial fibrosis as an early manifestation of hypertrophic cardiomyopathy. N. Engl. J. Med. 2010, 363, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Girolami, F.; Ho, C.Y.; Semsarian, C.; Baldi, M.; Will, M.L.; Baldini, K.; Torricelli, F.; Yeates, L.; Cecchi, F.; Ackerman, M.J.; et al. Clinical features and outcome of hypertrophic cardiomyopathy associated with triple sarcomere protein gene mutations. J. Am. Coll. Cardiol. 2010, 55, 1444–1453. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Y.; Zou, Y.; Sun, K.; Wang, Z.; Ding, H.; Yuan, J.; Wei, W.; Hou, Q.; Wang, H.; et al. Malignant effects of multiple rare variants in sarcomere genes on the prognosis of patients with hypertrophic cardiomyopathy. Eur. J. Heart Fail. 2014, 16, 950–957. [Google Scholar] [CrossRef] [PubMed]
- De Resende, M.M.; Kriegel, A.J.; Greene, A.S. Combined effects of low-dose spironolactone and captopril therapy in a rat model of genetic hypertrophic cardiomyopathy. J. Cardiovasc. Pharmacol. 2006, 48, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Gentry, J.L.; Mentz, R.J.; Hurdle, M.; Wang, A. Ranolazine for Treatment of Angina or Dyspnea in Hypertrophic Cardiomyopathy Patients (RHYME). J. Am. Coll. Cardiol. 2016, 68, 1815–1817. [Google Scholar] [CrossRef] [PubMed]
- Coppini, R.; Mazzoni, L.; Ferrantini, C.; Gentile, F.; Pioner, J.M.; Laurino, A.; Santini, L.; Bargelli, V.; Rotellini, M.; Bartolucci, G.; et al. Ranolazine Prevents Phenotype Development in a Mouse Model of Hypertrophic Cardiomyopathy. Circ. Heart Fail. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.S.; Lutucuta, S.; Bachireddy, P.; Youker, K.; Evans, A.; Entman, M.; Roberts, R.; Marian, A.J. Angiotensin II blockade reverses myocardial fibrosis in a transgenic mouse model of human hypertrophic cardiomyopathy. Circulation 2001, 103, 789–791. [Google Scholar] [CrossRef] [PubMed]
- Penicka, M.; Gregor, P.; Kerekes, R.; Marek, D.; Curila, K.; Krupicka, J. The effects of candesartan on left ventricular hypertrophy and function in nonobstructive hypertrophic cardiomyopathy: A pilot, randomized study. J. Mol. Diagn. 2009, 11, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Suzuki, J.-I.; Shimamoto, R.; Tsuji, T.; Ohmoto-Sekine, Y.; Ohtomo, K.; Nagai, R. A new therapeutic strategy for hypertrophic nonobstructive cardiomyopathy in humans. A randomized and prospective study with an Angiotensin II receptor blocker. Int. Heart J. 2007, 48, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Araujo, A.Q.; Arteaga, E.; Ianni, B.M.; Buck, P.C.; Rabello, R.; Mady, C. Effect of Losartan on left ventricular diastolic function in patients with nonobstructive hypertrophic cardiomyopathy. Am. J. Cardiol. 2005, 96, 1563–1567. [Google Scholar] [CrossRef] [PubMed]
- Sreeram, N.; Emmel, M.; de Giovanni, J.V. Percutaneous radiofrequency septal reduction for hypertrophic obstructive cardiomyopathy in children. J. Am. Coll. Cardiol. 2011, 58, 2501–2510. [Google Scholar] [CrossRef] [PubMed]
- Lawrenz, T.; Borchert, B.; Leuner, C.; Bartelsmeier, M.; Reinhardt, J.; Strunk-Mueller, C.; Meyer Zu Vilsendorf, D.; Schloesser, M.; Beer, G.; Lieder, F.; et al. Endocardial radiofrequency ablation for hypertrophic obstructive cardiomyopathy: Acute results and 6 months’ follow-up in 19 patients. J. Am. Coll. Cardiol. 2011, 57, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Green, E.M.; Wakimoto, H.; Anderson, R.L.; Evanchik, M.J.; Gorham, J.M.; Harrison, B.C.; Henze, M.; Kawas, R.; Oslob, J.D.; Rodriguez, H.M.; et al. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science 2016, 351, 617–621. [Google Scholar] [CrossRef] [PubMed]
- Stern, J.A.; Markova, S.; Ueda, Y.; Kim, J.B.; Pascoe, P.J.; Evanchik, M.J.; Green, E.M.; Harris, S.P. A Small Molecule Inhibitor of Sarcomere Contractility Acutely Relieves Left Ventricular Outflow Tract Obstruction in Feline Hypertrophic Cardiomyopathy. PLoS ONE 2016, 11, e0168407. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1868 | First pathological description by Vulpian [6] who reported it as idiopathic hypertrophic subaortic stenosis (IHSS). |
| 1957 | Brock [9] reported 3 cases of LVOT hypertrophy and attributed it to systemic hypertension. |
| 1958 | Teare [1] published series of 8 autopsy cases who had asymmetrical hypertrophy of the heart, 7 of whom died suddenly. Bercu et al. [10] published a case report on ‘pseudoaortic stenosis’. |
| 1961 | Brockenbrough et al. [11] described the Brockenbrough–Braunwald–Morrow sign. |
| 1963 | Nonobstructive form of HCM first described [13]. |
| 1964 | Morrow et al. [19] performed the first surgical myectomy for HCM. |
| 1965 | Bjork [20] postulated that SAM caused LVOTO. |
| 1969 | Moreyra et al. [21] pioneered the use of M-Mode echocardiography in HCM diagnosis. Shah et al. [22] described SAM in HCM using echocardiography. |
| 1972 | Introduction of cross-section/2D echocardiography [23]. |
| 1980 | First ICD implanted in a patient with HCM [24]. |
| 1990 | First pathogenic mutation implicated in HCM [25]. |
| 1995 | Introduction of alcohol septal ablation (ASA) as an alternative to surgical myectomy by Sigwart [26]. |
| 2000 | First efficacy study on ICD in the prevention of SCD in the HCM population [27]. |
| 2002 | ACC/AHA/NASPE guidelines [28] recommended (class IIb) the use of ICD in primary prevention of SCD in HCM. |
| Gene | Protein | Frequency (%) |
|---|---|---|
| Cardiac myosin-binding protein C | MYBPC3 | 30–40% |
| β cardiac myosin heavy chain | MYH7 | 20–30% |
| Cardiac troponin T | TNNT2 | 5–10% |
| Cardiac troponin I | TNNI3 | 4–8% |
| Regulatory myosin light chain | MYL2 | 2–4% |
| Essential myosin light chain | MYL3 | 1–2% |
| α tropomyosin | TPM1 | <1% |
| α cardiac actin | ACTC1 | <1% |
| Muscle LIM protein | CSRP3 | <1% |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liew, A.C.; Vassiliou, V.S.; Cooper, R.; Raphael, C.E. Hypertrophic Cardiomyopathy—Past, Present and Future. J. Clin. Med. 2017, 6, 118. https://doi.org/10.3390/jcm6120118
Liew AC, Vassiliou VS, Cooper R, Raphael CE. Hypertrophic Cardiomyopathy—Past, Present and Future. Journal of Clinical Medicine. 2017; 6(12):118. https://doi.org/10.3390/jcm6120118
Chicago/Turabian StyleLiew, Alphonsus C., Vassilios S. Vassiliou, Robert Cooper, and Claire E. Raphael. 2017. "Hypertrophic Cardiomyopathy—Past, Present and Future" Journal of Clinical Medicine 6, no. 12: 118. https://doi.org/10.3390/jcm6120118
APA StyleLiew, A. C., Vassiliou, V. S., Cooper, R., & Raphael, C. E. (2017). Hypertrophic Cardiomyopathy—Past, Present and Future. Journal of Clinical Medicine, 6(12), 118. https://doi.org/10.3390/jcm6120118