
Epicardial Epithelial-to-Mesenchymal Transition in Heart Development and Disease

{kind=link}
Abstract
:1. Introduction
2. Formation of the Embryonic Epicardium
3. Induction of Epicardial EMT
4. Transcriptional Regulation of Epicardial EMT
4.1. Wt1 and Tbx18
4.2. Snail and Slug
4.3. Tcf21 and Nfatc1
4.4. Twist1, Scleraxis, Sox9, and Hand2
4.5. Retinoic Acid
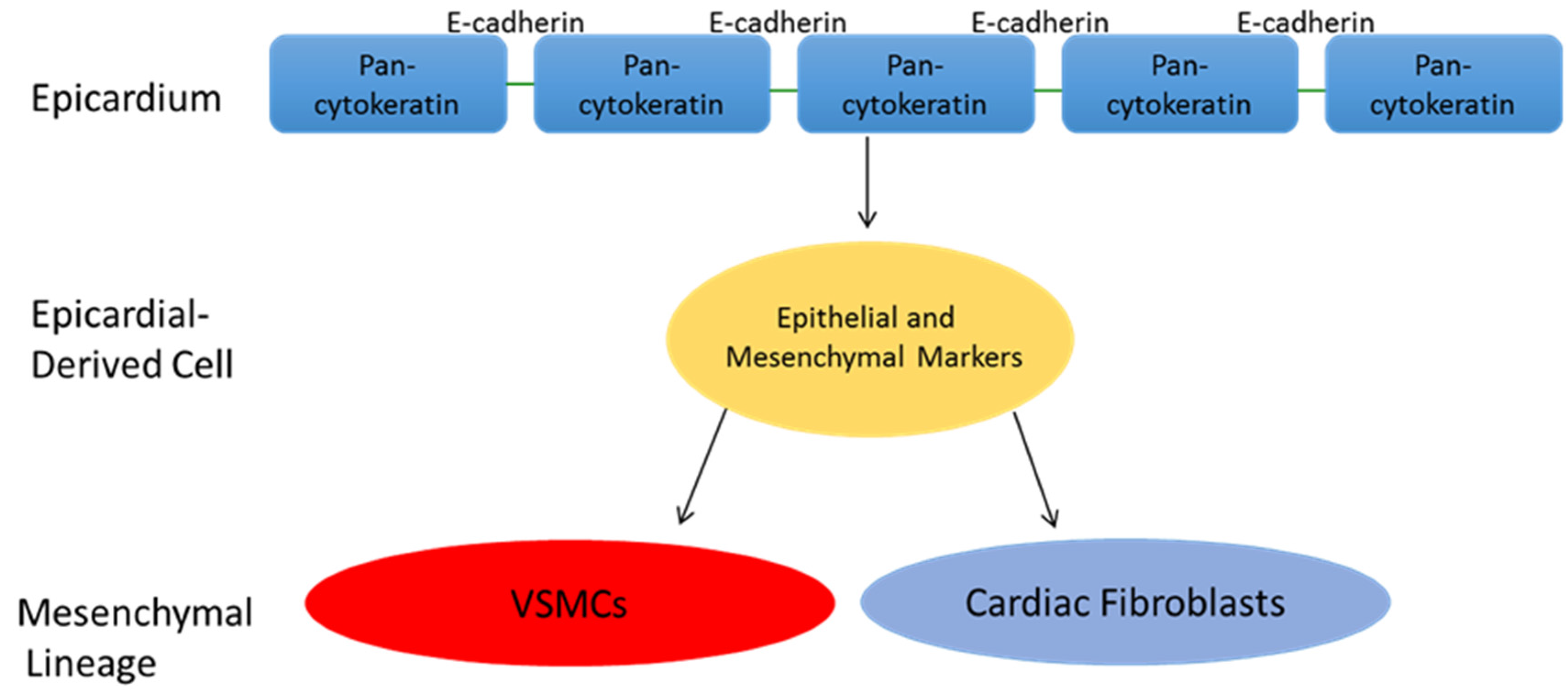
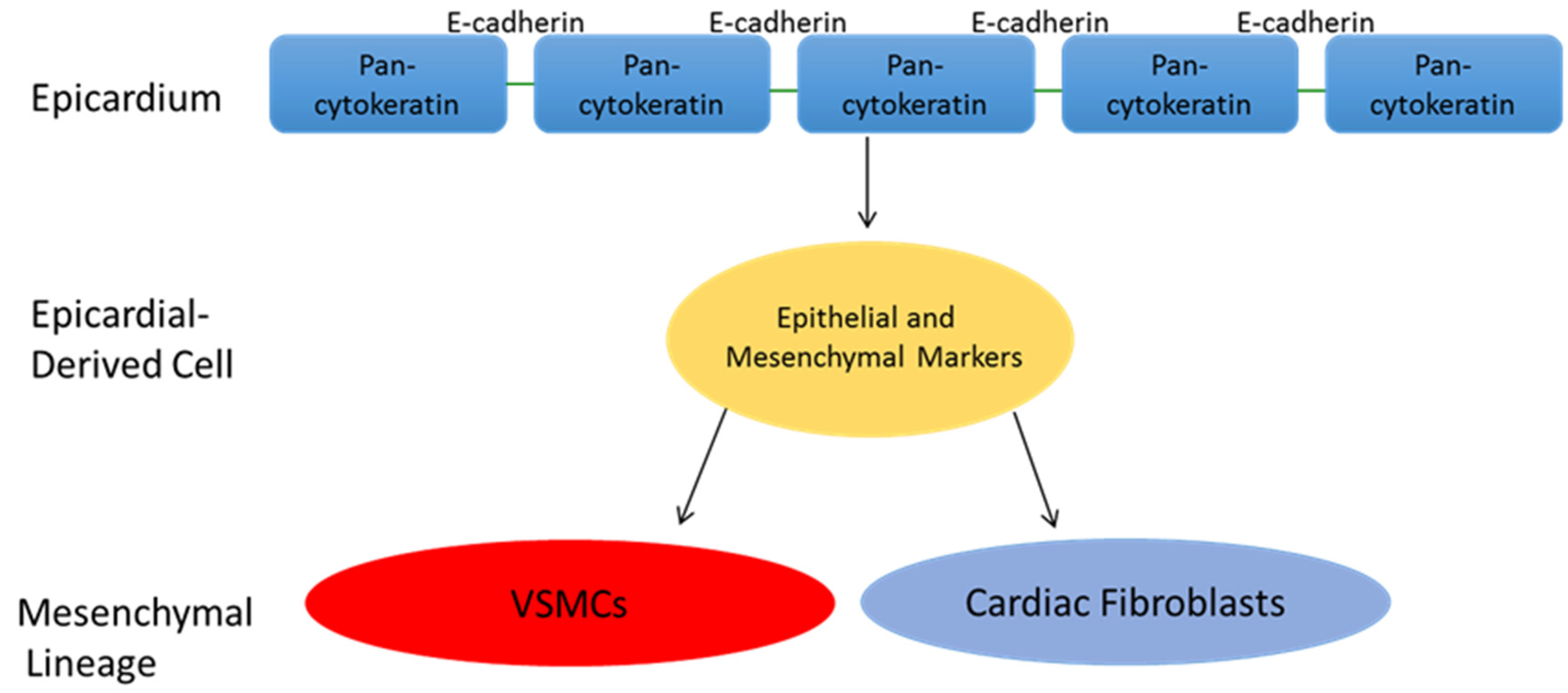
5. Epicardial Derived Cell Fate
6. Function of the Epicardium Following Myocardial Injury
7. Summary and Future Directions
Conflicts of Interest
References
- Li, P.; Cavallero, S.; Gu, Y.; Chen, T.H.P.; Hughes, J.; Bassim Hassan, A.; Brüning, J.C.; Pashmforoush, M.; Sucov, H.M. IGF signaling directs ventricular cardiomyocyte proliferation during embryonic heart development. Development 2011, 138, 1795–1805. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Harrison, M.R.; Osorio, A.; Kim, J.; Baugh, A.; Duan, C.; Sucov, H.M.; Lien, C.L. IGF signaling is required for cardiomyocyte proliferation during zebrafish heart development and regeneration. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Manner, J.; Perez-Pomares, J.M.; Macias, D.; Munoz-Chapuli, R. The origin, formation, and developmental significance of the epicardium: A review. Cells Tissues Organs 2001, 169, 89–103. [Google Scholar] [PubMed]
- Viragh, S.; Gittenberger-de Groot, A.C.; Poelmann, R.E.; Kalman, F. Early development of quail heart epicardium and associated vascular and glandular structures. Anat. Embryol. 1993, 188, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Komiyama, M.; Ito, K.; Shimada, Y. Origin and development of the epicardium in the mouse embryo. Anat. Embryol. 1987, 176, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Svensson, E.C. Deciphering the signals specifying the proepicardium. Circ. Res. 2010, 12, 1789–1790. [Google Scholar] [CrossRef] [PubMed]
- Schlueter, J.; Brand, T. Subpopulation of proepicardial cells is derived from the somatic mesoderm in the chick embryo. Circ. Res. 2013, 113, 1128–1137. [Google Scholar] [CrossRef] [PubMed]
- Perez-Pomares, J.M.; Phelps, A.; Munoz-Chapuli, R.; Wessels, A. The contribution of the proepicardium to avian cardiovascular development. Int. J. Dev. Biol. 2001, 45, S155–S156. [Google Scholar]
- Katz, T.C.; Singh, M.K.; Degenhardt, K.; Rivera-Feliciano, J.; Johnson, R.L.; Epstein, J.A.; Tabin, C.J. Distinct compartments of the Proepicardial organ give rise to coronary vascular endothelial cells. Dev. Cell 2012, 22, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Acharya, A.; Baek, S.T.; Huang, G.; Eskiocak, B.; Goetsch, S.; Sung, C.Y.; Banfi, S.; Sauer, M.F.; Olsen, G.S.; Duffield, J.S.; Olson, E.N.; Tallquist, M.D. The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development 2012, 139, 2139–2149. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, K.; Gupta, V.; Wang, J.; Holdway, J.E.; Wills, A.A.; Fang, Y.; Poss, K.D. Tcf21+ epicardial cells adopt non-myocardial fates during zebrafish heart development and regeneration. Development 2011, 138, 2895–2902. [Google Scholar] [CrossRef] [PubMed]
- Wessels, A.; Perez-Pomares, J.M. The epicardium and epicardially derived cells (EPDCs) as cardiac stem cells. Anat. Rec. Discov. Mol. Cell Evol. Biol. 2004, 267, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Morabito, C.J.; Dettman, R.W.; Kattan, J.; Collier, J.M.; Bristow, J. Positive and negative regulation of epicardial-mesenchymal transformation during avian heart development. Dev. Biol. 2001, 234, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Compton, L.A.; Potash, D.A.; Mundell, N.A.; Barnett, J.V. Transforming growth factor-beta induces loss of epithelial character and smooth muscle cell differentiation in epicardial cells. Dev. Dyn. 2006, 235, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Molin, D.G.; Bartram, U.; van der Heiden, K.; van Iperen, L.; Speer, C.P.; Hierck, B.P.; Poelmann, R.E.; Gittenberger-de-Groot, A.C. Expression patterns of Tgfbeta1-3 associate with myocardialisation of the outflow tract and the development of the epicardium and the fibrous heart. Dev. Dyn. 2003, 227, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Sridurongrit, S.; Larsson, J.; Schwartz, R.; Ruiz-Lozano, P.; Kaartinen, V. Signaling via the Tgf-beta type I receptor Alk5 in heart development. Dev. Biol. 2008, 322, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Langlois, D.; Hneino, M.; Bouazza, L.; Parlakian, A.; Sasaki, T.; Bricca, G.; Li, J.Y. Conditional inactivation of TGF-beta type II receptor in smooth muscle cells and epicardium causes lethal aortic and cardiac defects. Transgenic Res. 2010, 19, 1069–1082. [Google Scholar] [CrossRef] [PubMed]
- Jiao, K.; Langworthy, M.; Batts, L.; Brown, C.B.; Moses, H.L.; Baldwin, H.S. Tgfbeta signaling is required for atrioventricular cushion mesenchyme remodeling during in vivo cardiac development. Development 2006, 133, 4585–4593. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sanchez, N.S.; Hill, C.R.; Love, J.D.; Soslow, J.H.; Craig, E.; Austin, A.F.; Brown, C.B.; Czirok, A.; Camenisch, T.D.; Barnett, J.V. The cytoplasmic domain of TGFBR3 through its interaction with the scaffolding protein, GIPC, directs epicardial cell behavior. Dev. Biol. 2011, 2, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Vega-Hernandez, M.; Kovacs, A.; de Langhe, S.; Ornitz, D.M. FGF10/FGFR2b signaling is essential for cardiac fibroblast development and growth of the myocardium. Development 2011, 138, 3331–3340. [Google Scholar] [CrossRef] [PubMed]
- Lavine, K.J.; White, A.C.; Park, C.; Smith, C.S.; Choi, K.; Long, F.; Hui, C.C.; Ornitz, D.M. Fibroblast growth factor signals regulate a wave of Hedgehog activation that is essential for coronary vascular development. Genes Dev. 2006, 20, 1651–1666. [Google Scholar] [CrossRef] [PubMed]
- Lavine, K.J.; Long, F.; Choi, K.; Smith, C.; Ornitz, D.M. Hedgehog signaling to distinct cell types differentially regulates coronary artery and vein development. Development 2008, 135, 3161–3171. [Google Scholar] [CrossRef] [PubMed]
- Olivey, H.E.; Svensson, E.C. Epicardial-myocardial signaling directing coronary vasculogenesis. Circ Res. 2010, 106, 818–832. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.L.; Baek, S.T.; Sung, C.Y.; Tallquist, M.D. Epicardial-derived cell epithelial-to-mesenchymal transition and fate specification require PDGF receptor signaling. Circ Res. 2011, 108, e15–e26. [Google Scholar] [CrossRef] [PubMed]
- Von Gise, A.; Zhou, B.; Honor, L.B.; Ma, Q.; Petryk, A.; Pu, W.T. Wt1 regulates epicardial epithelial to mesenchymal transition through beta-catenin and retinoic acid signaling pathways. Dev. Biol. 2011, 356, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Zamora, M.; Manner, J.; Ruiz-Lozano, P. Epicardium-derived progenitor cells require beta-catenin for coronary artery formation. Proc. Natl. Acad. Sci. USA 2007, 104, 18109–18114. [Google Scholar] [CrossRef] [PubMed]
- Grieskamp, T.; Rudat, C.; Ludtke, T.H.; Norden, J.; Kispert, A. Notch signaling regulates smooth muscle differentiation of epicardium-derived cells. Circ. Res. 2011, 108, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Del Monte, G.; Casanova, J.C.; Guadix, J.A.; MacGrogan, D.; Burch, J.B.; Perez-Pomares, J.M.; de la Pompa, J.L. Differential Notch signaling in the epicardium is required for cardiac inflow development and coronary vessel morphogenesis. Circ. Res. 2011, 108, 824–836. [Google Scholar] [CrossRef] [PubMed]
- Braitsch, C.M.; Yutzey, K.E. Transcriptional control of cell lineage development in epicardium-derived cells. J. Dev. Biol. 2013, 1, 92–111. [Google Scholar] [CrossRef]
- Huff, V.; Miwa, H.; Haber, D.A.; Call, K.M.; Housman, D.; Strong, L.C.; Saunders, G.F. Evidence for WT1 as a Wilms tumor (WT) gene: Intragenic germinal deletion in bilateral WT. Am. J. Hum. Genet. 1991, 48, 997–1003. [Google Scholar] [PubMed]
- Armstrong, J.F.; Pritchard-Jones, K.; Bickmore, W.A.; Hastie, N.D.; Bard, J.B. The expression of the Wilms’ tumour gene, WT1, in the developing mammalian embryo. Mech. Dev. 1993, 40, 85–97. [Google Scholar] [CrossRef]
- Carmona, R.; Gonzalez-Iriarte, M.; Perez-Pomares, J.M.; Munoz-Chapuli, R. Localization of the Wilm’s tumour protein WT1 in avian embryos. Cell Tissue Res. 2001, 303, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.W.; McInnes, L.; Kreidberg, J.; Hastie, N.D.; Schedl, A. YAC complementation shows a requirement for Wt1 in the development of epicardium, adrenal gland and throughout nephrogenesis. Development 1999, 126, 1845–1857. [Google Scholar] [PubMed]
- Martinez-Estrada, O.M.; Lettice, L.A.; Essafi, A.; Guadix, J.A.; Slight, J.; Velecela, V.; Hall, E.; Reichmann, J.; Devenney, P.S.; Hohenstein, P.; et al. Wt1 is required for cardiovascular progenitor cell formation through transcriptional control of Snail and E-cadherin. Nat. Genet. 2010, 42, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Perez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor Snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef] [PubMed]
- De Herreros, A.G.; Peiro, S.; Nassour, M.; Savagner, P. Snail family regulation and epithelial mesenchymal transitions in breast cancer progression. J. Mammary Gland Biol. Neoplasia. 2010, 2, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Kraus, F.; Haenig, B.; Kispert, A. Cloning and expression analysis of the mouse T-box gene Tbx18. Mech. Dev. 2001, 100, 83–86. [Google Scholar] [CrossRef]
- Plageman, T.F., Jr.; Yutzey, K.E. T-box genes and heart development: Putting the "t" in heart. Dev. Dyn. 2005, 232, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Airik, R.; Bussen, M.; Singh, M.K.; Petry, M.; Kispert, A. Tbx18 regulates the development of the ureteral mesenchyme. J. Clin. Invest. 2006, 116, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Shelton, E.L.; Yutzey, K.E. Twist1 function in endocardial cushion cell proliferation, migration, and differentiation during heart valve development. Dev. Biol. 2008, 317, 282–295. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.L.; Zhou, W.; Yang, L.; Bu, L.; Qyang, Y.; Zhang, X.; Li, X.; Rosenfeld, M.G.; Chen, J.; Evans, S. T-box genes coordinate regional rates of proliferation and regional specification during cardiogenesis. Development 2005, 132, 2475–2487. [Google Scholar] [CrossRef] [PubMed]
- Shelton, E.L.; Yutzey, K.E. Tbx20 regulation of endocardial cushion cell proliferation and extracellular matrix gene expression. Dev. Biol. 2007, 302, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Takeichi, M.; Nimura, K.; Mori, M.; Nakagami, H.; Kaneda, Y. The transcription factors Tbx18 and Wt1 control the epicardial epithelial-mesenchymal transition through bi-directional regulation of Slug in murine primary epicardial cells. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Sancho, E.; Franci, C.; Dominguez, D.; Monfar, M.; Baulida, J.; Garcia de Herreros, A. The transcription factor Snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Tao, G.; Miller, L.J.; Lincoln, J. Snai1 is important for avian epicardial cell transformation and motility. Dev. Dyn. 2013, 242, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Solanas, G.; Porta-de-la-Riva, M.; Agusti, C.; Casagolda, D.; Sanchez-Aguilera, F.; Larriba, M.J.; Pons, F.; Peiro, S.; Escriva, M.; Munoz, A.; Dunach, M.; de Herreros, A.G.; Baulida, J. E-cadherin controls beta-catenin and NF-kappaB transcriptional activity in mesecnhymal gene expression. J. Cell Sci. 2008, 121, 2224–2234. [Google Scholar] [CrossRef] [PubMed]
- Hidai, H.; Bardales, R.; Goodwin, R.; Quertermous, T.; Quertermous, E.E. Cloning of capsulin, a basic helix-loop-helix factor expressed in progenitor cells of the pericardium and the coronary arteries. Mech. Dev. 1998, 73, 33–43. [Google Scholar] [CrossRef]
- Lu, J.; Richardson, J.A.; Olson, E.N. Capsulin: A novel bHLH transcription factor expressed in epicardial progenitors and mesenchyme of visceral organs. Mech. Dev. 1998, 73, 23–32. [Google Scholar] [CrossRef]
- Braitsch, C.M.; Combs, M.D.; Quaggin, S.E.; Yutzey, K.E. Pod1/Tcf21 is regulated by retinoic acid signaling and inhibits differentiation of epicardium-derived cells into smooth muscle in the developing heart. Dev. Biol. 2012, 368, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Combs, M.D.; Braitsch, C.M.; Lange, A.W.; James, J.F.; Yutzey, K.E. Nfatc1 promotes epicardium-derived cell invasion into myocardium. Development 2011, 138, 1747–1757. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.D.; Corsa, C.A.; Biswas, H.; Aft, R.L.; Longmore, G.D. Temporal and spatial cooperation of Snail1 and Twist1 during epithelial-mesenchymal transition predicts for human breast cancer recurrence. Mol. Cancer Res. 2011, 12, 1644–1657. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; von Gise, A.; Ma, Q.; Hu, Y.W.; Pu, W.T. Genetic fate mapping demonstrates contribution of epicardium-derived cells to the annulus fibrosis of the mammalian heart. Dev. Biol. 2010, 338, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Von Gise, A.; Pu, W.T. Endocardial and epicardial epithelial to mesenchymal transitions in heart development and disease. Circ. Res. 2012, 110, 1628–1645. [Google Scholar] [CrossRef] [PubMed]
- Lincoln, J.; Alfieri, C.M.; Yutzey, K.E. BMP and FGF regulatory pathways control cell lineage diversification of heart valve precursor cells. Dev. Biol. 2006, 292, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Wessels, A.; van den Hoff, M.J.; Adamo, R.F.; Phelps, A.L.; Lockhart, M.M.; Sauls, K.; Briggs, L.E.; Norris, R.A.; van Wijk, B.; Perez-Pomares, J.M.; Dettman, R.W.; Burch, J.B. Epicardially derived fibroblasts preferentially contribute to the parietal leaflets of the atrioventricular valves in the murine heart. Dev. Biol. 2012, 366, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Espira, L.; Lamoureux, L.; Jones, S.C.; Gerard, R.D.; Dixon, I.M.; Czubryt, M.P. The basic helix-loop-helix transcription factor Scleraxis regulates fibroblast collagen synthesis. J. Mol. Cell. Cardiol. 2009, 47, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Levay, A.K.; Peacock, J.D.; Lu, Y.; Kock, M.; Hinton, R.B., Jr.; Kadler, K.E.; Lincoln, J. Scleraxis is required for cell lineage differentiation and extracellular matrix remodeling during murine heart valve formation in vivo. Circ. Res. 2008, 103, 948–956. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.L.; Baek, S.T.; Sung, C.Y.; Tallquist, M.D. Epicardial-derived cell epithelial-to-mesenchymal transition and fate specification require PDGF receptor signaling. Circ. Res. 2011, 108, e15–e26. [Google Scholar] [CrossRef] [PubMed]
- Barnes, R.M.; Firulli, B.A.; VanDusen, N.J.; Morikawa, Y.; Conway, S.J.; Cserjesi, P.; Vincentz, J.W.; Firulli, A.B. Hand2 loss-of-function in Hand1-expressing cells reveals distinct roles in epicardial and coronary vessel development. Circ. Res. 2011, 108, 940–949. [Google Scholar] [CrossRef] [PubMed]
- Stuckmann, I.; Evans, S.; Lassar, A.B. Erythopoietin and retinoic acid, secreted from the epicardium, are required for cardiac myocyte proliferation. Dev. Biol. 2003, 255, 334–349. [Google Scholar] [CrossRef]
- Perez-Pomares, J.M.; Phelps, A.; Sedmerova, M.; Carmona, R.; Gonzalez-Iriarte, M.; Munoz-Chapuli, R.; Wessels, A. Experimental studies on the spatiotemporal expression of WT1 and RALDH2 in the embryonic avian heart: A model for the regulation of myocardial and valvuloseptal development by epicardially derived cells (EPDCs). Dev. Biol. 2002, 247, 307–326. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.H.; Chang, T.C.; Kang, J.O.; Choudhary, B.; Makita, T.; Tran, C.M.; Burch, J.B.; Eid, H.; Sucov, H.M. Epicardial induction of fetal cardiomyocyte proliferation via a retinoic acid inducible trophic factor. Dev. Biol. 2002, 250, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.O.; Sucov, H.M. Convergent proliferative response and divergent morphogenic pathways induced by epicardial and endocardial signaling in fetal heart development. Mech. Dev. 2005, 122, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Merki, E.; Zamora, M.; Raya, A.; Kawakami, Y.; Wang, J.; Zhang, X.; Burch, J.; Kubalak, S.W.; Kaliman, P.; Belmonte, J.C.; Chien, K.R.; Ruiz-Lozano, P. Epicardial retinoid X receptor alpha is required for myocardial growth and coronary artery formation. Proc. Natl. Acad. Sci. USA 2005, 102, 18455–18460. [Google Scholar] [CrossRef] [PubMed]
- Lavine, K.J.; Yu, K.; White, A.C.; Zhang, X.; Smith, C.; Partanen, J.; Ornitz, D.M. Endocardial and epicardial derived FGF signals regulate myocardial proliferation and differentiation in vivo. Dev. Cell 2005, 1, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Guadix, J.A.; Ruiz-Villalba, A.; Lettice, L.; Velecela, V.; Munoz-Chapuli, R.; Hastie, N.D.; Perez-Pomares, J.M.; Martinez-Estrada, O.M. Wt1 controls retinoic acid signalling in embryonic epicardium through transcriptional activation of Raldh2. Development 2011, 138, 1093–1097. [Google Scholar] [CrossRef] [PubMed]
- Dettman, R.W.; Denetclaw, W., Jr.; Ordahl, C.P.; Bristow, J. Common epicardial origin of coronary vascular smooth muscle, perivascular fibroblasts, and intermyocardial fibroblasts in the avian heart. Dev. Biol. 1998, 193, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Gittenberger-de Groot, A.C.; Vrancken Peeters, M.P.; Mentink, M.M.; Gourdie, R.G.; Poelmann, R.E. Epicardium-derived cells contribute a novel population to the myocardial wall and the atrioventricular cushions. Circ. Res. 1998, 82, 1043–1052. [Google Scholar] [CrossRef] [PubMed]
- Lie-Venema, H.; van den Akker, N.M.; Bax, N.A.; Winter, E.M.; Maas, S.; Kekarainen, T.; Hoeben, R.C.; de Ruiter, M.C.; Poelmann, R.E.; Gittenberger-de Groot, A.C. Origin, fate, and function of epicardium-derived cells (EPDCs) in normal and abnormal cardiac development. Sci. World J. 2007, 7, 1777–1798. [Google Scholar] [CrossRef] [PubMed]
- Kolditz, D.P.; Wijffels, M.C.; Blom, N.A.; van der Laarse, A.; Hahurij, N.D.; Lie-Venema, H.; Markwald, R.R.; Poelmann, R.E.; Schalij, M.J.; Gittenberger-de Groot, A.C. Epicardium-derived cells in development of annulus fibrosis and persistence of accessory pathways. Circulation 2008, 117, 1508–1517. [Google Scholar] [CrossRef] [PubMed]
- Eralp, I.; Lie-Venema, H.; Bax, N.A.; Wijffels, M.C.; van der Laarse, A.; Deruiter, M.C.; Bogers, A.J.; van den Akker, N.M.; Gourdie, R.G.; Schalij, M.J.; Poelmann, R.E.; Gittenberger-De Groot, A.C. Epicardium-derived cells are important for correct development of the Purkinje fibers in the avian heart. Anat. Rec. A Discov. Mol. Cell Evol. Biol. 2006, 288, 1272–1280. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.L.; Martin, J.C.; Sun, Y.; Cui, L.; Wang, L.; Ouyang, K.; Yang, L.; Bu, L.; Liang, X.; Zhang, X.; et al. A myocardial lineage derives from Tbx18 epicardial cells. Nature 2008, 454, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Ma, Q.; Rajagopal, S.; Wu, S.M.; Domian, I.; Rivera-Feliciano, J.; Jiang, D.; von Gise, A.; Ikeda, S.; Chien, K.R.; et al. Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature 2008, 454, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Christoffels, V.M.; Grieskamp, T.; Norden, J.; Mommersteeg, M.T.M.; Rudat, C.; Kispert, A. Tbx18 and the fate of epicardial progenitors. Nature 2009, 458, E8–E9. [Google Scholar] [CrossRef] [PubMed]
- Manner, J. Does the subepicardial mesenchyme contribute myocardioblasts to the myocardium of the chick embryo heart? A quail-chick chimera study tracing the fate of the epicardial primordium. Anat Rec. 1999, 255, 212–226. [Google Scholar] [CrossRef]
- Perez-Pomares, J.M.; Carmona, R.; Gonzalez-Iriarte, M.; Atencia, G.; Wessels, A.; Munoz-Chapuli, R. Origin of coronary endothelial cells from epicardial mesothelium in avian embryos. Int. J. Dev. Biol. 2002, 46, 1005–1013. [Google Scholar] [PubMed]
- Guadix, J.A.; Carmona, R.; Munoz-Chapuli, R.; Perez-Pomares, J.M. In vivo and in vitro analysis of the vasculogenic potential of avian proepicardial and epicardial cells. Dev. Dyn. 2006, 235, 1014–1026. [Google Scholar] [CrossRef] [PubMed]
- Peralta, M.; Gonzalez-Rosa, J.M.; Marques, I.J.; Mercader, N. The epicardium in the embryonic and adult zebrafish. J. Dev. Biol. 2014, 2, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Lepilina, A.; Coon, A.N.; Kikuchi, K.; Holdway, J.E.; Roberts, R.W.; Burns, C.G.; Poss, K.D. A dynamic epicardial injury response supports progenitor cell activity during zebrafish heart regeneration. Cell 2006, 127, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rosa, J.M.; Martin, V.; Peralta, M.; Torres, M.; Mercader, N. Extensive scar formation and regression during heart regeneration after cryoinjury in the zebrafish. Development 2011, 138, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Schnabel, K.; Wu, C.C.; Kurth, T.; Weidinger, G. Regeneration of cryoinjury induced necrotic heart lesions in zebrafish is associated with epicardial activation and cardiomyocyte proliferation. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rosa, J.M.; Peralta, M.; Mercader, N. Pan-epicardial lineage tracing reveals epicardium derived cells give rise to myofibroblasts and perivascular cells during zebrafish heart regeneration. Dev. Biol. 2012, 370, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, K.; Holdway, J.E.; Major, R.J.; Blum, N.; Dahn, R.D.; Begemann, G.; Poss, K.D. Retinoic acid production by endocardium and epicardium is an injury response essential for zebrafish heart regeneration. Dev. Cell. 2011, 20, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Itou, J.; Oishi, I.; Kawakami, H.; Glass, T.J.; Richter, J.; Johnson, A.; Lund, T.C.; Kawakami, Y. Migration of cardiomyocytes is essential for heart regeneration in zebrafish. Development 2012, 139, 4133–4142. [Google Scholar] [CrossRef] [PubMed]
- Gittenberger-de Groot, A.C.; Winter, E.M.; Poelmann, R.E. Epicardium-derived cells (EPDCs)in development, cardiac disease and repair of ischemia. J. Cell. Mol. Med. 2010, 14, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Limana, F.; Bertolami, C.; Mangoni, A.; Di Carlo, A.; Avitabile, D.; Mocini, D.; Iannelli, P.; De-Mori, R.; Marchetti, C.; Pozzoli, O.; et al. Myocardial infarction induces embryonic reprogramming of epicardial c-kit+ cells: Role of the pericardial fluid. J. Mol. Cell. Cardiol. 2010, 48, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Di Meglio, F.; Castaldo, C.; Nurzynska, D.; Romano, V.; Miraglia, R.; Bancone, C.; Langella, G.; Vosa, C.; Montagnani, S. Epithelial-mesenchymal transition of epicardial mesothelium is a source of cardiac CD117-positive stem cells in adult human heart. J. Mol. Cell. Cardiol. 2010, 49, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Gherghe, C.; Liu, D.; Hamlett, E.; Srikantha, L.; Rodgers, L.; Regan, J.N.; Rojas, M.; Willis, M.; Leask, A.; Majesky, M.; Deb, A. Wnt1/beta catenin injury response activates the epicardium and cardiac fibroblasts to promote cardiac repair. EMBO J. 2012, 31, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.L.; Goetsch, S.C.; Gaiano, N.R.; Hill, J.A.; Olson, E.N.; Schneider, J.W. A dynamic Notch injury response activates epicardium and contributes to fibrosis repair. Circ. Res. 2011, 108, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Honor, L.B.; He, H.; Ma, Q.; Oh, J.H.; Butterfield, C.; Lin, R.Z.; Melero-Martin, J.M.; Dolmatova, E.; Duffy, H.S.; et al. Adult mouse epicardium modulates myocardial injury by secreting paracrine factors. J. Clin. Investig. 2011, 121, 1894–1904. [Google Scholar] [CrossRef] [PubMed]
- Smart, N.; Bollini, S.; Dube, K.N.; Vieira, J.M.; Zhou, B.; Davidson, S.; Yellon, D.; Riegler, J.; Price, A.N.; Lythgoe, M.F.; Pu, W.T.; Riley, P.R. De novo cardiomyocytes from within the activated adult heart after injury. Nature 2011, 474, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Smart, N.; Risebro, C.A.; Melville, A.A.; Moses, K.; Schwartz, R.J.; Chien, K.R.; Riley, P.R. Thymosin beta4 induces adult epicardial progenitor mobilization and neovascularization. Nature 2007, 445, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Honor, L.B.; Ma, Q.; Oh, J.H.; Lin, R.Z.; Melero-Martin, J.M.; von Gise, A.; Zhou, P.; Hu, T.; He, L.; et al. Thymosin beta 4 treatment after myocardial infarction does not reprogram epicardial cell into cardiomyocytes. J. Mol. Cell. Cardiol. 2012, 52, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Nam, Y.J.; Luo, X.; Qi, X.; Tan, W.; Huang, G.N.; Acharya, A.; Smith, C.L.; Tallquist, M.D.; Neilson, E.G.; Hill, J.A.; Bassel-Duby, R.; Olson, E.N. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 2012, 485, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Huang, Y.; Spencer, C.I.; Foley, A.; Vedantham, V.; Liu, L.; Conway, S.J.; Fu, J.D.; Srivastava, D. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature 2012, 485, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Inagawa, K.; Miyamoto, K.; Yamakawa, H.; Muraoka, N.; Sadahiro, T.; Umei, T.; Wada, R.; Katsumata, Y.; Kaneda, R.; Nakade, K.; et al. Induction of cardiomyocyte-like cells in infarct hearts by gene transfer of Gata4, Mef2c, and Tbx5. Circ. Res. 2012, 111, 1147–1156. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krainock, M.; Toubat, O.; Danopoulos, S.; Beckham, A.; Warburton, D.; Kim, R. Epicardial Epithelial-to-Mesenchymal Transition in Heart Development and Disease. J. Clin. Med. 2016, 5, 27. https://doi.org/10.3390/jcm5020027
Krainock M, Toubat O, Danopoulos S, Beckham A, Warburton D, Kim R. Epicardial Epithelial-to-Mesenchymal Transition in Heart Development and Disease. Journal of Clinical Medicine. 2016; 5(2):27. https://doi.org/10.3390/jcm5020027
Chicago/Turabian StyleKrainock, Michael, Omar Toubat, Soula Danopoulos, Allison Beckham, David Warburton, and Richard Kim. 2016. "Epicardial Epithelial-to-Mesenchymal Transition in Heart Development and Disease" Journal of Clinical Medicine 5, no. 2: 27. https://doi.org/10.3390/jcm5020027
APA StyleKrainock, M., Toubat, O., Danopoulos, S., Beckham, A., Warburton, D., & Kim, R. (2016). Epicardial Epithelial-to-Mesenchymal Transition in Heart Development and Disease. Journal of Clinical Medicine, 5(2), 27. https://doi.org/10.3390/jcm5020027