
MicroRNAs and Osteolytic Bone Metastasis: The Roles of MicroRNAs in Tumor-Induced Osteoclast Differentiation

Abstract
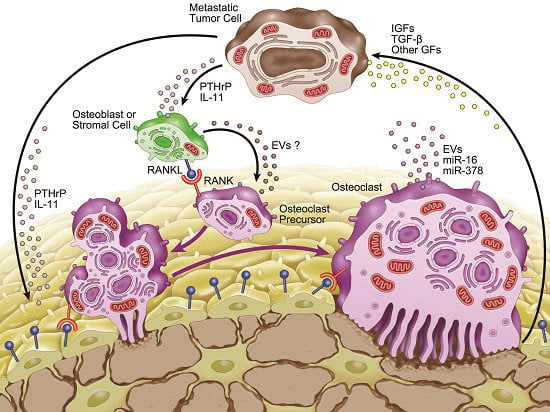
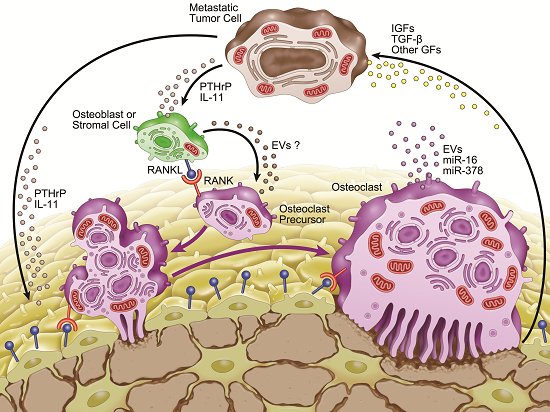
:
1. Introduction
2. Bone Metastasis
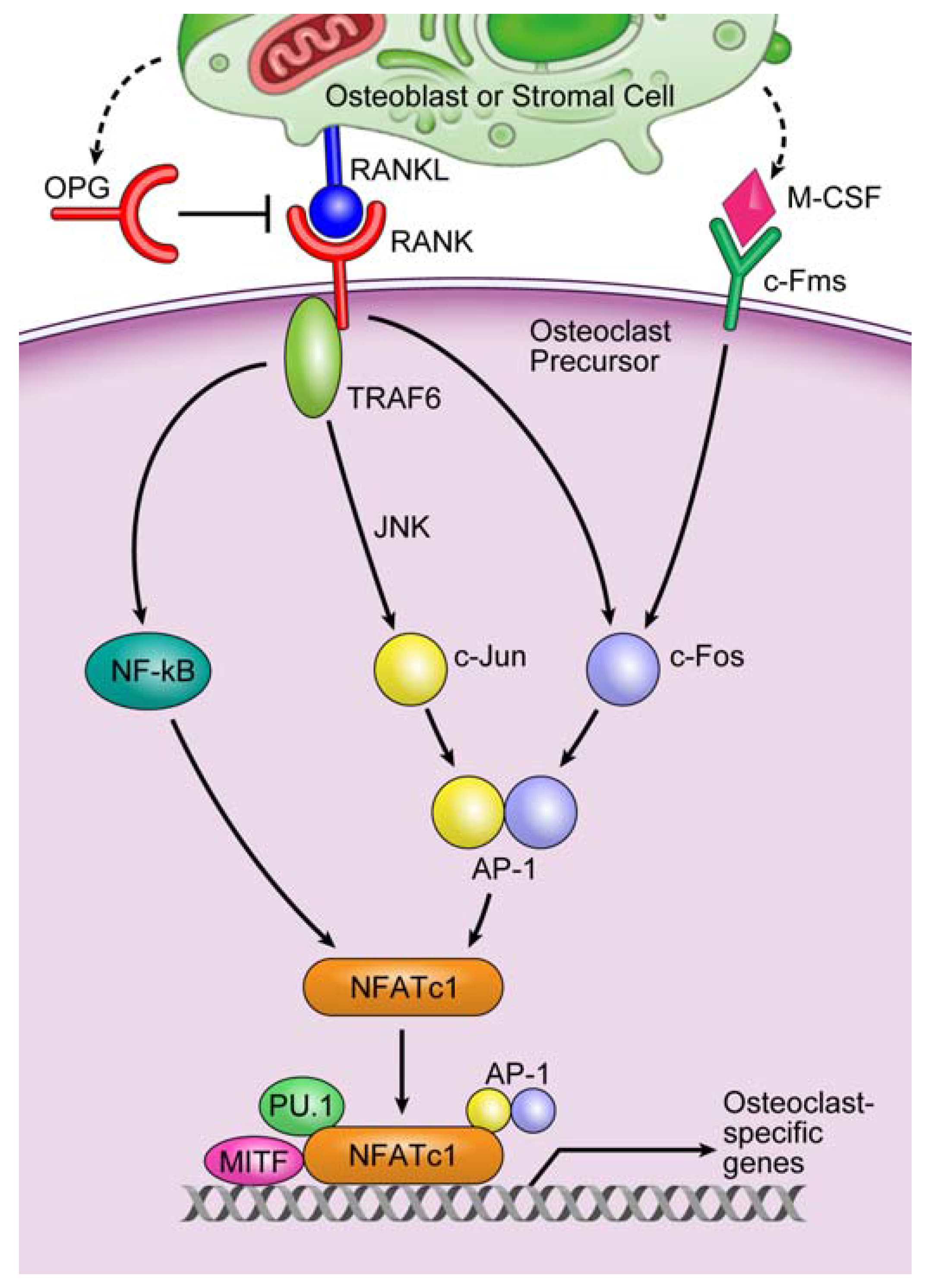
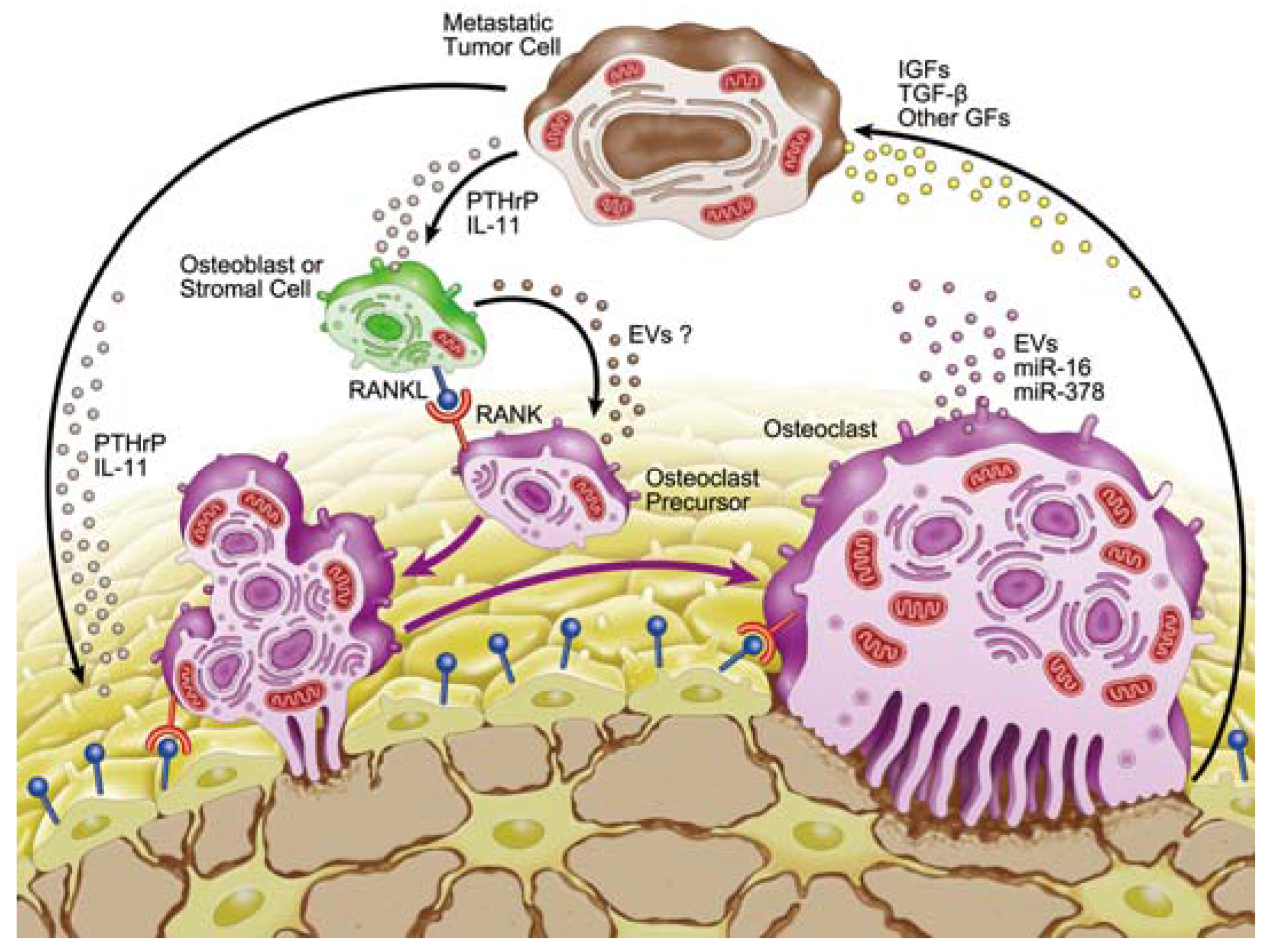
3. Microenvironment of Osteolytic Lesions
3.1. Growth Factors in the Microenvironment of Osteolytic Lesions
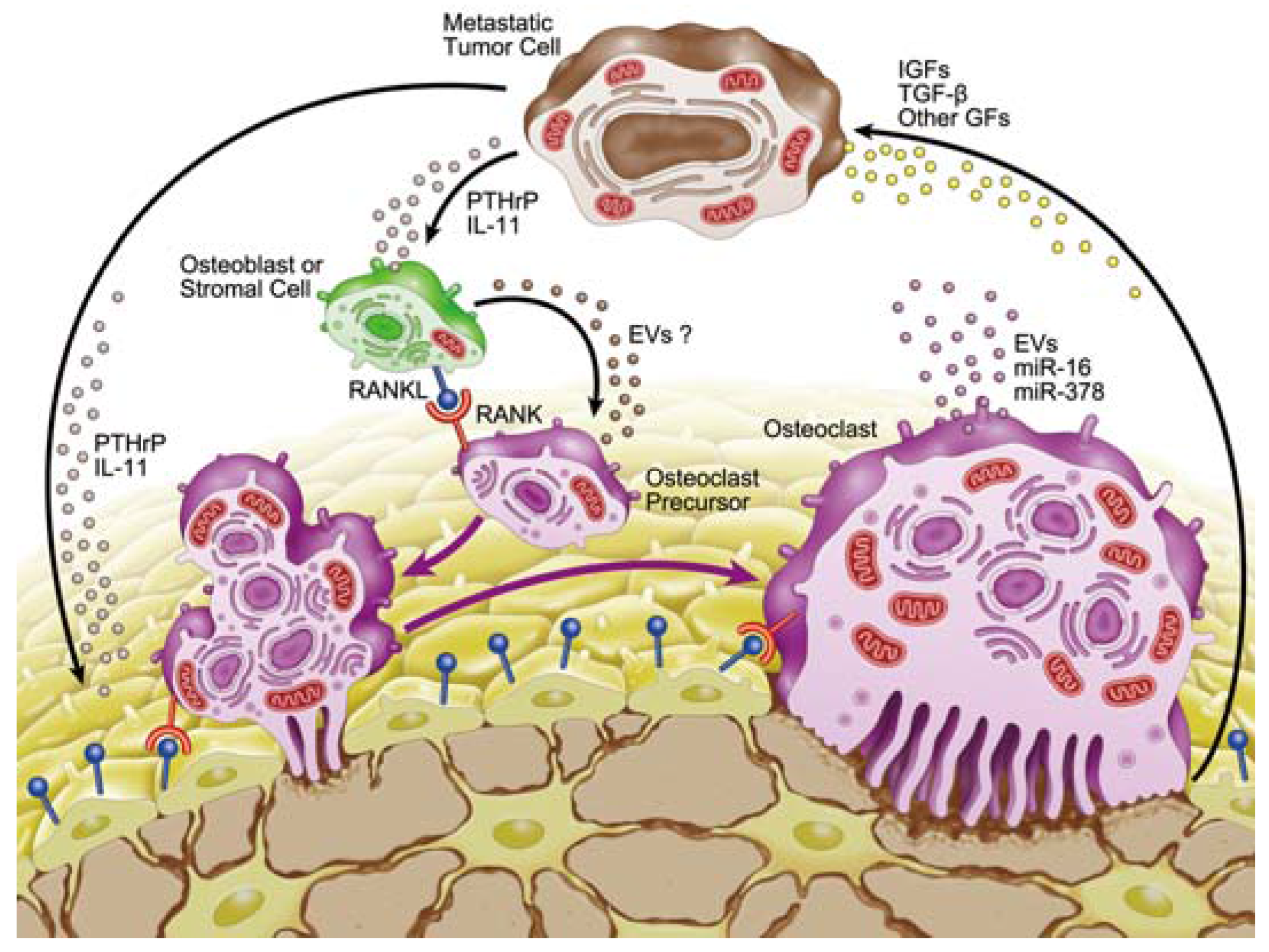
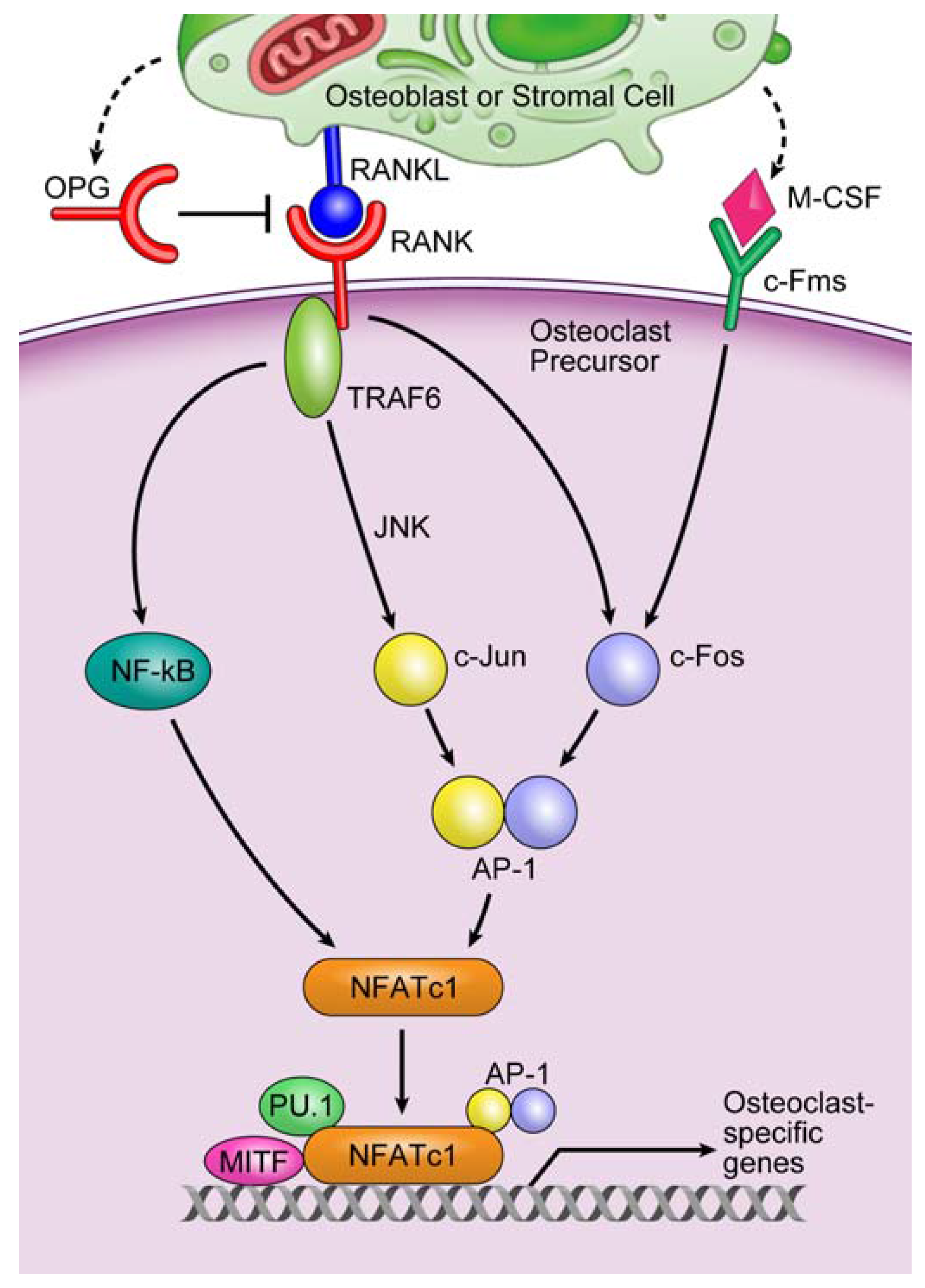
3.2. Involvement of microRNAs in Tumor-Induced Osteoclast Differentiation
4. Involvement of Extracellular Vesicles in Osteolytic Bone Metastasis

{kind=link}
{kind=link}
{kind=link}
| miRNA | Function(s) | Reference(s) |
|---|---|---|
| miR-16 | Potential circulating biomarker for bone metastasis | [3] |
| miR-21 | Functions as an oncogene | [32] |
| Highly expressed during osteoclastogenesis | [24] | |
| Highly detected in osteoclast EVs | [6] | |
| miR-31 | Inhibits breast cancer metastasis | [33] |
| Promotes ring-shaped mature osteoclast formation | [34] | |
| miR-33a | Inhibits bone metastasis by targeting PTHrP | [31] |
| Downregulated during osteoclastogenesis | [3] | |
| miR-34a | Inhibits osteoclast differentiation by targeting Tgif2 | [29] |
| Attenuates bone metastasis | [29] | |
| miR-125a | Tumor suppressor in breast cancer | [32] |
| Upregulated during osteoclastogenesis | [28] | |
| Inhibits osteoclast differentiation by targeting TRAF6 | [35] | |
| miR-133a | Inhibits osteoclast differentiation by targeting Mitf and Mmp14 | [3] |
| miR-141 | Inhibits osteoclast differentiation by targeting Mitf and Calcr | [3] |
| miR-155 | Highly expressed in invasive tumors | [32] |
| Inhibits osteoclastogenesis by repressing MITF and PU.1 | [25] | |
| Deficiency promotes tumor growth in vivo | [36] | |
| miR-190 | Inhibits osteoclast differentiation by targeting Calcr | [3] |
| miR-192 | Inhibits angiogenesis and decreases bone metastasis | [37] |
| miR-219 | Inhibits osteoclast differentiation by targeting Mitf and Traf6 | [3] |
| miR-223 | Inhibits murine osteoclast differentiation | [21,22] |
| Decreases breast cancer cell proliferation | [38] | |
| miR-326 | Potential circulating biomarker for bone metastasis | [39] |
| miR-378 | Potential circulating biomarker for bone metastasis | [3] |
| Highly detected in osteoclast EVs | [6] | |
| Promotes cell survival, tumor growth, and angiogenesis | [40] | |
| miR-204/211/379 | Inhibits TGF-β-induced IL-11 production | [30] |
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Krzeszinski, J.Y.; Wan, Y. New therapeutic targets for cancer bone metastasis. Trends Pharmacol. Sci. 2015, 36, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Browne, G.; Taipaleenmaki, H.; Stein, G.S.; Stein, J.L.; Lian, J.B. MicroRNAs in the control of metastatic bone disease. Trends Endocrinol. Metab. 2014, 25, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Ell, B.; Mercatali, L.; Ibrahim, T.; Campbell, N.; Schwarzenbach, H.; Pantel, K.; Amadori, D.; Kang, Y. Tumor-induced osteoclast miRNA changes as regulators and biomarkers of osteolytic bone metastasis. Cancer Cell 2013, 24, 542–556. [Google Scholar] [CrossRef] [PubMed]
- Waning, D.L.; Mohammad, K.S.; Guise, T.A. Cancer-associated osteoclast differentiation takes a good look in the miR(NA)ror. Cancer Cell 2013, 24, 407–409. [Google Scholar] [CrossRef] [PubMed]
- Kagiya, T.; Nakamura, S. Expression profiling of microRNAs in RAW264.7 cells treated with a combination of tumor necrosis factor alpha and RANKL during osteoclast differentiation. J. Periodontal Res. 2013, 48, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Kagiya, T.; Taira, M. Expression of MicroRNAs in the Extracellular Microvesicles of Murine Osteoclasts. J. Oral Tissue Engin. 2013, 10, 142–150. [Google Scholar]
- Itzstein, C.; Coxon, F.P.; Rogers, M.J. The regulation of osteoclast function and bone resorption by small GTPases. Small GTPases 2011, 2, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, T.; Takayanagi, H. Osteoclasts and the immune system. J. Bone Miner. Metab. 2009, 27, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Weilbaecher, K.N.; Guise, T.A.; McCauley, L.K. Cancer to bone: a fatal attraction. Nat. Rev. Cancer 2011, 11, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.H.; Nakashima, T.; Sanchez, O.H.; Kozieradzki, I.; Komarova, S.V.; Sarosi, I.; Morony, S.; Rubin, E.; Sarao, R.; Hojilla, C.V.; et al. Regulation of cancer cell migration and bone metastasis by RANKL. Nature 2006, 440, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Kingsley, L.A.; Fournier, P.G.; Chirgwin, J.M.; Guise, T.A. Molecular biology of bone metastasis. Mol. Cancer Ther. 2007, 6, 2609–2617. [Google Scholar] [CrossRef] [PubMed]
- Croset, M.; Santini, D.; Iuliani, M.; Fioramonti, M.; Zoccoli, A.; Vincenzi, B.; Tonini, G.; Pantano, F. MicroRNAs and bone metastasis: a new challenge. Molecules 2014, 19, 10115–10128. [Google Scholar] [CrossRef] [PubMed]
- Sugino, T.; Yamaguchi, T.; Hoshi, N.; Kusakabe, T.; Ogura, G.; Goodison, S.; Suzuki, T. Sinusoidal tumor angiogenesis is a key component in hepatocellular carcinoma metastasis. Clin. Exp. Metastasis 2008, 25, 835–841. [Google Scholar] [CrossRef] [PubMed]
- Sugino, T.; Kusakabe, T.; Hoshi, N.; Yamaguchi, T.; Kawaguchi, T.; Goodison, S.; Sekimata, M.; Homma, Y.; Suzuki, T. An invasion-independent pathway of blood-borne metastasis: a new murine mammary tumor model. Am. J. Pathol. 2002, 160, 1973–1980. [Google Scholar] [CrossRef]
- Kats-Ugurlu, G.; Roodink, I.; de Weijert, M.; Tiemessen, D.; Maass, C.; Verrijp, K.; van der Laak, J.; de Waal, R.; Mulders, P.; Oosterwijk, E.; et al. Circulating tumour tissue fragments in patients with pulmonary metastasis of clear cell renal cell carcinoma. J. Pathol. 2009, 219, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Al-Mehdi, A.B.; Tozawa, K.; Fisher, A.B.; Shientag, L.; Lee, A.; Muschel, R.J. Intravascular origin of metastasis from the proliferation of endothelium-attached tumor cells: a new model for metastasis. Nat. Med. 2000, 6, 100–102. [Google Scholar] [PubMed]
- Suva, L.J.; Washam, C.; Nicholas, R.W.; Griffin, R.J. Bone metastasis: mechanisms and therapeutic opportunities. Nat. Rev. Endocrinol. 2011, 7, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Hiraga, T.; Myoui, A.; Hashimoto, N.; Sasaki, A.; Hata, K.; Morita, Y.; Yoshikawa, H.; Rosen, C.J.; Mundy, G.R.; Yoneda, T. Bone-derived IGF mediates crosstalk between bone and breast cancer cells in bony metastases. Cancer Res. 2012, 72, 4238–4249. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.J. Manipulating the environment of cancer cells in bone: a novel therapeutic approach. J. Clin. Invest. 2002, 110, 1399–1401. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.U.; Miyazaki, H.; Ochiya, T. The Roles of MicroRNAs in Breast Cancer. Cancers 2015, 7, 598–616. [Google Scholar] [CrossRef] [PubMed]
- Sugatani, T.; Hruska, K.A. MicroRNA-223 is a key factor in osteoclast differentiation. J. Cell Biochem. 2007, 101, 996–999. [Google Scholar] [CrossRef] [PubMed]
- Sugatani, T.; Hruska, K.A. Impaired micro-RNA pathways diminish osteoclast differentiation and function. J. Biol. Chem. 2009, 284, 4667–4678. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kim, H.J.; Park, C.K.; Kim, Y.G.; Lee, H.J.; Kim, J.Y.; Kim, H.H. MicroRNA-124 regulates osteoclast differentiation. Bone 2013, 56, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Sugatani, T.; Vacher, J.; Hruska, K.A. A microRNA expression signature of osteoclastogenesis. Blood 2011, 117, 3648–3657. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.; Barad, O.; Agami, R.; Geiger, B.; Hornstein, E. miRNA-based mechanism for the commitment of multipotent progenitors to a single cellular fate. Proc. Natl. Acad. Sci. USA 2010, 107, 15804–15809. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Pitari, M.R.; Amodio, N.; Di Martino, M.T.; Conforti, F.; Leone, E.; Botta, C.; Paolino, F.M.; Del Giudice, T.; Iuliano, E.; et al. miR-29b negatively regulates human osteoclastic cell differentiation and function: implications for the treatment of multiple myeloma-related bone disease. J. Cell Physiol. 2013, 228, 1506–1515. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Cheng, P.; Xie, H.; Zhou, H.D.; Wu, X.P.; Liao, E.Y.; Luo, X.H. MiR-503 regulates osteoclastogenesis via targeting RANK. J. Bone Miner. Res. 2014, 29, 338–347. [Google Scholar] [CrossRef] [PubMed]
- De la Rica, L.; Garcia-Gomez, A.; Comet, N.R.; Rodriguez-Ubreva, J.; Ciudad, L.; Vento-Tormo, R.; Company, C.; Alvarez-Errico, D.; Garcia, M.; Gomez-Vaquero, C.; et al. NF-kappaB-direct activation of microRNAs with repressive effects on monocyte-specific genes is critical for osteoclast differentiation. Genome Biol. 2015, 16, 2. [Google Scholar] [CrossRef] [PubMed]
- Krzeszinski, J.Y.; Wei, W.; Huynh, H.; Jin, Z.; Wang, X.; Chang, T.C.; Xie, X.J.; He, L.; Mangala, L.S.; Lopez-Berestein, G.; et al. miR-34a blocks osteoporosis and bone metastasis by inhibiting osteoclastogenesis and Tgif2. Nature 2014, 512, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Pollari, S.; Leivonen, S.K.; Perala, M.; Fey, V.; Kakonen, S.M.; Kallioniemi, O. Identification of microRNAs inhibiting TGF-beta-induced IL-11 production in bone metastatic breast cancer cells. PLoS One 2012, 7, e37361. [Google Scholar] [CrossRef] [PubMed]
- Kuo, P.L.; Liao, S.H.; Hung, J.Y.; Huang, M.S.; Hsu, Y.L. MicroRNA-33a functions as a bone metastasis suppressor in lung cancer by targeting parathyroid hormone related protein. Biochim. Biophys. Acta. 2013, 1830, 3756–3766. [Google Scholar] [CrossRef] [PubMed]
- O’Day, E.; Lal, A. MicroRNAs and their target gene networks in breast cancer. Breast Cancer Res. 2010, 12, 201. [Google Scholar] [CrossRef] [PubMed]
- Valastyan, S.; Weinberg, R.A. miR-31: a crucial overseer of tumor metastasis and other emerging roles. Cell Cycle 2010, 9, 2124–2129. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, F.; Murakami, Y.; Saito, T.; Miyasaka, N.; Kohsaka, H. miR-31 controls osteoclast formation and bone resorption by targeting RhoA. Arthritis Res. Ther. 2013, 15, R102. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.J.; Liao, L.; Yang, L.; Li, Y.; Jiang, T.J. MiR-125a TNF receptor-associated factor 6 to inhibit osteoclastogenesis. Exp. Cell Res. 2014, 321, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yu, F.; Jia, X.; Iwanowycz, S.; Wang, Y.; Huang, S.; Ai, W.; Fan, D. MicroRNA-155 deficiency enhances the recruitment and functions of myeloid-derived suppressor cells in tumor microenvironment and promotes solid tumor growth. Int. J. Cancer 2015, 136, E602–E613. [Google Scholar] [CrossRef] [PubMed]
- Valencia, K.; Luis-Ravelo, D.; Bovy, N.; Anton, I.; Martinez-Canarias, S.; Zandueta, C.; Ormazabal, C.; Struman, I.; Tabruyn, S.; Rebmann, V.; et al. miRNA cargo within exosome-like vesicle transfer influences metastatic bone colonization. Mol. Oncol. 2014, 8, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Lim, P.K.; Bliss, S.A.; Patel, S.A.; Taborga, M.; Dave, M.A.; Gregory, L.A.; Greco, S.J.; Bryan, M.; Patel, P.S.; Rameshwar, P. Gap junction-mediated import of microRNA from bone marrow stromal cells can elicit cell cycle quiescence in breast cancer cells. Cancer Res. 2011, 71, 1550–1560. [Google Scholar] [CrossRef] [PubMed]
- Valencia, K.; Martin-Fernandez, M.; Zandueta, C.; Ormazabal, C.; Martinez-Canarias, S.; Bandres, E.; de la Piedra, C.; Lecanda, F. miR-326 associates with biochemical markers of bone turnover in lung cancer bone metastasis. Bone 2013, 52, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Deng, Z.; Wang, C.H.; Yang, B.B. MicroRNA-378 promotes cell survival, tumor growth, and angiogenesis by targeting SuFu and Fus-1 expression. Proc. Natl Acad Sci USA 2007, 104, 20350–20355. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell. Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, N.; Iguchi, H.; Yoshioka, Y.; Takeshita, F.; Matsuki, Y.; Ochiya, T. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J. Biol. Chem. 2010, 285, 17442–17452. [Google Scholar] [CrossRef] [PubMed]
- Carandini, T.; Colombo, F.; Finardi, A.; Casella, G.; Garzetti, L.; Verderio, C.; Furlan, R. Microvesicles: What is the Role in Multiple Sclerosis? Front Neurol. 2015, 6, 111. [Google Scholar] [CrossRef] [PubMed]
- Nishida-Aoki, N.; Ochiya, T. Interactions between cancer cells and normal cells via miRNAs in extracellular vesicles. Cell. Mol. Life Sci. 2015, 72, 1849–1861. [Google Scholar] [CrossRef] [PubMed]
- Crescitelli, R.; Lasser, C.; Szabo, T.G.; Kittel, A.; Eldh, M.; Dianzani, I.; Buzas, E.I.; Lotvall, J. Distinct RNA profiles in subpopulations of extracellular vesicles: apoptotic bodies, microvesicles and exosomes. J. Extracell Vesicles 2013, 2, 20677. [Google Scholar] [CrossRef] [PubMed]
- Gould, S.J.; Raposo, G. As we wait: coping with an imperfect nomenclature for extracellular vesicles. J. Extracell Vesicles 2013, 2, 20389. [Google Scholar] [CrossRef] [PubMed]
- Kagiya, T.; Taira, M. A New Application for Microarrays: Analysis of Global MicroRNA Expression Profiles in the Extracellular Microvesicles of Human Macrophage-like Cells. In Microarrays: Principles, Applications and Technologies; Rogers, J.V., Ed.; Nova Science Publishers: New York, NY, USA, 2014; pp. 69–80. [Google Scholar]
- Arroyo, J.D.; Chevillet, J.R.; Kroh, E.M.; Ruf, I.K.; Pritchard, C.C.; Gibson, D.F.; Mitchell, P.S.; Bennett, C.F.; Pogosova-Agadjanyan, E.L.; Stirewalt, D.L.; et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc. Natl. Acad. Sci. USA 2011, 108, 5003–5008. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, L.; De Luca, A.; Amodio, N.; Manno, M.; Raccosta, S.; Taverna, S.; Bellavia, D.; Naselli, F.; Fontana, S.; Schillaci, O.; et al. Involvement of multiple myeloma cell-derived exosomes in osteoclast differentiation. Oncotarget 2015, 6, 13772–13789. [Google Scholar] [PubMed]
- Deng, L.; Wang, Y.; Peng, Y.; Wu, Y.; Ding, Y.; Jiang, Y.; Shen, Z.; Fu, Q. Osteoblast-derived microvesicles: A novel mechanism for communication between osteoblasts and osteoclasts. Bone 2015, 79, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Selbach, M.; Schwanhausser, B.; Thierfelder, N.; Fang, Z.; Khanin, R.; Rajewsky, N. Widespread changes in protein synthesis induced by microRNAs. Nature 2008, 455, 58–63. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kagiya, T. MicroRNAs and Osteolytic Bone Metastasis: The Roles of MicroRNAs in Tumor-Induced Osteoclast Differentiation. J. Clin. Med. 2015, 4, 1741-1752. https://doi.org/10.3390/jcm4091741
Kagiya T. MicroRNAs and Osteolytic Bone Metastasis: The Roles of MicroRNAs in Tumor-Induced Osteoclast Differentiation. Journal of Clinical Medicine. 2015; 4(9):1741-1752. https://doi.org/10.3390/jcm4091741
Chicago/Turabian StyleKagiya, Tadayoshi. 2015. "MicroRNAs and Osteolytic Bone Metastasis: The Roles of MicroRNAs in Tumor-Induced Osteoclast Differentiation" Journal of Clinical Medicine 4, no. 9: 1741-1752. https://doi.org/10.3390/jcm4091741
APA StyleKagiya, T. (2015). MicroRNAs and Osteolytic Bone Metastasis: The Roles of MicroRNAs in Tumor-Induced Osteoclast Differentiation. Journal of Clinical Medicine, 4(9), 1741-1752. https://doi.org/10.3390/jcm4091741