
Hypopituitarism in Traumatic Brain Injury—A Critical Note

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Hypopituitarism in TBI—Pathophysiology
3. What is the Reported Prevalence of Acute Anterior Pituitary Hormone Deficiency?
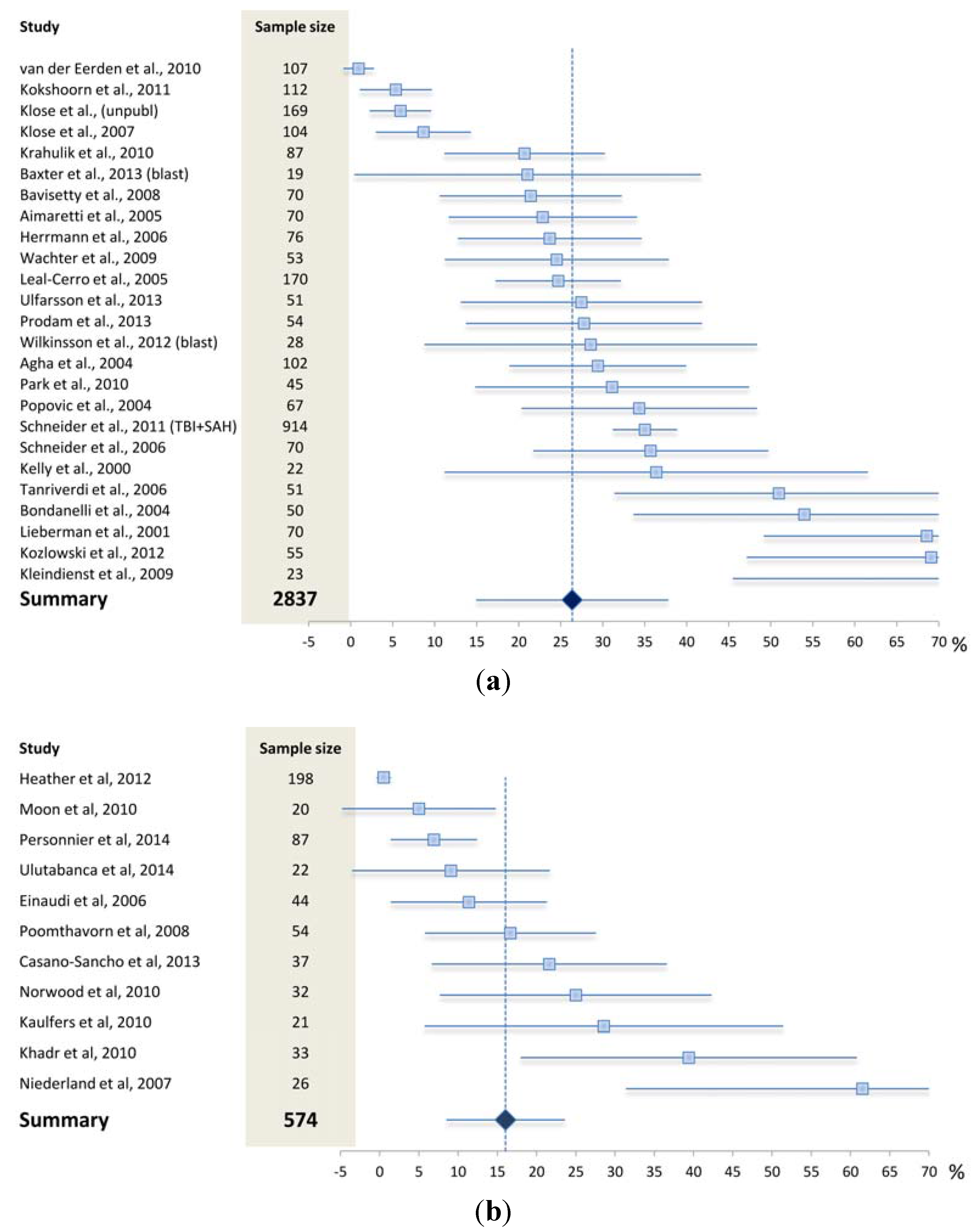
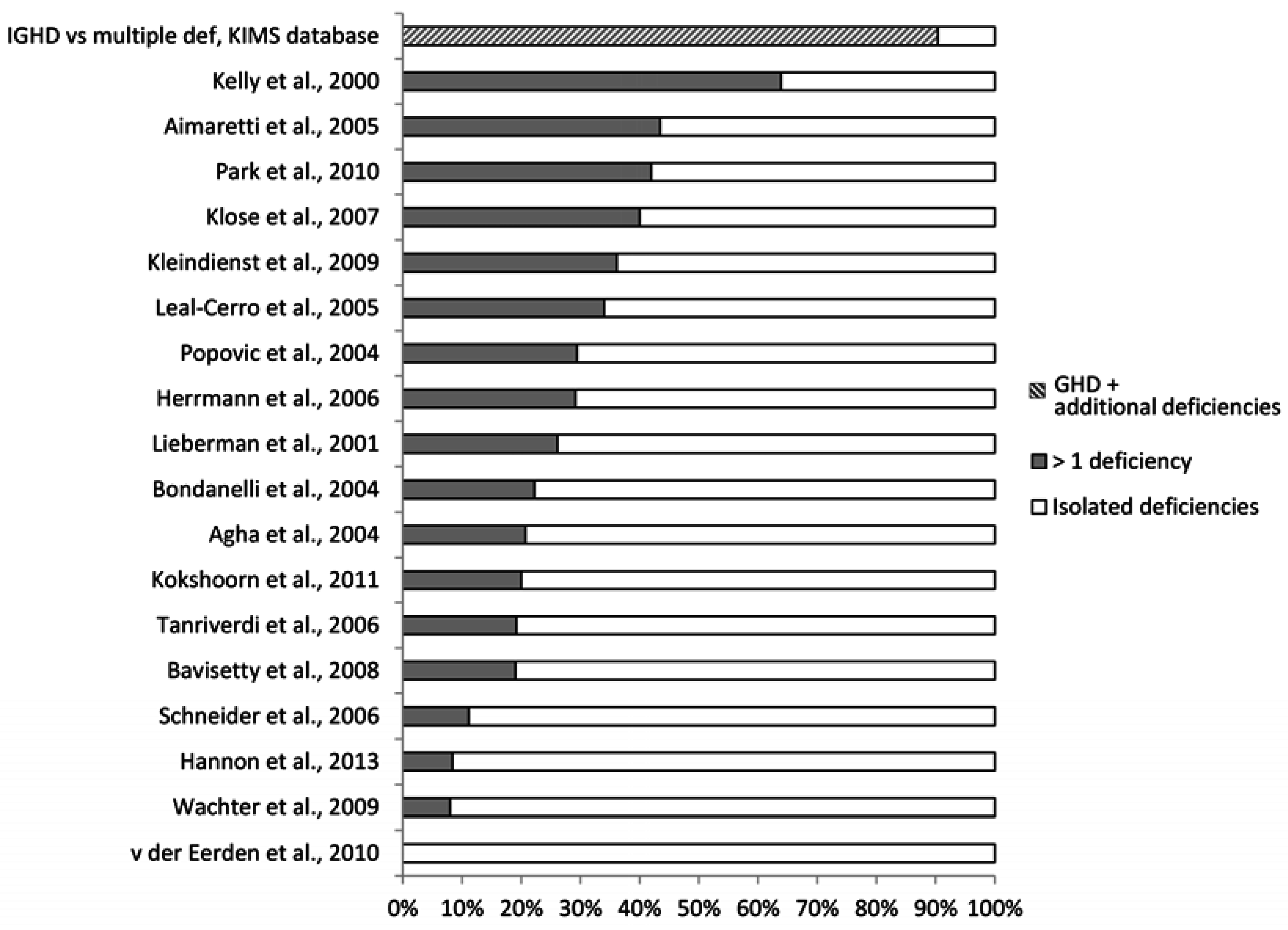
4. What is the Reported Prevalence of Chronic Anterior Pituitary Hormone Deficiency?
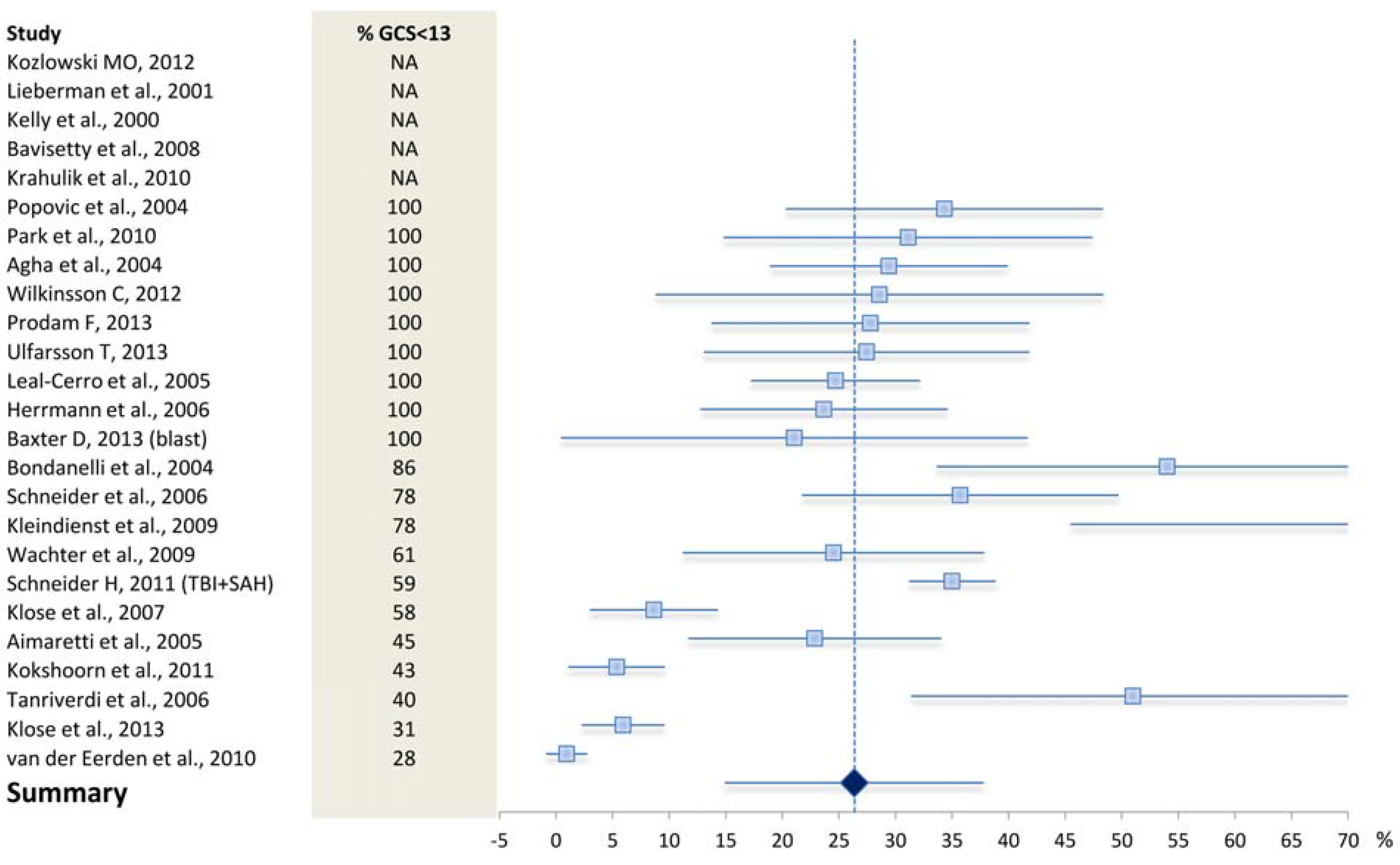
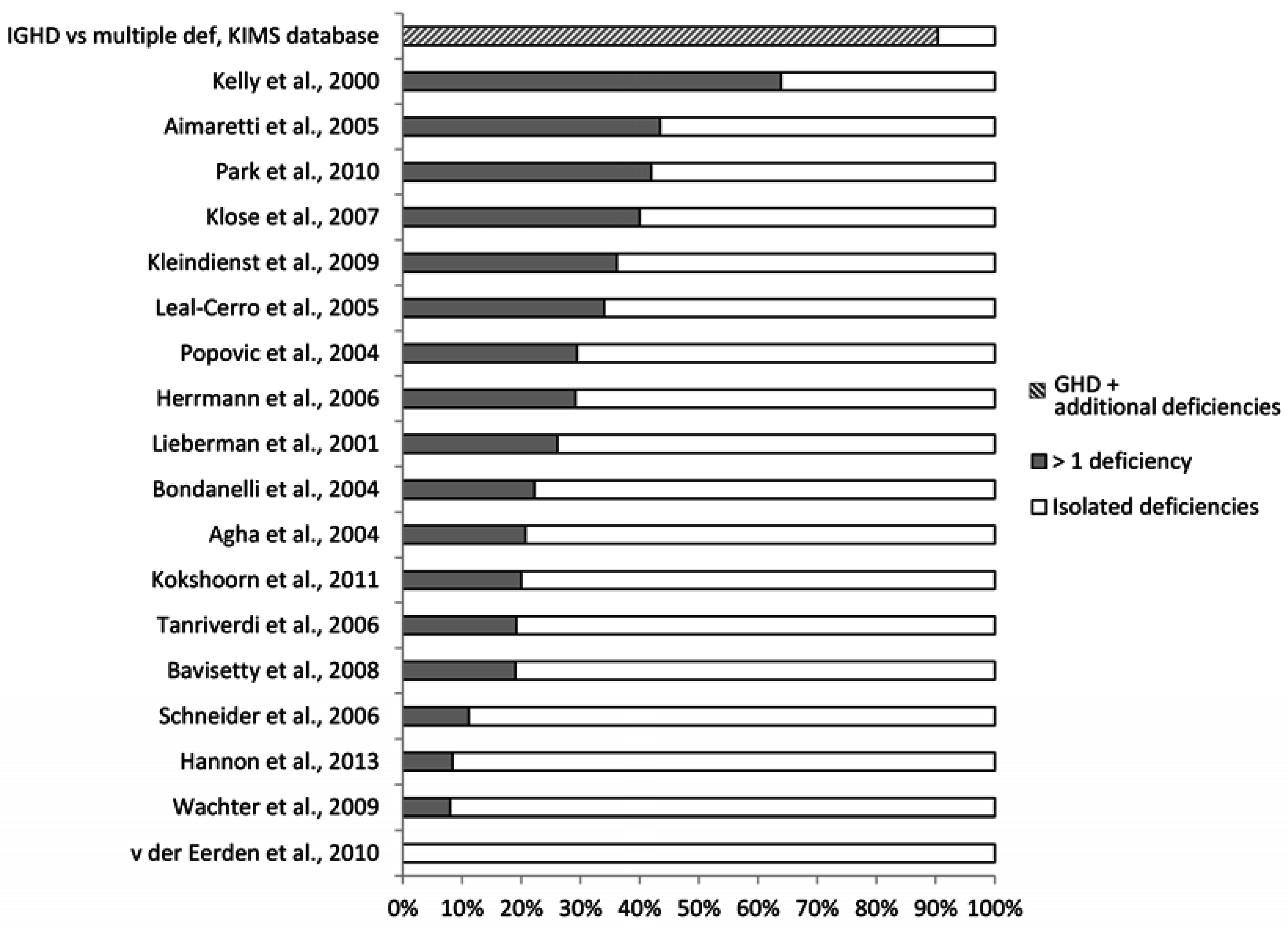
4.1. What May Affect the Reported Prevalence Rate—Patient Selection?
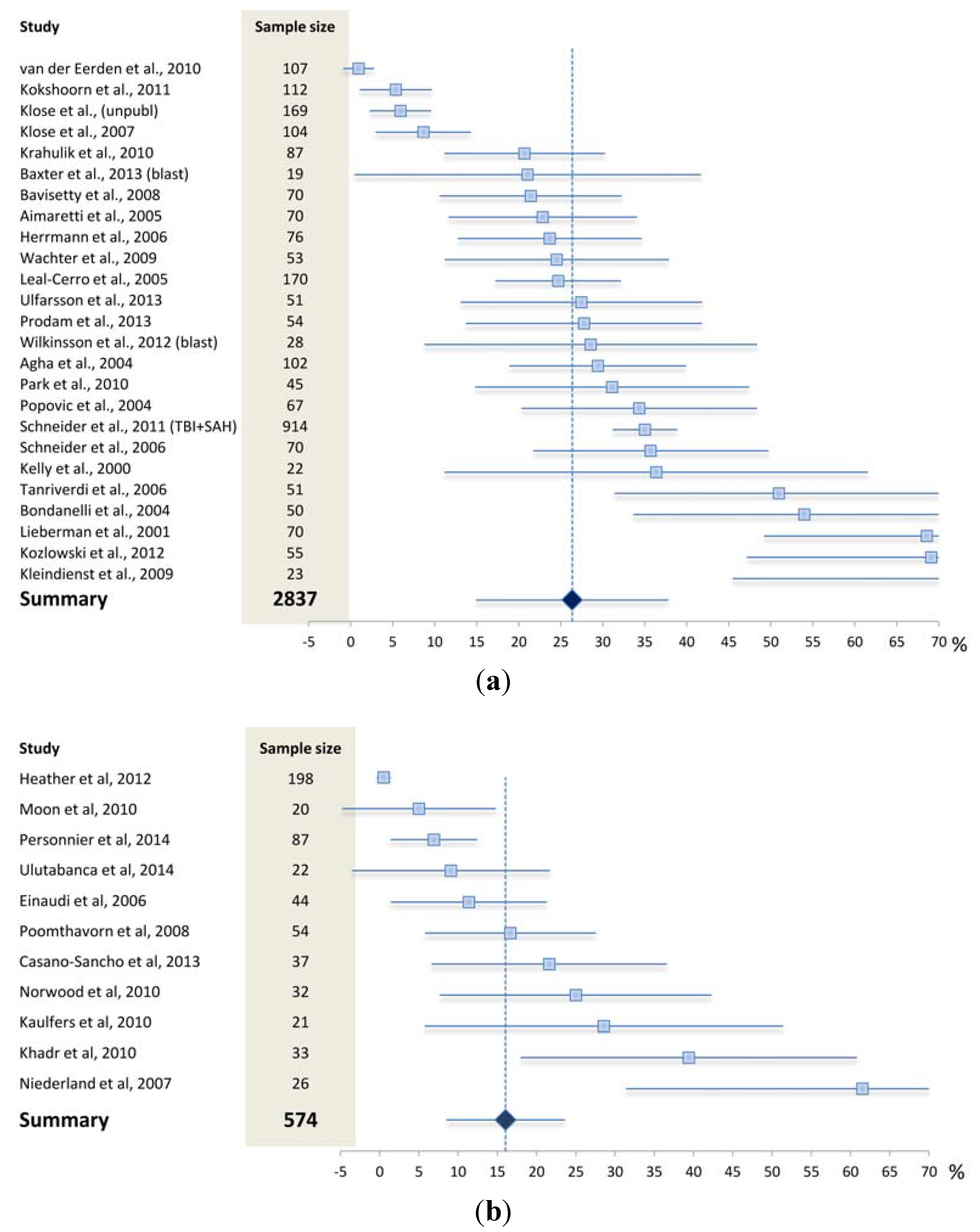
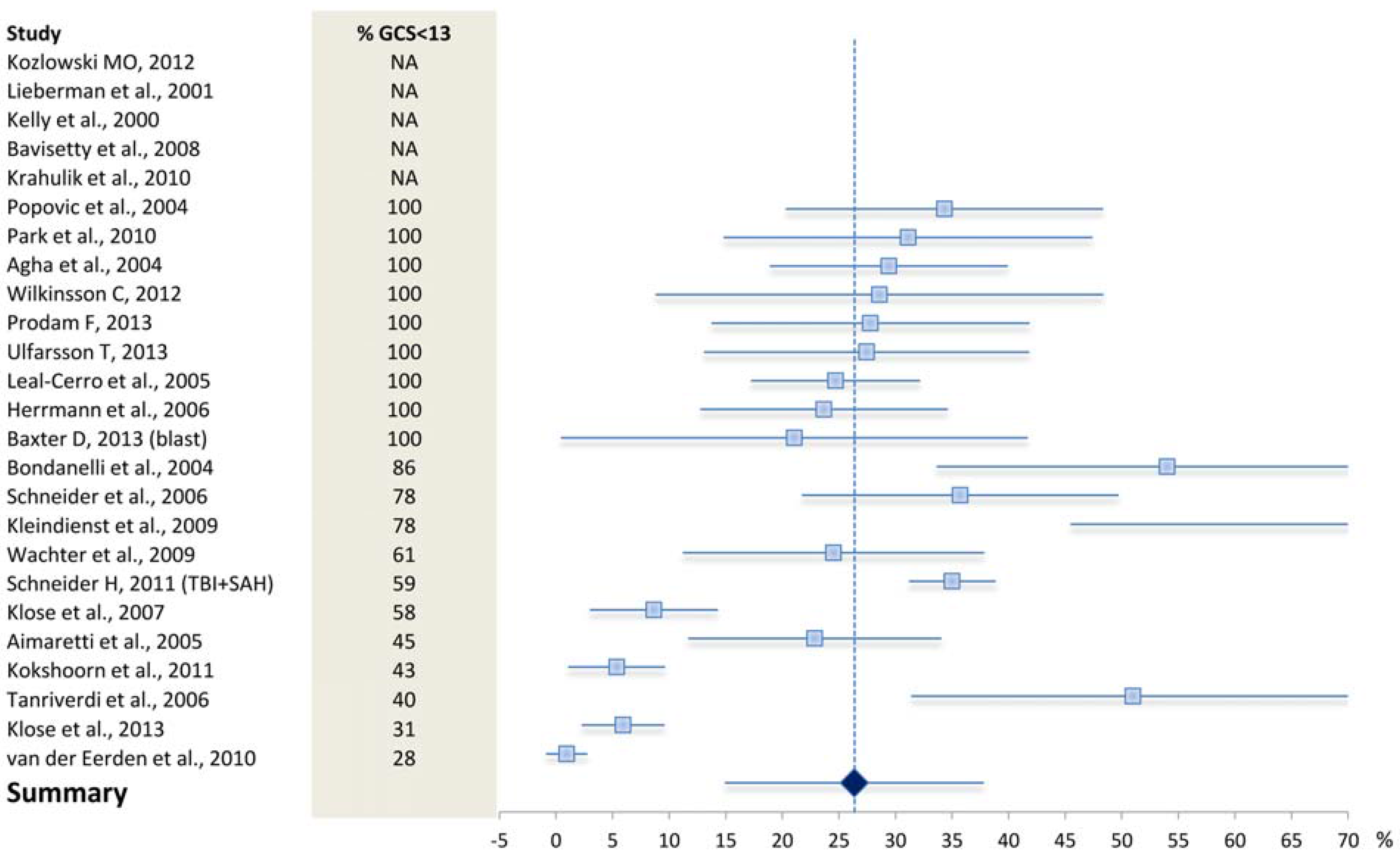
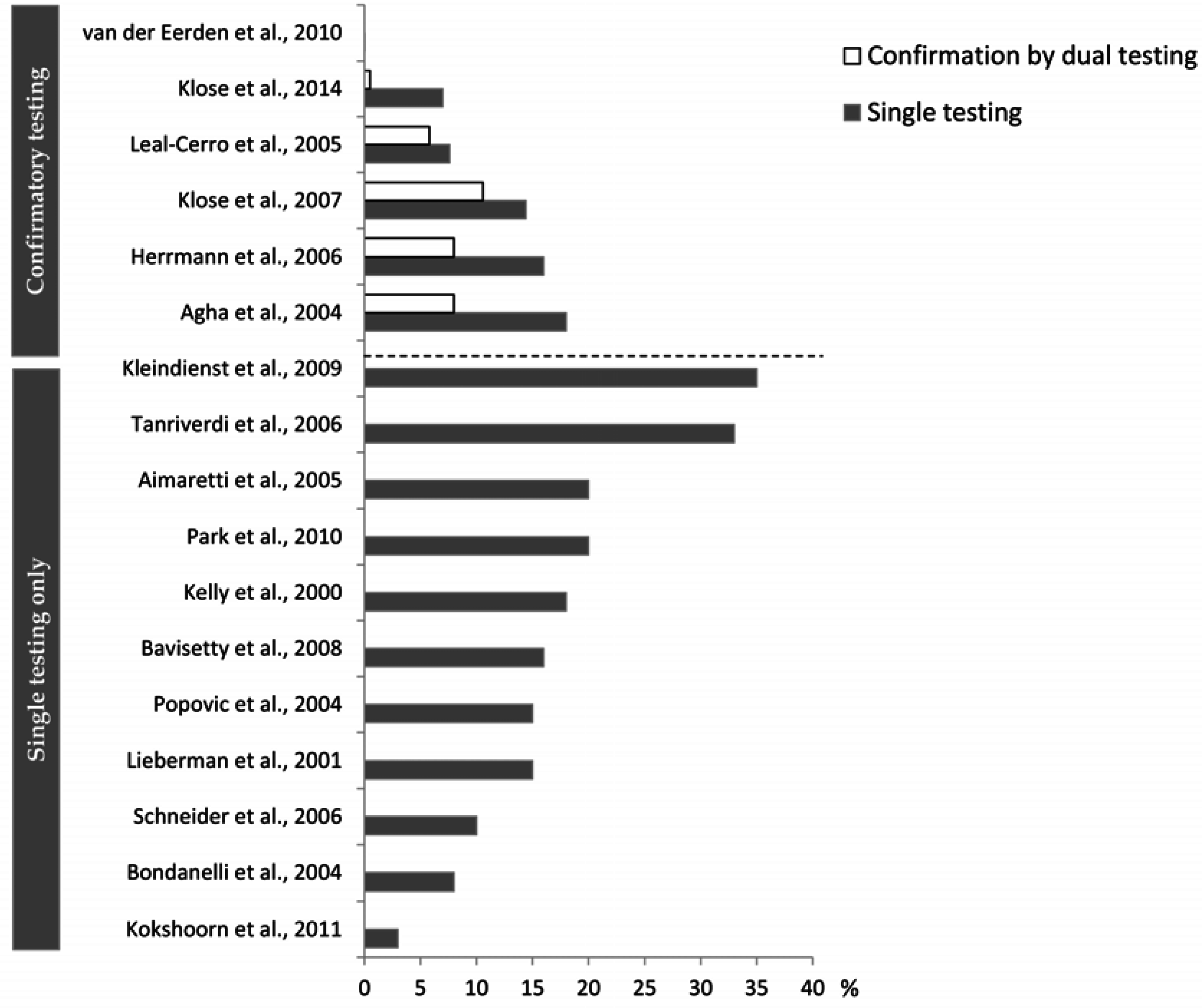
4.2. What May Affect the Reported Prevalence Rate—Diagnostic Challenges?
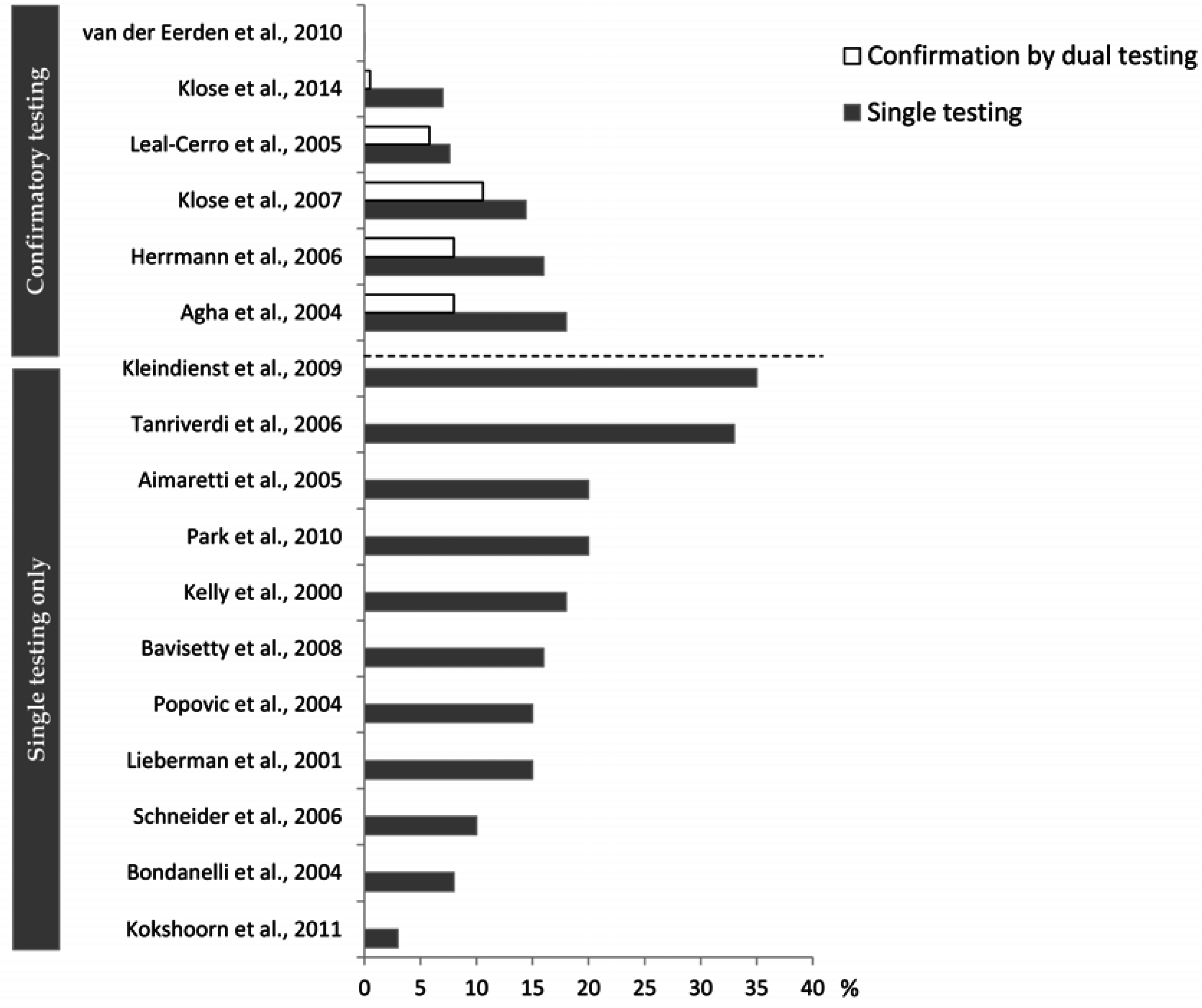
5. Implication of Posttraumatic Hypopituitarism
6. Treatment of Posttraumatic Hypopituitarism
7. Conclusions
Acknowledgments
Author Contributions
Conflict of Interests
References
- Schneider, H.J.; Kreitschmann-Andermahr, I.; Ghigo, E.; Stalla, G.K.; Agha, A. Hypothalamopituitary dysfunction following traumatic brain injury and aneurysmal subarachnoid hemorrhage: A systematic review. JAMA 2007, 298, 1429–1438. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.K. Consensus guidelines for the diagnosis and treatment of adults with GH deficiency II: A statement of the GH Research Society in association with the European Society for Pediatric Endocrinology, Lawson Wilkins Society, European Society of Endocrinology, Japan Endocrine Society, and Endocrine Society of Australia. Eur. J. Endocrinol. 2007, 157, 695–700. [Google Scholar] [PubMed]
- Molitch, M.E.; Clemmons, D.R.; Malozowski, S.; Merriam, G.R.; Vance, M.L. Evaluation and treatment of adult growth hormone deficiency: An Endocrine Society clinical practice guideline. J. Clin. Endocrinol. Metab. 2011, 96, 1587–1609. [Google Scholar] [CrossRef] [PubMed]
- Crompton, M.R. Hypothalamic lesions following closed head injury. Brain 1971, 94, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Salehi, F.; Kovacs, K.; Scheithauer, B.W.; Pfeifer, E.A.; Cusimano, M. Histologic study of the human pituitary gland in acute traumatic brain injury. Brain Injury 2007, 21, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Van Den Berghe, G. Endocrine evaluation of patients with critical illness. Endocrinol. Metab. Clin. N. Am. 2003, 32, 385–410. [Google Scholar] [CrossRef]
- Cohan, P.; Wang, C.; McArthur, D.L.; Cook, S.W.; Dusick, J.R.; Armin, B.; Swerdloff, R.; Vespa, P.; Muizelaar, J.P.; Cryer, H.G.; et al. Acute secondary adrenal insufficiency after traumatic brain injury: A prospective study. Crit. Care Med. 2005, 33, 2358–2366. [Google Scholar] [CrossRef] [PubMed]
- Hannon, M.J.; Crowley, R.K.; Behan, L.A.; O’Sullivan, E.P.; O'Brien, M.M.; Sherlock, M.; Rawluk, D.; O’Dwyer, R.; Tormey, W.; Thompson, C.J. Acute glucocorticoid deficiency and diabetes insipidus are common after acute traumatic brain injury and predict mortality. J. Clin. Endocrinol. Metab. 2013, 98, 3229–3237. [Google Scholar] [CrossRef] [PubMed]
- Hamrahian, A.H.; Oseni, T.S.; Arafah, B.M. Measurements of serum free cortisol in critically ill patients. N. Engl. J. Med. 2004, 350, 1629–1638. [Google Scholar] [CrossRef] [PubMed]
- Kleindienst, A.; Brabant, G.; Bock, C.; Maser-Gluth, C.; Buchfelder, M. Neuroendocrine function following traumatic brain injury and subsequent intensive care treatment: A prospective longitudinal evaluation. J. Neurotrauma 2009, 26, 1435–1446. [Google Scholar] [CrossRef] [PubMed]
- Annane, D.; Sebille, V.; Troche, G.; Raphael, J.C.; Gajdos, P.; Bellissant, E. A 3-level prognostic classification in septic shock based on cortisol levels and cortisol response to corticotropin. JAMA 2000, 283, 1038–1045. [Google Scholar] [CrossRef] [PubMed]
- Boonen, E.; Vervenne, H.; Meersseman, P.; Andrew, R.; Mortier, L.; Declercq, P.E.; Vanwijngaerden, Y.M.; Spriet, I.; Wouters, P.J.; Vander, P.S.; et al. Reduced cortisol metabolism during critical illness. N. Engl. J. Med. 2013, 368, 1477–1488. [Google Scholar] [CrossRef] [PubMed]
- Beishuizen, A.; Thijs, L.G.; Vermes, I. Patterns of corticosteroid-binding globulin and the free cortisol index during septic shock and multitrauma. Intensive Care Med. 2001, 27, 1584–1591. [Google Scholar] [CrossRef] [PubMed]
- Molijn, G.J.; Spek, J.J.; van Uffelen, J.C.; de Jong, F.H.; Brinkmann, A.O.; Bruining, H.A.; Lamberts, S.W.; Koper, J.W. Differential adaptation of glucocorticoid sensitivity of peripheral blood mononuclear leukocytes in patients with sepsis or septic shock. J. Clin. Endocrinol. Metab. 1995, 80, 1799–1803. [Google Scholar] [PubMed]
- Gehlbach, B.K.; Chapotot, F.; Leproult, R.; Whitmore, H.; Poston, J.; Pohlman, M.; Miller, A.; Pohlman, A.S.; Nedeltcheva, A.; Jacobsen, J.H.; et al. Temporal disorganization of circadian rhythmicity and sleep-wake regulation in mechanically ventilated patients receiving continuous intravenous sedation. Sleep 2012, 35, 1105–1114. [Google Scholar] [CrossRef] [PubMed]
- Llompart-Pou, J.A.; Perez, G.; Raurich, J.M.; Riesco, M.; Brell, M.; Ibanez, J.; Perez-Barcena, J.; Abadal, J.M.; Homar, J.; Burguera, B. Loss of cortisol circadian rhythm in patients with traumatic brain injury: A microdialysis evaluation. Neurocrit. Care 2010, 13, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.S.; Stewart, P.M. Corticosteroid insufficiency in acutely ill patients. N. Engl. J. Med. 2003, 348, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Agha, A.; Phillips, J.; O’Kelly, P.; Tormey, W.; Thompson, C.J. The natural history of post-traumatic hypopituitarism: Implications for assessment and treatment. Am. J. Med. 2005, 118, 1416. [Google Scholar] [CrossRef] [PubMed]
- Tanriverdi, F.; Senyurek, H.; Unluhizarci, K.; Selcuklu, A.; Casanueva, F.F.; Kelestimur, F. High risk of hypopituitarism after traumatic brain injury: A prospective investigation of anterior pituitary function in the acute phase and 12 months after trauma. J. Clin. Endocrinol. Metab. 2006, 91, 2105–2111. [Google Scholar] [CrossRef] [PubMed]
- Klose, M.; Juul, A.; Struck, J.; Morgenthaler, N.G.; Kosteljanetz, M.; Feldt-Rasmussen, U. Acute and long-term pituitary insufficiency in traumatic brain injury: A prospective single-centre study. Clin. Endocrinol. 2007, 67, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Van den, B.G.; de Zegher, F.; Bouillon, R. Clinical review 95: Acute and prolonged critical illness as different neuroendocrine paradigms. J. Clin. Endocrinol. Metab. 19 1998, 83, 1827–1834. [Google Scholar]
- Leal-Cerro, A.; Flores, J.M.; Rincon, M.; Murillo, F.; Pujol, M.; Garcia-Pesquera, F.; Dieguez, C.; Casanueva, F.F. Prevalence of hypopituitarism and growth hormone deficiency in adults long-term after severe traumatic brain injury. Clin. Endocrinol. 2005, 62, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, B.L.; Rehder, J.; Kahlke, S.; Wiedemayer, H.; Doerfler, A.; Ischebeck, W.; Laumer, R.; Forsting, M.; Stolke, D.; Mann, K. Hypopituitarism following severe traumatic brain injury. Exp. Clin. Endocrinol. Diabetes 2006, 114, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Klose, M.; Stochholm, K.; Janukonyte, J.; Lehman, C.L.; Frystyk, J.; Andersen, M.; Laurberg, P.; Christiansen, J.S.; Feldt-Rasmussen, U. Prevalence of posttraumatic growth hormone deficiency is highly dependent on the diagnostic set-up: Results from The Danish National Study on Posttraumatic Hypopituitarism. J. Clin. Endocrinol. Metab. 2014, 99, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Van der Eerden, A.W.; Twickler, M.T.; Sweep, F.C.; Beems, T.; Hendricks, H.T.; Hermus, A.R.; Vos, P.E. Should anterior pituitary function be tested during follow-up of all patients presenting at the emergency department because of traumatic brain injury? Eur. J. Endocrinol. 2010, 162, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Kokshoorn, N.; Smit, J.W.; Nieuwlaat, W.A.; Tiemensma, J.; Bisschop, P.; Groote, V.R.; Roelfsema, F.; Franken, A.A.; Wassenaar, M.; Biermasz, N.R.; et al. Low prevalence of hypopituitarism after traumatic brain injury—A multi-center study. Eur. J. Endocrinol. 2011, 165, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Klose, M.; Juul, A.; Poulsgaard, L.; Kosteljanetz, M.; Brennum, J.; Feldt-Rasmussen, U. Prevalence and predictive factors of post-traumatic hypopituitarism. Clin. Endocrinol. 2007, 67, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.F.; Gonzalo, I.T.; Cohan, P.; Berman, N.; Swerdloff, R.; Wang, C. Hypopituitarism following traumatic brain injury and aneurysmal subarachnoid hemorrhage: A preliminary report. J. Neurosurg. 2000, 93, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Bondanelli, M.; de Marinis, L.; Ambrosio, M.R.; Monesi, M.; Valle, D.; Zatelli, M.C.; Fusco, A.; Bianchi, A.; Farneti, M.; degli Uberti, E.C. Occurrence of pituitary dysfunction following traumatic brain injury. J. Neurotrauma 2004, 21, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Schneider, H.J.; Yassouridis, A.; Saller, B.; von Rosen, F.; Stalla, G.K. Predictors of anterior pituitary insufficiency after traumatic brain injury. Clin. Endocrinol. 2008, 68, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Bavisetty, S.; Bavisetty, S.; McArthur, D.L.; Dusick, J.R.; Wang, C.; Cohan, P.; Boscardin, W.J.; Swerdloff, R.; Levin, H.; Chang, D.J.; et al. Chronic hypopituitarism after traumatic brain injury: Risk assessment and relationship to outcome. Neurosurgery 2008, 62, 1080–1093. [Google Scholar] [CrossRef] [PubMed]
- Aimaretti, G.; Ambrosio, M.R.; di Somma, C.; Gasperi, M.; Cannavo, S.; Scaroni, C.; Fusco, A.; Del Monte, P.; de Menis, E.; Faustini-Fustini, M.; et al. Residual pituitary function after brain injury-induced hypopituitarism: A prospective 12-month study. J. Clin. Endocrinol. Metab. 2 2005, 90, 6085–6092. [Google Scholar] [CrossRef] [PubMed]
- Agha, A.; Rogers, B.; Sherlock, M.; O’Kelly, P.; Tormey, W.; Phillips, J.; Thompson, C.J. Anterior pituitary dysfunction in survivors of traumatic brain injury. J. Clin. Endocrinol. Metab. 2004, 89, 4929–4936. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; He, B.; Tong, W.S. Decrease in pituitary apparent diffusion coefficient in normal appearing brain correlates with hypopituitarism following traumatic brain injury. J. Endocrinol. Investig. 2014, 37, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Tanriverdi, F.; De, B.A.; Bizzarro, A.; Sinisi, A.A.; Bellastella, G.; Pane, E.; Bellastella, A.; Unluhizarci, K.; Selcuklu, A.; Casanueva, F.F.; Kelestimur, F. Antipituitary antibodies after traumatic brain injury: Is head trauma-induced pituitary dysfunction associated with autoimmunity? Eur. J. Endocrinol. 2008, 159, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Neyeloff, J.L.; Fuchs, S.C.; Moreira, L.B. Meta-analyses and Forest plots using a microsoft excel spreadsheet: Step-by-step guide focusing on descriptive data analysis. BMC Res. Notes 2012, 5, 52. [Google Scholar] [CrossRef] [PubMed]
- Feinsteinm, AR. The inadequacy of binary models for the clinical reality of three-zone diagnostic decisions. J. Clin. Epidemiol. 1990, 43, 109–113. [Google Scholar] [CrossRef]
- Bhasin, S.; Cunningham, G.R.; Hayes, F.J.; Matsumoto, A.M.; Snyder, P.J.; Swerdloff, R.S.; Montori, V.M. Testosterone therapy in men with androgen deficiency syndromes: An Endocrine Society clinical practice guideline. J. Clin. Endocrinol. Metab. 2010, 95, 2536–2559. [Google Scholar] [CrossRef] [PubMed]
- Vestergaard, P.; Hoeck, H.C.; Jakobsen, P.E.; Laurberg, P. Reproducibility of growth hormone and cortisol responses to the insulin tolerance test and the short ACTH test in normal adults. Horm. Metab. Res. 1997, 29, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Veldhuis, J.D.; Iranmanesh, A.; Ho, K.K.; Waters, M.J.; Johnson, M.L.; Lizarralde, G. Dual defects in pulsatile growth hormone secretion and clearance subserve the hyposomatotropism of obesity in man. J. Clin. Endocrinol. Metab. 1991, 72, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, M.H.; Hvidberg, A.; Juul, A.; Main, K.M.; Gotfredsen, A.; Skakkebaek, N.E.; Hilsted, J. Massive weight loss restores 24-h growth hormone release profiles and serum insulin-like growth factor-I levels in obese subjects. J. Clin. Endocrinol. Metab. 1995, 8, 1407–1415. [Google Scholar] [CrossRef] [PubMed]
- Lange, M.; Svendsen, O.L.; Skakkebaek, N.E.; Muller, J.; Juul, A.; Schmiegelow, M.; Feldt-Rasmussen, U. An audit of the insulin-tolerance test in 255 patients with pituitary disease. Eur. J. Endocrinol. 2002, 147, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Fisker, S.; Jorgensen, J.O.; Orskov, H.; Christiansen, J.S. l-arginine and insulin-tolerance tests in the diagnosis of adult growth hormone deficiency: Influence of confounding factors. Clin. Endocrinol. 1998, 48, 109–115. [Google Scholar] [CrossRef]
- Hoeck, H.C.; Jakobsen, P.E.; Vestergaard, P.; Falhof, J.; Laurberg, P. Differences in reproducibility and peak growth hormone responses to repeated testing with various stimulators in healthy adults. Growth Horm. IGF Res. 1999, 9, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Corneli, G.; di Somma, C.; Baldelli, R.; Rovere, S.; Gasco, V.; Croce, C.G.; Grottoli, S.; Maccario, M.; Colao, A.; Lombardi, G.; et al. The cut-off limits of the GH response to GH-releasing hormone-arginine test related to body mass index. Eur. J. Endocrinol. 2005, 153, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Kelestimur, F.; Popovic, V.; Leal, A.; van Dam, P.S.; Torres, E.; Perez Mendez, L.F.; Greenman, Y.; Koppeschaar, H.P.; Dieguez, C.; Casanueva, F.F. Effect of obesity and morbid obesity on the growth hormone (GH) secretion elicited by the combined GHRH + GHRP-6 test. Clin. Endocrinol. 2006, 64, 667–671. [Google Scholar] [CrossRef] [PubMed]
- Dichtel, L.E.; Yuen, K.C.; Bredella, M.A.; Gerweck, A.V.; Russell, B.M.; Riccio, A.D.; Gurel, M.H.; Sluss, P.M.; Biller, B.M.; Miller, K.K. Overweight/Obese adults with pituitary disorders require lower peak growth hormone cutoff values on glucagon stimulation testing to avoid overdiagnosis of growth hormone deficiency. J. Clin. Endocrinol. Metab. 2014, 99, 4712–4719. [Google Scholar] [CrossRef] [PubMed]
- Klose, M.; Feldt-Rasmussen, U. Does the type and severity of brain injury predict hypothalamo-pituitary dysfunction? Does post-traumatic hypopituitarism predict worse outcome? Pituitary 2008, 11, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Kokshoorn, N.E.; Wassenaar, M.J.; Biermasz, N.R.; Roelfsema, F.; Smit, J.W.; Romijn, J.A.; Pereira, A.M. Hypopituitarism following traumatic brain injury: Prevalence is affected by the use of different dynamic tests and different normal values. Eur. J. Endocrinol. 2010, 162, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Morley, J.E. The endocrinology of the opiates and opioid peptides. Metabolism 1981, 30, 195–209. [Google Scholar] [CrossRef]
- Bauer, J.; Blumenthal, S.; Reuber, M.; Stoffel-Wagner, B. Epilepsy syndrome, focus location, and treatment choice affect testicular function in men with epilepsy. Neurology 2004, 62, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Abs, R.; Mattsson, A.F.; Bengtsson, B.A.; Feldt-Rasmussen, U.; Goth, M.I.; Koltowska-Haggstrom, M.; Monson, J.P.; Verhelst, J.; Wilton, P. Isolated growth hormone (GH) deficiency in adult patients: Baseline clinical characteristics and responses to GH replacement in comparison with hypopituitary patients. A sub-analysis of the KIMS database. Growth Horm. IGF Res. 2005, 15, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Klose, M.; Lange, M.; Rasmussen, A.K.; Skakkebaek, N.E.; Hilsted, L.; Haug, E.; Andersen, M.; Feldt-Rasmussen, U. Factors influencing the adrenocorticotropin test: Role of contemporary cortisol assays, body composition, and oral contraceptive agents. J. Clin. Endocrinol. Metab. 2007, 92, 1326–1333. [Google Scholar] [CrossRef] [PubMed]
- Klose, M.; Brennum, J.; Poulsgaard, L.; Kosteljanetz, M.; Wagner, A.; Feldt-Rasmussen, U. Hypopituitarism is uncommon after aneurysmal subarachnoid haemorrhage. Clin. Endocrinol. 2010, 73, 95–101. [Google Scholar]
- Gardner, C.J.; Javadpour, M.; Stoneley, C.; Purthuran, M.; Biswas, S.; Daousi, C.; MacFarlane, I.A.; Cuthbertson, D.J. Low prevalence of hypopituitarism after Subarachnoid Haemorrhage using confirmatory testing and with BMI-specific growth hormone cut-off levels. Eur. J. Endocrinol. 2012, 168, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Hannon, M.J.; Behan, L.A.; O’Brien, M.M.; Tormey, W.; Javadpour, M.; Sherlock, M.; Thompson, C.J. Chronic hypopituitarism is uncommon in survivors of aneurysmal subarachnoid haemorrhage. Clin. Endocrinol. 2015, 82, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.F.; McArthur, D.L.; Levin, H.; Swimmer, S.; Dusick, J.R.; Cohan, P.; Wang, C.; Swerdloff, R. Neurobehavioral and quality of life changes associated with growth hormone insufficiency after complicated mild, moderate, or severe traumatic brain injury. J. Neurotrauma 2006, 23, 928–942. [Google Scholar] [CrossRef] [PubMed]
- Wachter, D.; Gundling, K.; Oertel, M.F.; Stracke, H.; Boker, D.K. Pituitary insufficiency after traumatic brain injury. J. Clin. Neurosci. 2009, 16, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Klose, M.; Watt, T.; Brennum, J.; Feldt-Rasmussen, U. Posttraumatic hypopituitarism is associated with an unfavorable body composition and lipid profile, and decreased quality of life 12 months after injury. J. Clin. Endocrinol. Metab. 2007, 92, 3861–3868. [Google Scholar] [CrossRef] [PubMed]
- Klose, M.; Stochholm, K.; Janukonyte, J.; Christensen, L.L.; Cohen, A.; Wagner, A.; Laurberg, P.; Christiansen, J.S.; Andersen, M.; Feldt-Rasmussen, U. Patient reported out-come in posttraumatic pituitary deficiency: Results from The Danish National Study on Posttraumatic Hypopituitarism. Eur. J. Endocrinol. 2015, 172, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Gibby, C.C.; Aday, L.A.; Anderson, K.O.; Mendoza, T.R.; Cleeland, C.S. Pain, depression, and fatigue in community-dwelling adults with and without a history of cancer. J. Pain Symptom Manage 2006, 32, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Naess, H.; Lunde, L.; Brogger, J. The effects of fatigue, pain, and depression on quality of life in ischemic stroke patients: The Bergen Stroke Study. Vasc. Health Risk Manag. 2012, 8, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Bondanelli, M.; Ambrosio, M.R.; Cavazzini, L.; Bertocchi, A.; Zatelli, M.C.; Carli, A.; Valle, D.; Basaglia, N.; Uberti, E.C. Anterior pituitary function may predict functional and cognitive outcome in patients with traumatic brain injury undergoing rehabilitation. J. Neurotrauma 2007, 24, 1687–1697. [Google Scholar] [CrossRef] [PubMed]
- Pavlovic, D.; Pekic, S.; Stojanovic, M.; Zivkovic, V.; Djurovic, B.; Jovanovic, V.; Miljic, N.; Medic-Stojanoska, M.; Doknic, M.; Miljic, D.; Djurovic, M.; Casanueva, F.; Popovic, V. Chronic cognitive sequelae after traumatic brain injury are not related to growth hormone deficiency in adults. Eur. J. Neurol. 2010, 17, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Mossberg, K.A.; Masel, B.E.; Gilkison, C.R.; Urban, R.J. Aerobic capacity and growth hormone deficiency after traumatic brain injury. J. Clin. Endocrinol. Metab. 2008, 93, 2581–2587. [Google Scholar] [CrossRef] [PubMed]
- Bellone, S.; Einaudi, S.; Caputo, M.; Prodam, F.; Busti, A.; Belcastro, S.; Parlamento, S.; Zavattaro, M.; Verna, F.; Bondone, C.; et al. Measurement of height velocity is an useful marker for monitoring pituitary function in patients who had traumatic brain injury. Pituitary 2013, 16, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Personnier, C.; Crosnier, H.; Meyer, P.; Chevignard, M.; Flechtner, I.; Boddaert, N.; Breton, S.; Mignot, C.; Dassa, Y.; Souberbielle, J.C.; et al. Prevalence of pituitary dysfunction after severe traumatic brain injury in children and adolescents: A large prospective study. J. Clin. Endocrinol. Metab. 2014, 99, 2052–2060. [Google Scholar] [CrossRef] [PubMed]
- Heather, N.L.; Jefferies, C.; Hofman, P.L.; Derraik, J.G.; Brennan, C.; Kelly, P.; Hamill, J.K.; Jones, R.G.; Rowe, D.L.; Cutfield, W.S. Permanent hypopituitarism is rare after structural traumatic brain injury in early childhood. J. Clin. Endocrinol. Metab. 2012, 97, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Kopczak, A.; Kilimann, I.; Von, R.F.; Krewer, C.; Schneider, H.J.; Stalla, G.K.; Schneider, M. Screening for hypopituitarism in 509 patients with traumatic brain injury or subarachnoid hemorrhage. J. Neurotrauma 2014, 31, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Kreitschmann-Andermahr, I.; Poll, E.M.; Reineke, A.; Gilsbach, J.M.; Brabant, G.; Buchfelder, M.; Fassbender, W.; Faust, M.; Kann, P.H.; Wallaschofski, H. Growth hormone deficient patients after traumatic brain injury—Baseline characteristics and benefits after growth hormone replacement—An analysis of the German KIMS database. Growth Horm. IGF Res. 2008, 18, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Casanueva, F.F.; Leal, A.; Koltowska-Haggstrom, M.; Jonsson, P.; Goth, M.I. Traumatic brain injury as a relevant cause of growth hormone deficiency in adults: A KIMS-based study. Arch. Phys. Med. Rehabil. 2005, 86, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Gardner, C.J.; Mattsson, A.F.; Daousi, C.; Korbonits, M.; Koltowska-Haggstrom, M.; Cuthbertson, D.J. Growth Hormone Deficiency after Traumatic Brain Injury: Improvement in quality of life with GH therapy—Analysis of the KIMS database. Eur. J. Endocrinol. 2015, 172, 371–381. [Google Scholar]
- High, W.M., Jr.; Briones-Galang, M.; Clark, J.A.; Gilkison, C.; Mossberg, K.A.; Zgaljardic, D.J.; Masel, B.E.; Urban, R.J. Effect of growth hormone replacement therapy on cognition after traumatic brain injury. J. Neurotrauma 2010, 27, 1565–1575. [Google Scholar]
- Maric, N.P.; Doknic, M.; Pavlovic, D.; Pekic, S.; Stojanovic, M.; Jasovic-Gasic, M.; Popovic, V. Psychiatric and neuropsychological changes in growth hormone-deficient patients after traumatic brain injury in response to growth hormone therapy. J. Endocrinol. Investig. 2010, 33, 770–775. [Google Scholar]
- Reimunde, P.; Quintana, A.; Castanon, B.; Casteleiro, N.; Vilarnovo, Z.; Otero, A.; Devesa, A.; Otero-Cepeda, X.L.; Devesa, J. Effects of growth hormone (GH) replacement and cognitive rehabilitation in patients with cognitive disorders after traumatic brain injury. Brain Injury 2011, 25, 65–73. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klose, M.; Feldt-Rasmussen, U. Hypopituitarism in Traumatic Brain Injury—A Critical Note. J. Clin. Med. 2015, 4, 1480-1497. https://doi.org/10.3390/jcm4071480
Klose M, Feldt-Rasmussen U. Hypopituitarism in Traumatic Brain Injury—A Critical Note. Journal of Clinical Medicine. 2015; 4(7):1480-1497. https://doi.org/10.3390/jcm4071480
Chicago/Turabian StyleKlose, Marianne, and Ulla Feldt-Rasmussen. 2015. "Hypopituitarism in Traumatic Brain Injury—A Critical Note" Journal of Clinical Medicine 4, no. 7: 1480-1497. https://doi.org/10.3390/jcm4071480
APA StyleKlose, M., & Feldt-Rasmussen, U. (2015). Hypopituitarism in Traumatic Brain Injury—A Critical Note. Journal of Clinical Medicine, 4(7), 1480-1497. https://doi.org/10.3390/jcm4071480