
Diabetes Insipidus after Traumatic Brain Injury

{kind=link}
Abstract
:1. Introduction
- ○
- Neurosurgical interventions
- ○
- Traumatic brain injury (TBI)
- ○
- (Para)sellar tumors (e.g., craniopharyngioma, germinoma, meningioma, germ cell tumor)
- ○
- Metastases to pituitary gland (especially from breast/lung malignancies)
- ○
- Infections (e.g., meningitis, abscess, encephalitis)
- ○
- Infiltrative diseases (e.g., sarcoidosis, histiocytosis, lymphocytic hypophysitis)
- ○
- Vascular abnormalities (aneurysm)
- ○
- Autoimmune
- ○
- Genetic (AD, AR, X-linked recessive, DIDMOAD)
- ○
- Congenital (e.g., septo-optic dysplasia, holoprosencephaly, midline defects)
2. Pathophysiology of Posttraumatic DI (PTDI)
3. Epidemiology of PTDI
4. Natural History
5. Clinical Manifestations
6. Diagnosis
7. Imaging
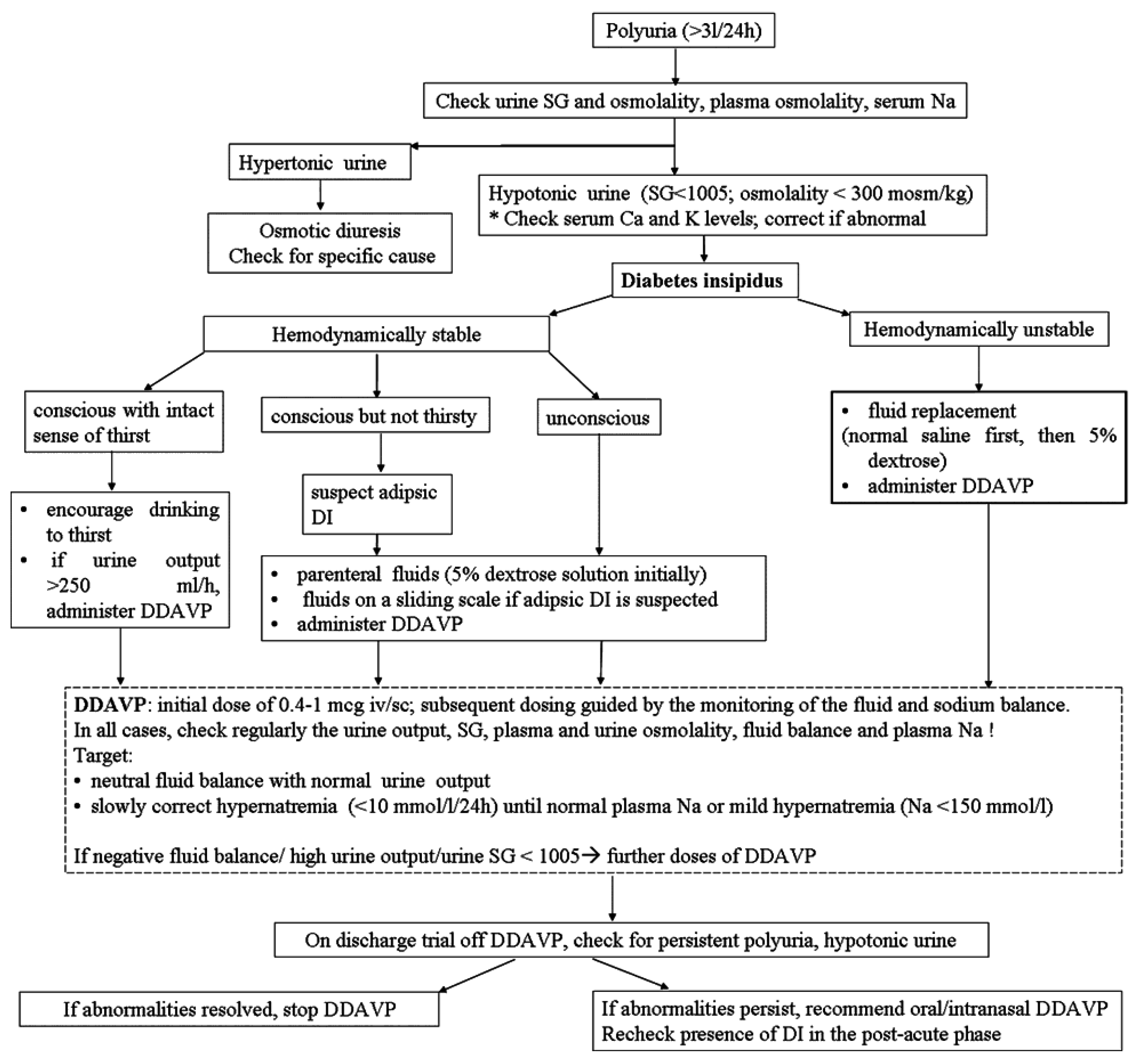
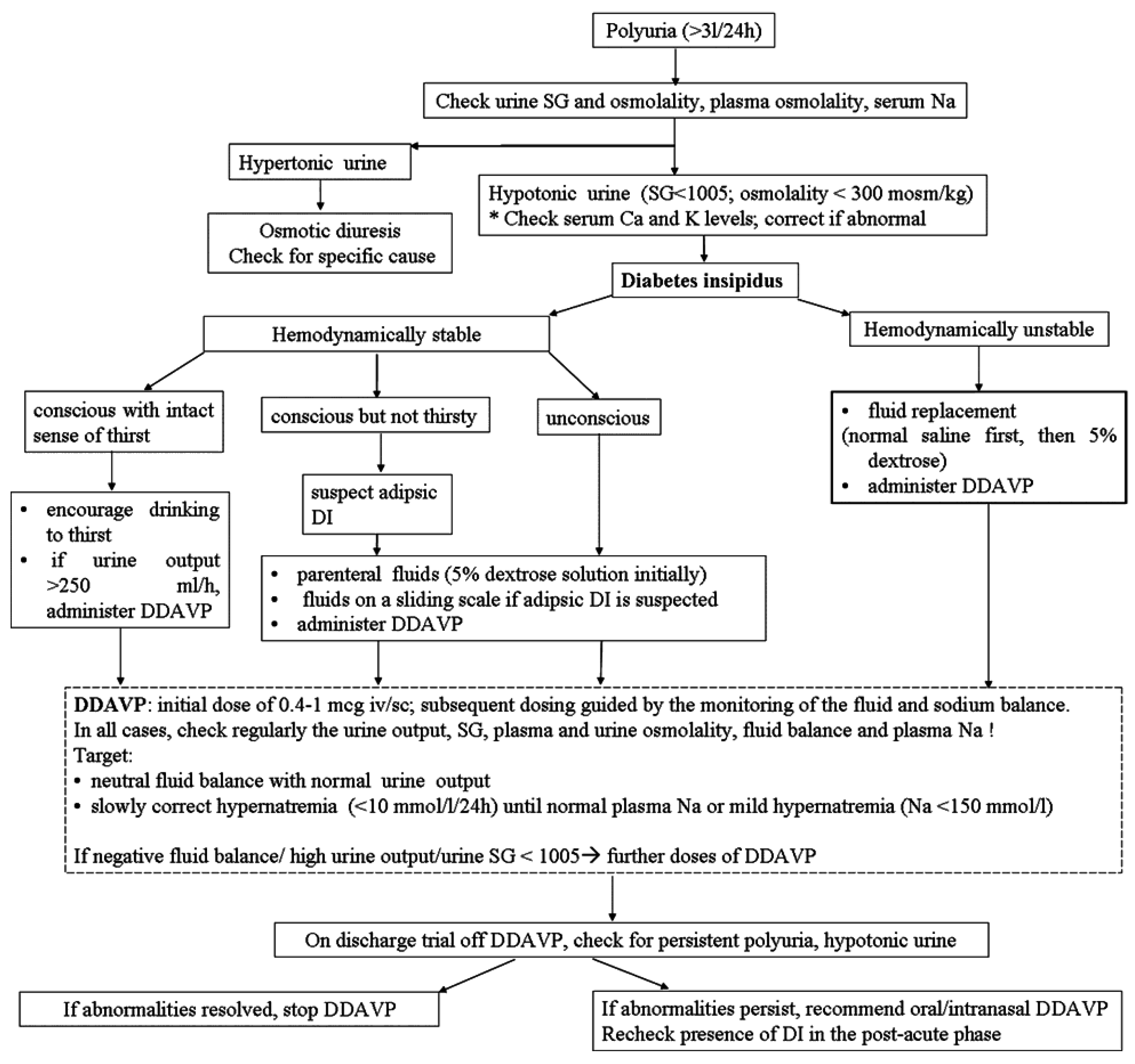
8. Management
9. Prognosis
10. Conclusions
Author Contributions
Conflicts of Interest
References
- Treschan, T.A.; Peters, J. The vasopressin system: physiology and clinical strategies. Anesthesiology 2006, 105, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Saifan, C.; Nasr, R.; Mehta, S.; Sharma, A.P.; Perrera, I.; Faddoul, G.; Nalluri, N.; Kesavan, M.; Azzi, Y.; El-Sayegh, S. Diabetes insipidus: A challenging diagnosis with new drug therapies. ISRN. Nephrol. 2013, 2013, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Oiso, Y.; Robertson, G.L.; Norgaard, J.P.; Juul, K.V. Clinical review: Treatment of neurohypophyseal diabetes insipidus. J. Clin. Endocrinol. Metab. 2013, 98, 3958–3967. [Google Scholar] [CrossRef] [PubMed]
- Rutland-Brown, W.; Langlois, J.A.; Thomas, K.E.; Xi, Y.L. Incidence of traumatic brain injury in the United States, 2003. J. Head Trauma Rehabil. 2006, 21, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Zaloshnja, E.; Miller, T.; Langlois, J.A.; Selassie, A.W. Prevalence of long-term disability from traumatic brain injury in the civilian population of the United States, 2005. J. Head Trauma Rehabil. 2008, 23, 394–400. [Google Scholar] [CrossRef] [PubMed]
- King, B.S.; Gupta, R.; Narayan, R.K. The early assessment and intensive care unit management of patients with severe traumatic brain and spinal cord injuries. Surg. Clin. N. Am. 2000, 80, 855–870. [Google Scholar] [CrossRef]
- Coronado, V.G.; Xu, L.; Basavaraju, S.V.; McGuire, L.C.; Wald, M.M.; Faul, M.D.; Guzman, B.R.; Hemphill, J.D. Surveillance for traumatic brain injury-related deaths: United States, 1997–2007. MMWR Surveill Summ. 2011, 60, 1–32. [Google Scholar] [PubMed]
- Boughey, J.C.; Yost, M.J.; Bynoe, R.P. Diabetes insipidus in the head-injured patient. Am. Surg. 2004, 70, 500–503. [Google Scholar] [PubMed]
- Cyran, E. Hypophysenschaedigung durch schaedelbasisfraktur. Deutsch Med. Wochenschr. 1918, 44, 1261. [Google Scholar]
- Holborn, A.H.S. Mechanics of head injury. Lancet 1943, 2, 438–441. [Google Scholar] [CrossRef]
- Porter, R.J.; Miller, R.A. Diabetes insipidus following closed head injury. J. Neurol. Neurosurg. Psych. 1948, 11, 258–262. [Google Scholar] [CrossRef]
- Maghnie, M.; Cosi, G.; Genovese, E.; Manca-Bitti, M.L.; Cohen, A.; Zecca, S.; Tinelli, C.; Gallucci, M.; Bernasconi, S.; Boscherini, B.; et al. Central diabetes insipidus in children and young adults. N. Engl. J. Med. 2000, 343, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.Q.; Wade, C.E. Neuroendocrine abnormalities in patients with traumatic brain injury. Front. Neuroendocrinol. 1991, 12, 209–230. [Google Scholar] [PubMed]
- Crowley, R.K.; Sherlock, M.; Agha, A.; Smith, D.; Thompson, C.J. Clinical insights into adipsic diabetes insipidus: A large case series. Clin. Endocrinol. (Oxf.) 2007, 66, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Labib, M.; McPhate, G.; Marks, V. Post-traumatic diabetes insipidus combined with primary polydipsia. Postgrad. Med. J. 1987, 63, 33–35. [Google Scholar] [CrossRef] [PubMed]
- Crowley, R.K.; Hamnvik, O.P.; O’Sullivan, E.P.; Behan, L.A.; Smith, D.; Agha, A.; Thompson, C.J. Morbidity and mortality in patients with craniopharyngioma after surgery. Clin Endocrinol (Oxf.) 2010, 73, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Harper, C.G.; Doyle, D.; Adams, J.H.; Graham, D.I. Analysis of abnormalities in pituitary gland in non-missile head injury: Study of 100 consecutive cases. J. Clin. Pathol. 1986, 39, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Treip, C.S. Hypothalamic and pituitary injury. J. Clin. Pathol. Suppl (R. Coll. Pathol.) 1970, 4, 178–186. [Google Scholar] [CrossRef]
- Hensen, J.; Henig, A.; Fahlbusch, R.; Meyer, M.; Boehnert, M.; Buchfelder, M. Prevalence, predictors and patterns of postoperative polyuria and hyponatraemia in the immediate course after transsphenoidal surgery for pituitary adenomas. Clin. Endocrinol. (Oxf.) 1999, 50, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Agha, A.; Thompson, C.J. Anterior pituitary dysfunction following traumatic brain injury (TBI). Clin. Endocrinol. (Oxf.) 2006, 64, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Kleindienst, A.; Brabant, G.; Bock, C.; Maser-Gluth, C.; Buchfelder, M. Neuroendocrine function following traumatic brain injury and subsequent intensive care treatment: A prospective longitudinal evaluation. J. Neurotrauma 2009, 26, 1435–1446. [Google Scholar] [CrossRef] [PubMed]
- Powner, D.J.; Boccalandro, C.; Alp, M.S.; Vollmer, D.G. Endocrine failure after traumatic brain injury in adults. Neurocrit. Care 2006, 5, 61–70. [Google Scholar] [CrossRef]
- Haddad, S.H.; Arabi, Y.M. Critical care management of severe traumatic brain injury in adults. Scand. J. Trauma Resusc. Emerg. Med. 2012, 20, 12. [Google Scholar] [CrossRef] [PubMed]
- Hannon, M.J.; Crowley, R.K.; Behan, L.A.; O’Sullivan, E.P.; O’Brien, M.M.; Sherlock, M.; Rawluk, D.; O’Dwyer, R.; Tormey, W.; Thompson, C.J. Acute glucocorticoid deficiency and diabetes insipidus are common after acute traumatic brain injury and predict mortality. J. Clin. Endocrinol. Metab. 2013, 98, 3229–3237. [Google Scholar] [CrossRef] [PubMed]
- Agha, A.; Thornton, E.; O’Kelly, P.; Tormey, W.; Phillips, J.; Thompson, C.J. Posterior pituitary dysfunction after traumatic brain injury. J. Clin. Endocrinol. Metab. 2004, 89, 5987–5992. [Google Scholar] [CrossRef] [PubMed]
- Hadjizacharia, P.; Beale, E.O.; Inaba, K.; Chan, L.S.; Demetriades, D. Acute diabetes insipidus in severe head injury: A prospective study. J. Am. Coll. Surg. 2008, 207, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Karali, V.; Massa, E.; Vassiliadou, G.; Chouris, I.; Rodin, I.; Bitzani, M. Evaluation of development of diabetes insipidus in the early phase following traumatic brain injury in critically ill patients. Crit. Care 2008, 12, S51–S52. [Google Scholar] [CrossRef]
- Benvenga, S.; Campenni, A.; Ruggeri, R.M.; Trimarchi, F. Clinical review 113: Hypopituitarism secondary to head trauma. J. Clin. Endocrinol. Metab. 2000, 85, 1353–1361. [Google Scholar] [CrossRef] [PubMed]
- Su, D.H.; Chang, Y.C.; Chang, C.C. Post-traumatic anterior and posterior pituitary dysfunction. J. Formos. Med. Assoc. 2005, 104, 463–467. [Google Scholar] [PubMed]
- Agha, A.; Sherlock, M.; Phillips, J.; Tormey, W.; Thompson, C.J. The natural history of post-traumatic neurohypophysial dysfunction. Eur. J. Endocrinol. 2005, 152, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Chou, Y.C.; Wang, T.Y.; Yang, P.Y.; Meng, N.H.; Chou, L.W. Permanent central diabetes insipidus after mild traumatic brain injury. Brain Inj. 2009, 23, 1095–1098. [Google Scholar] [CrossRef] [PubMed]
- Seckl, J.R.; Dunger, D.B.; Lightman, S.L. Neurohypophyseal peptide function during early postoperative diabetes insipidus. Brain 1987, 110, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Bohnen, N.; Twijnstra, A.; Jolles, J. Water metabolism and postconcussional symptoms 5 weeks after mild head injury. Eur. Neurol. 1993, 33, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Aimaretti, G.; Ambrosio, M.R.; Di, S.C.; Gasperi, M.; Cannavo, S.; Scaroni, C.; Fusco, A.; Del, M.P.; De, M.E.; Faustini-Fustini, M.; et al. Residual pituitary function after brain injury-induced hypopituitarism: a prospective 12-month study. J. Clin. Endocrinol. Metab. 2005, 90, 6085–6092. [Google Scholar] [CrossRef] [PubMed]
- Aimaretti, G.; Ambrosio, M.R.; Di, S.C.; Gasperi, M.; Cannavo, S.; Scaroni, C.; De, M.L.; Baldelli, R.; Bona, G.; Giordano, G.; et al. Hypopituitarism induced by traumatic brain injury in the transition phase. J. Endocrinol. Investig. 2005, 28, 984–989. [Google Scholar] [CrossRef]
- Lieberman, S.A.; Oberoi, A.L.; Gilkison, C.R.; Masel, B.E.; Urban, R.J. Prevalence of neuroendocrine dysfunction in patients recovering from traumatic brain injury. J. Clin. Endocrinol. Metab. 2001, 86, 2752–2756. [Google Scholar] [CrossRef] [PubMed]
- Klose, M.; Juul, A.; Poulsgaard, L.; Kosteljanetz, M.; Brennum, J.; Feldt-Rasmussen, U. Prevalence and predictive factors of post-traumatic hypopituitarism. Clin. Endocrinol. (Oxf.) 2007, 67, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Teasdale, G.; Jennett, B. Assessment of coma and impaired consciousness. A practical scale. Lancet 1974, 2, 81–84. [Google Scholar] [CrossRef]
- Wood, P.H. Appreciating the consequences of disease: The international classification of impairments, disabilities, and handicaps. WHO Chron. 1980, 34, 376–380. [Google Scholar] [PubMed]
- Helmy, A.; Vizcaychipi, M.; Gupta, A.K. Traumatic brain injury: Intensive care management. Br. J. Anaesth. 2007, 99, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Adrogue, H.J.; Madias, N.E. Hypernatremia. N. Engl. J. Med. 2000, 342, 1493–1499. [Google Scholar] [CrossRef] [PubMed]
- Stern, R.H. Disorders of plasma sodium. N. Engl. J. Med. 2015, 372, 55–65. [Google Scholar]
- Overgaard-Steensen, C.; Ring, T. Clinical review: Practical approach to hyponatraemia and hypernatraemia in critically ill patients. Crit. Care 2013, 17, 206. [Google Scholar] [CrossRef] [PubMed]
- Alaca, R.; Yilmaz, B.; Gunduz, S. Anterior hypopituitarism with unusual delayed onset of diabetes insipidus after penetrating head injury. Am. J. Phys. Med. Rehabil. 2002, 81, 788–791. [Google Scholar] [CrossRef] [PubMed]
- Hadani, M.; Findler, G.; Shaked, I.; Sahar, A. Unusual delayed onset of diabetes insipidus following closed head trauma. Case report. J. Neurosurg. 1985, 63, 456–458. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, J.; Shiroozu, A.; Zaitsu, A.; Imazono, Y.; Kohrogi, T.; Yokohata, K.; Kishikawa, H. Diabetes insipidus after traumata of two extremes in severity. Yonsei Med. J. 1990, 31, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Robertson, G.L.; Aycinena, P.; Zerbe, R.L. Neurogenic disorders of osmoregulation. Am. J. Med. 1982, 72, 339–353. [Google Scholar] [CrossRef]
- Yang, Y.H.; Lin, J.J.; Hsia, S.H.; Wu, C.T.; Wang, H.S.; Hung, P.C.; Chou, M.L.; Hsieh, M.Y.; Lin, K.L. Central diabetes insipidus in children with acute brain insult. Pediatr. Neurol. 2011, 45, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Hannon, M.J.; Finucane, F.M.; Sherlock, M.; Agha, A.; Thompson, C.J. Clinical review: Disorders of water homeostasis in neurosurgical patients. J. Clin. Endocrinol. Metab. 2012, 97, 1423–1433. [Google Scholar] [CrossRef] [PubMed]
- Zerbe, R.L.; Robertson, G.L. A comparison of plasma vasopressin measurements with a standard indirect test in the differential diagnosis of polyuria. N. Engl. J. Med. 1981, 305, 1539–1546. [Google Scholar] [CrossRef] [PubMed]
- Jochberger, S.; Morgenthaler, N.G.; Mayr, V.D.; Luckner, G.; Wenzel, V.; Ulmer, H.; Schwarz, S.; Hasibeder, W.R.; Friesenecker, B.E.; Dünser, M.W. Copeptin and arginine vasopressin concentrations in critically ill patients. J. Clin Endocrinol Metab. 2006, 9, 4381–4386. [Google Scholar] [CrossRef] [PubMed]
- Katan, M.; Morgenthaler, N.G.; Dixit, K.C.; Rutishauser, J.; Brabant, G.E.; Muller, B.; Christ-Crain, M. Anterior and posterior pituitary function testing with simultaneous insulin tolerance test and a novel copeptin assay. J. Clin. Endocrinol. Metab. 2007, 92, 2640–2643. [Google Scholar] [CrossRef] [PubMed]
- Kleindienst, A.; Brabant, G.; Morgenthaler, N.G.; Dixit, K.C.; Parsch, H.; Buchfelder, M. Following brain trauma, copeptin, a stable peptide derived from the AVP precusor, does not reflect osmoregulation but correlates with injury severity. Acta Neurochir Suppl. 2010, 106, 221–2244. [Google Scholar] [PubMed]
- Lin, J.J.; Lin, K.L.; Hsia, S.H.; Wu, C.T.; Wang, H.S. Combined central diabetes insipidus and cerebral salt wasting syndrome in children. Pediatr. Neurol. 2009, 40, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Saatman, K.E.; Duhaime, A.C.; Bullock, R.; Maas, A.I.; Valadka, A.; Manley, G.T. Classification of traumatic brain injury for targeted therapies. J. Neurotrauma 2008, 25, 719–738. [Google Scholar] [CrossRef] [PubMed]
- Roguski, M.; Morel, B.; Sweeney, M.; Talan, J.; Rideout, L.; Riesenburger, R.I.; Madan, N.; Hwang, S. Magnetic resonance imaging as an alternative to computed tomography in select patients with traumatic brain injury: A retrospective comparison. J. Neurosurg. Pediatr. 2015, 1, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Maiya, B.; Newcombe, V.; Nortje, J.; Bradley, P.; Bernard, F.; Chatfield, D.; Outtrim, J.; Hutchinson, P.; Matta, B.; Antoun, N.; et al. Magnetic resonance imaging changes in the pituitary gland following acute traumatic brain injury. Intensive Care Med. 2008, 34, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Kawai, K.; Aoki, M.; Nakayama, H.; Kobayashi, K.; Sano, K.; Tamura, A. Posterior pituitary hematoma in a case of posttraumatic diabetes insipidus. Case report. J. Neurosurg. 1995, 83, 368–371. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Lee, H.K.; Choi, C.G.; Suh, D.C.; Kim, C.J.; Hong, S.K.; Na, D.G. MR imaging of central diabetes insipidus: A pictorial essay. Korean J. Radiol. 2001, 2, 222–230. [Google Scholar] [CrossRef] [PubMed]
- John, C.A.; Day, M.W. Central neurogenic diabetes insipidus, syndrome of inappropriate secretion of antidiuretic hormone, and cerebral salt-wasting syndrome in traumatic brain injury. Crit. Care Nurse 2012, 32, e1–e7. [Google Scholar] [CrossRef] [PubMed]
- Clifton, G.L.; Miller, E.R.; Choi, S.C.; Levin, H.S. Fluid thresholds and outcome from severe brain injury. Crit. Care Med. 2002, 30, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Wright, W.L. Sodium and fluid management in acute brain injury. Curr. Neurol. Neurosci. Rep. 2012, 12, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Tisdall, M.; Crocker, M.; Watkiss, J.; Smith, M. Disturbances of sodium in critically ill adult neurologic patients: A clinical review. Neurosurg. Anesthesiol. 2006, 18, 57–63. [Google Scholar] [CrossRef]
- Lindner, G.; Funk, G.C. Hypernatremia in critically ill patients. J. Crit. Care. 2013, 28, e11–e20. [Google Scholar] [CrossRef] [PubMed]
- Alharfi, I.M.; Stewart, T.C.; Kelly, S.H.; Morrison, G.C.; Fraser, D.D. Hypernatremia is associated with increased risk of mortality in pediatric severe traumatic brain injury. J. Neurotrauma 2013, 30, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Vokes, T.J.; Gaskill, M.B.; Robertson, G.L. Antibodies to vasopressin in patients with diabetes insipidus. Implications for diagnosis and therapy. Ann. Intern. Med. 1988, 108, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Maggiore, U.; Picetti, E.; Antonucci, E.; Parenti, E.; Regolisti, G.; Mergoni, M.; Vezzani, A.; Cabassi, A.; Fiaccadori, E. The relation between the incidence of hypernatremia and mortality in patients with severe traumatic brain injury. Crit. Care 2009, 13, R110. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.S.; Wat, M.S.; Choi, K.L.; Ip, T.P.; Pang, R.W.; Kumana, C.R. Pharmacokinetics, pharmacodynamics, long-term efficacy and safety of oral 1-deamino-8-D-arginine vasopressin in adult patients with central diabetes insipidus. Br. J. Clin. Pharmacol. 1996, 42, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, K.; Simth, M. Disorders of sodium balance after brain injury. Critical Care Pain 2008, 8, 129–133. [Google Scholar] [CrossRef]
- Hwang, J.J.; Hwang, D.Y. Treatment of endocrine disorders in the neuroscience intensive care unit. Curr. Treat. Options. Neurol. 2014, 16, 271. [Google Scholar] [CrossRef] [PubMed]
- Rembratt, A.; Graugaard-Jensen, C.; Senderovitz, T.; Norgaard, J.P.; Djurhuus, J.C. Pharmacokinetics and pharmacodynamics of desmopressin administered orally versus intravenously at daytime versus night-time in healthy men aged 55–70 years. Eur. J. Clin. Pharmacol. 2004, 60, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Juul, K.V.; Bichet, D.G.; Norgaard, J.P. Desmopressin duration of antidiuretic action in patients with central diabetes insipidus. Endocrine 2011, 40, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Tsagarakis, S.; Tzanela, M.; Dimopoulou, I. Diabetes insipidus, secondary hypoadrenalism and hypothyroidism after traumatic brain injury: Clinical implications. Pituitary 2005, 8, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Green, R.P.; Landt, M. Home sodium monitoring in patients with diabetes insipidus. J. Pediatr. 2002, 141, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Osterberg, O.; Savic, R.M.; Karlsson, M.O.; Simonsson, U.S.; Norgaard, J.P.; Walle, J.V.; Agerso, H. Pharmacokinetics of desmopressin administrated as an oral lyophilisate dosage form in children with primary nocturnal enuresis and healthy adults. J. Clin. Pharmacol. 2006, 46, 1204–1211. [Google Scholar] [CrossRef] [PubMed]
- Arima, H.; Oiso, Y.; Juul, K.V.; Norgaard, J.P. Efficacy and safety of desmopressin orally disintegrating tablet in patients with central diabetes insipidus: Results of a multicenter open-label dose-titration study. Endocr. J. 2013, 60, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- Leal-Cerro, A.; Flores, J.M.; Rincon, M.; Murillo, F.; Pujol, M.; Garcia-Pesquera, F.; Dieguez, C.; Casanueva, F.F. Prevalence of hypopituitarism and growth hormone deficiency in adults long-term after severe traumatic brain injury. Clin. Endocrinol. (Oxf.) 2005, 62, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Ranasinghe, A.M.; Bonser, R.S. Endocrine changes in brain death and transplantation. Best. Pract. Res. Clin. Endocrinol. Metab. 2011, 25, 799–812. [Google Scholar] [CrossRef] [PubMed]
- Sigounas, D.G.; Sharpless, J.L.; Cheng, D.M.; Johnson, T.G.; Senior, B.A.; Ewend, M.G. Predictors and incidence of central diabetes insipidus after endoscopic pituitary surgery. Neurosurgery 2008, 62, 71–78. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capatina, C.; Paluzzi, A.; Mitchell, R.; Karavitaki, N. Diabetes Insipidus after Traumatic Brain Injury. J. Clin. Med. 2015, 4, 1448-1462. https://doi.org/10.3390/jcm4071448
Capatina C, Paluzzi A, Mitchell R, Karavitaki N. Diabetes Insipidus after Traumatic Brain Injury. Journal of Clinical Medicine. 2015; 4(7):1448-1462. https://doi.org/10.3390/jcm4071448
Chicago/Turabian StyleCapatina, Cristina, Alessandro Paluzzi, Rosalid Mitchell, and Niki Karavitaki. 2015. "Diabetes Insipidus after Traumatic Brain Injury" Journal of Clinical Medicine 4, no. 7: 1448-1462. https://doi.org/10.3390/jcm4071448
APA StyleCapatina, C., Paluzzi, A., Mitchell, R., & Karavitaki, N. (2015). Diabetes Insipidus after Traumatic Brain Injury. Journal of Clinical Medicine, 4(7), 1448-1462. https://doi.org/10.3390/jcm4071448