
Endothelin Blockade in Diabetic Kidney Disease


Abstract
:1. Introduction
2. Endothelin Receptors in the Kidney
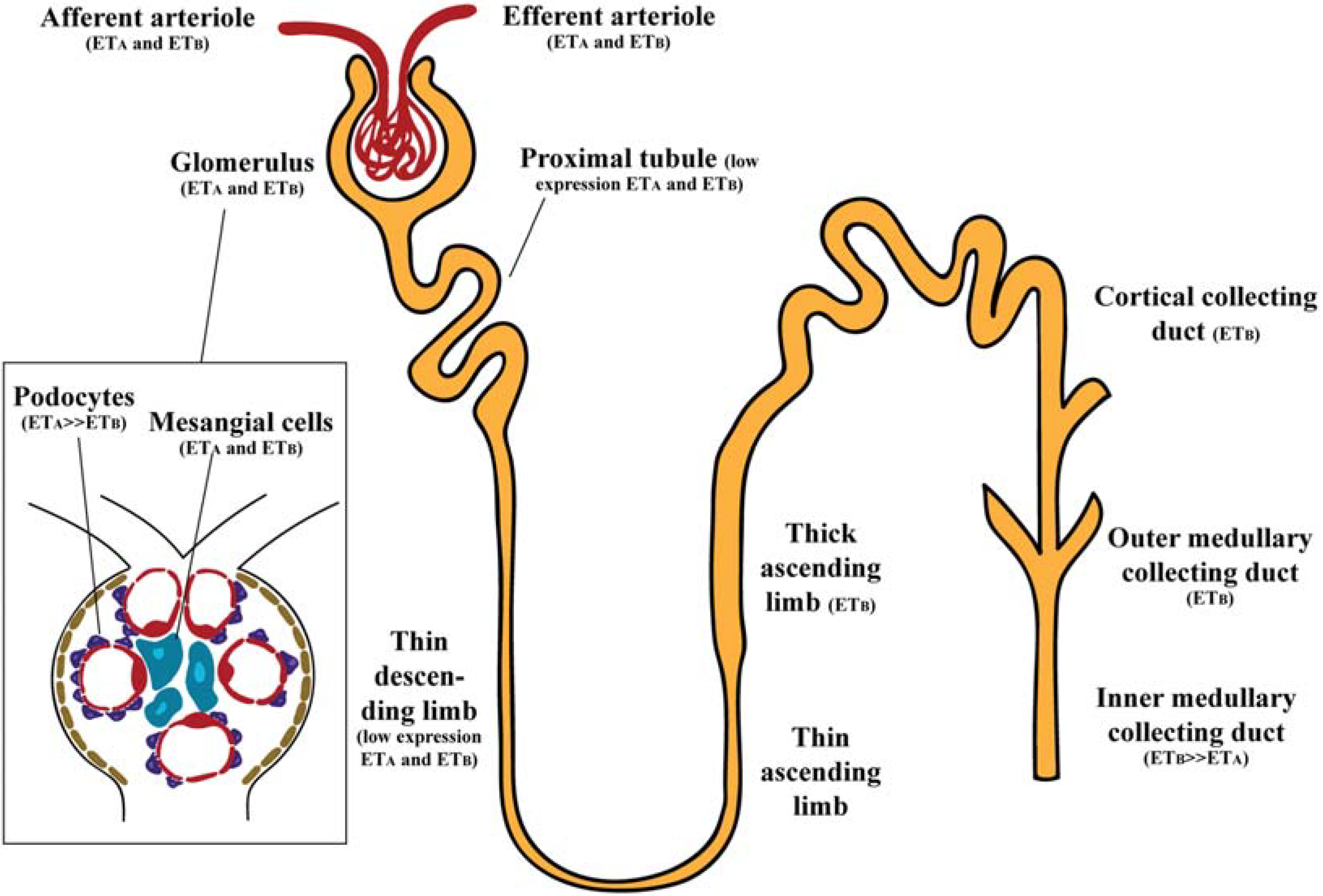
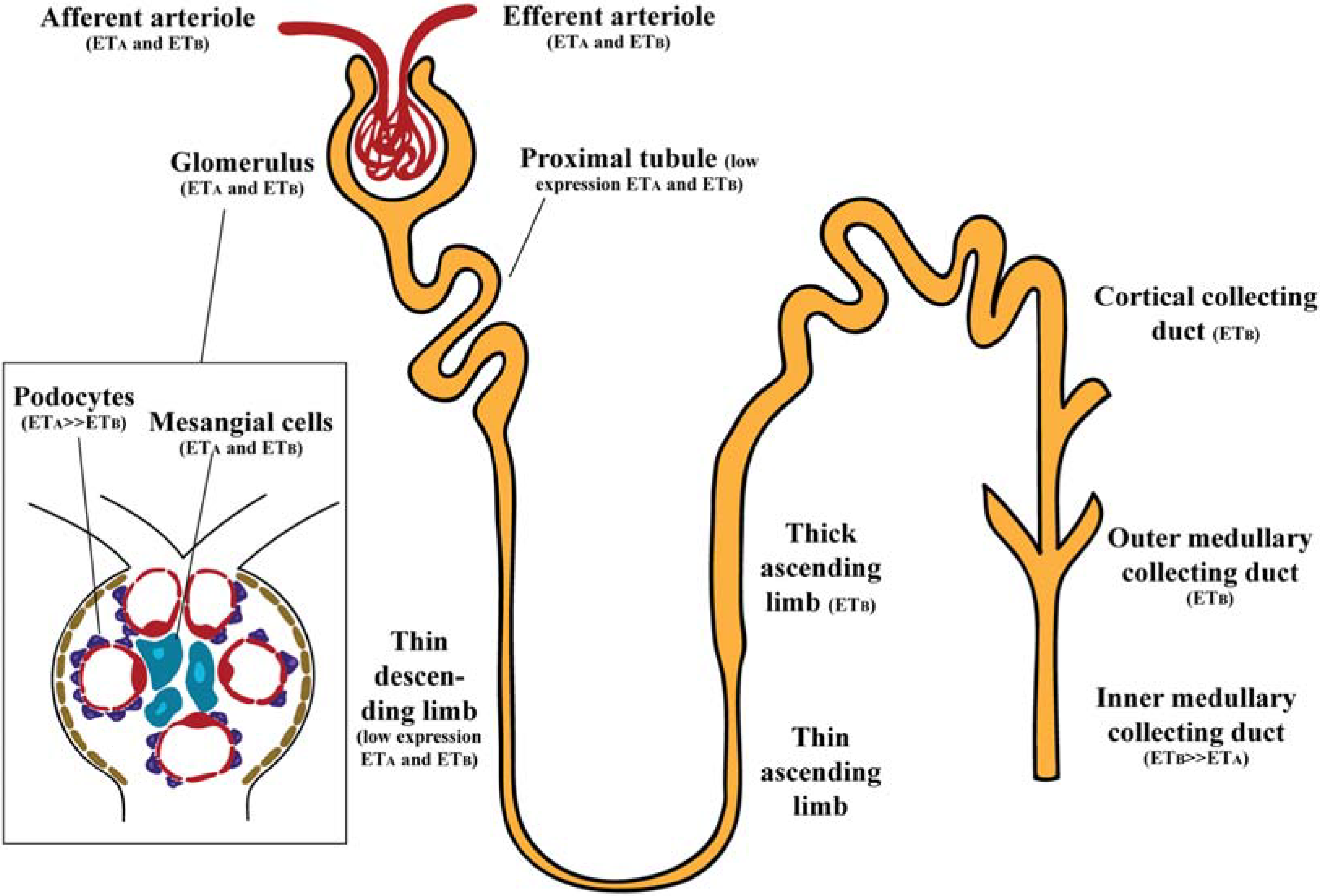
2.1. Glomerulus
2.2. Renal Vasculature
2.3. Renal Tubules
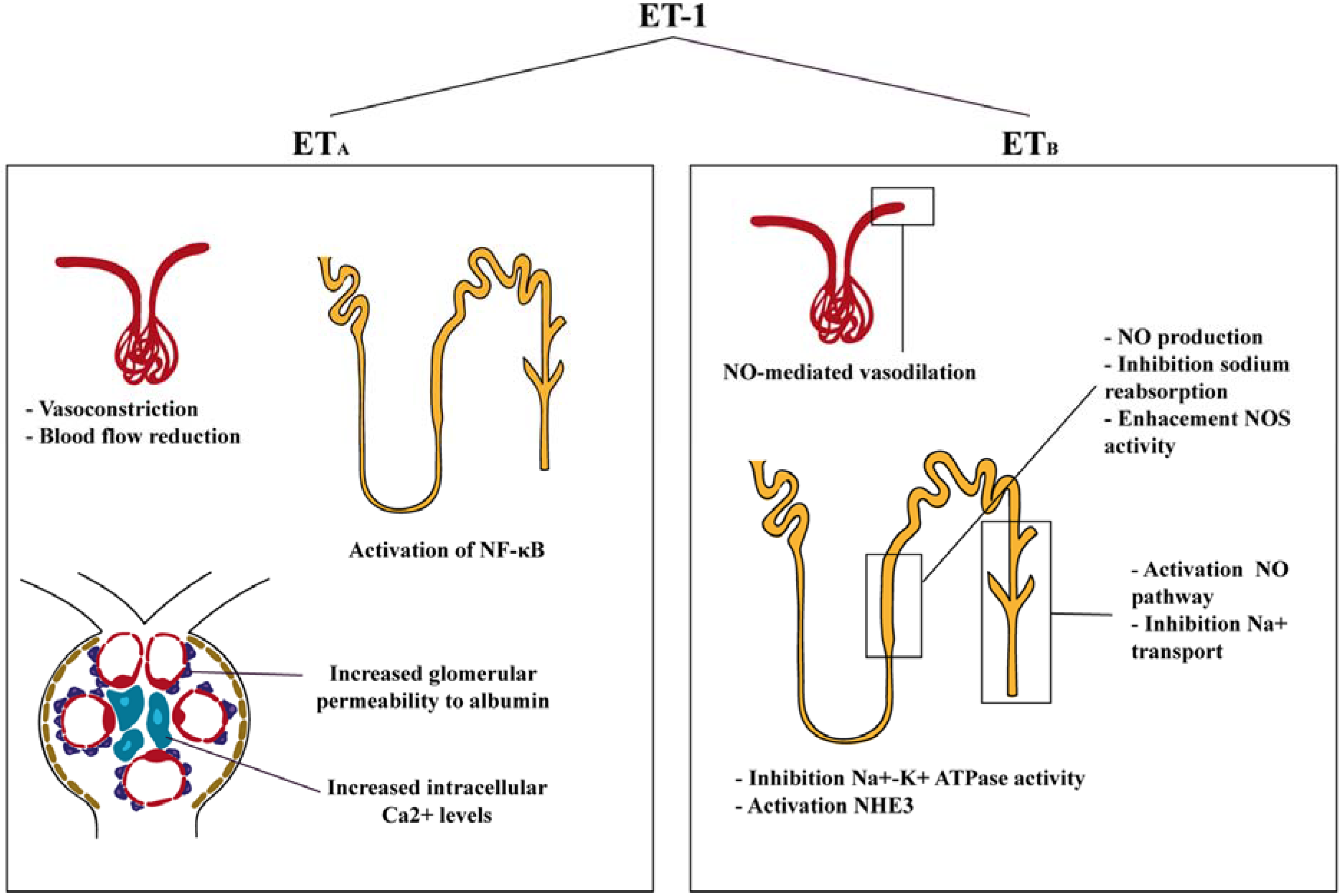
3. Functions of Endothelin in the Kidney
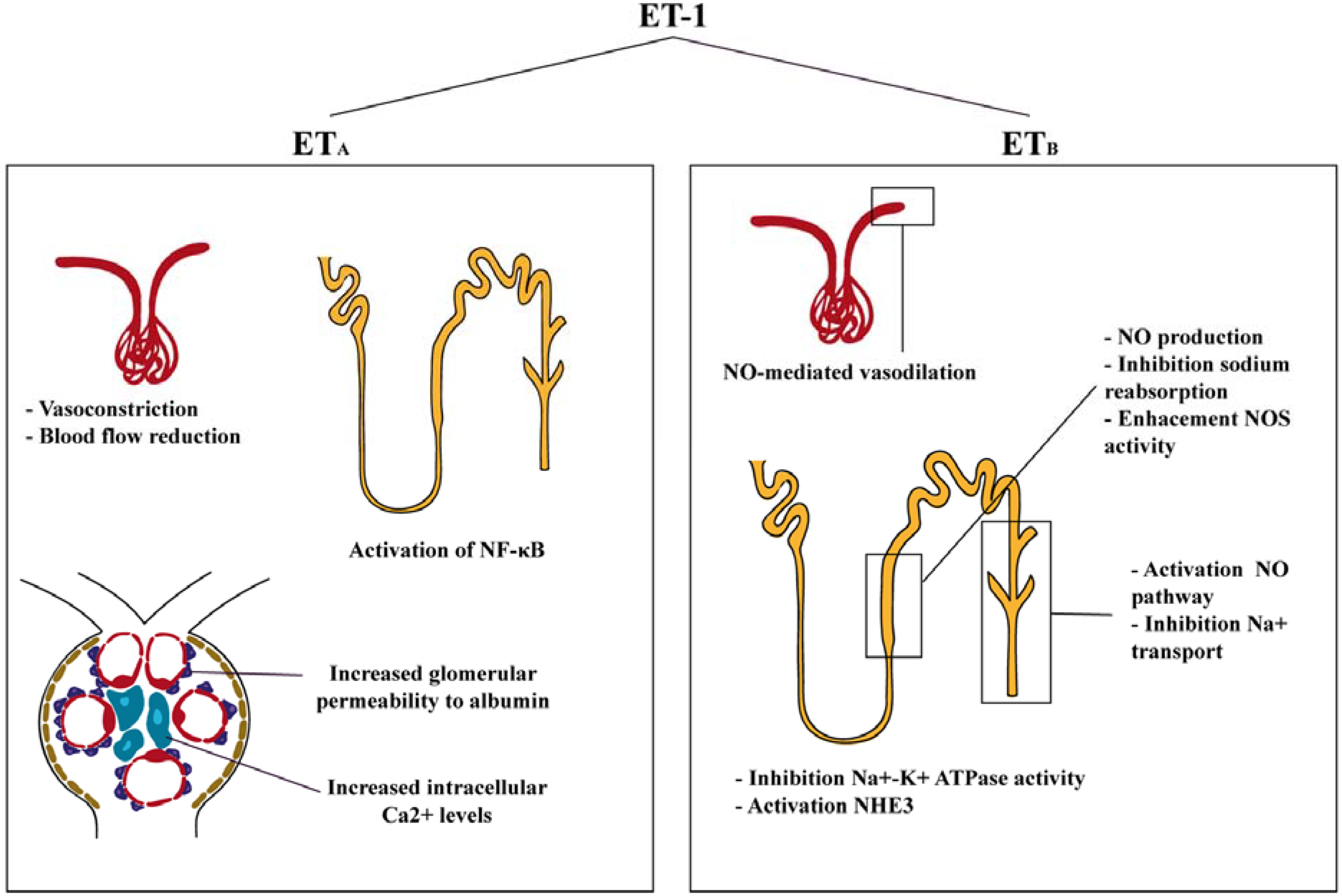
4. Endothelin Antagonists in Diabetic Nephropathy
4.1. Experimental Studies
4.1.1. BQ-123
4.1.2. BQ-788
4.1.3. Bosentan

{kind=link}
{kind=link}
| Drug | ETA/ETB Affinity | Source | Type of Study | Experimental Model | Type of Diabetes | Main Outcomes |
|---|---|---|---|---|---|---|
| BQ-123 | ETA | Simonson et al., 1990 [40] | In vitro | Rat mesangial cells | - | Reduction of albumin permeability |
| Granstam et al., 2011 [72] | In vivo | STZ-induced diabetic Sprague Dawley rats | Type 1 | No alteration on renal blood flow | ||
| Tang et al., 2014 [73] | In vitro | Rat tubular epithelial cells | - | Prevention of changes in E-cadherin and vimentin (epithelial-mesenchymal transition) | ||
| BQ-778 | ETB | Saleh et al., 2011a [37] | In vitro | STZ-induced diabetic Sprague Dawley rats | Type 1 | No effect on elevated albumin permeability |
| Reduction of albumin permeability in combination with BQ-123 | ||||||
| Bosentan | ETA/ETB | Kelly et al., 2000 [75] | In vivo | STZ-induced diabetic Ren-2 rats | Type 1 | Increased albuminuria |
| Attenuation of decrease in GFR | ||||||
| Severe glomerulosclerosis and tubulointerstitial damage | ||||||
| Tikkanen et al., 2002 [104] | In vivo | STZ-induced diabetic Sprague Dawley rats | Type 1 | No reduction in albuminuria | ||
| Chen et al., 2002 [78] | In vivo | STZ-induced diabetic hypertensive rats | Type 1 | Prevention of urinary protein excretion | ||
| Cosenzi et al., 2003 [76] | STZ-induced diabetic Wistar Kyoto rats | Reduction in diabetes-induced fibrosis | ||||
| Ding et al., 2003 [77] | Uninephrectomized STZ-induced diabetic rats | Prevention of renal injury | ||||
| Darusentan | ETA | Hocher et al., 1998 [80] | In vivo | STZ-induced diabetic rats | Type 1 | Reduction in urinary protein excretion |
| Dhein et al., 2000 [81] | STZ-induced diabetic Wistar Kyoto rats | Prevention of glomerulosclerosis index, tubulointerstitial damage index and glomerular volume | ||||
| Gross et al., 2004a/b [82,83] | SHR/N-corpulent rats | Ineffective in prevention of podocyte loss and damage | ||||
| Avosentan | ETA | Gagliardini et al., 2009 [84] | In vivo | STZ-induced diabetic Sprague Dawley rats | Type 1 | Reduction in urinary protein excretion |
| Reduction of glomerulosclerosis, tubulointerstitial damage and mesangial expansion | ||||||
| Reduction of accumulation of inflammatory cells and staining of TGFβ and collagen deposition | ||||||
| No reduction of glomerular hypertrophy Increase in nephrin protein expression | ||||||
| Watson et al., 2010 [85] | In vivo | STZ-induced diabetic ApoE KO mice | Type 1 | Reduction in urinary protein excretion | ||
| Reduction on gene expression levels of fibronectin, collagen IV, TGFβ and α.SMA | ||||||
| Sitaxentan | ETA | Zoja et al., 2011 [86] | In vivo | Zucker Diabetic Fatty rats | Type 2 | No effect on albuminuria and glomerulosclerosis |
| Decrease in systolic blood pressure Reduction in protein matrix accumulation | ||||||
| Atrasentan | ETA | Sasser et al., 2007 [89] | In vivo | STZ-induced diabetic Sprague Dawley rats | Type 1 | Attenuation of urinary excretion of TGFβ |
| No effects in reactive oxygen species production | ||||||
| Saleh et al., 2011a [37] | In vivo/In vitro | STZ-induced diabetic Sprague Dawley rats/Isolated glomeruli | Type 1 | Reduction in proteinuria and albumin permeability | ||
| Saleh et al., 2011b [88] | In vivo/In vitro | STZ-induced diabetic Sprague Dawley rats/Isolated glomeruli | Type 1 | Reduction in proteinuria and albumin permeability | ||
| Prevention of proinflammatory molecules increase | ||||||
| Increase in gene expression levels of nephrin, ZO-1 and podocin |
| Drug | ETA/ETB Affinity | Source | Type of Study | Subjects (Completed Study) | Type of Diabetes | Dosage | Main Outcomes | Adverse Effects |
|---|---|---|---|---|---|---|---|---|
| Bosentan | ETA/ETB | Rafnsson et al., 2012 [92] | Randomized, double-blind, placebo-control trial | 46 | Type 2 | 62.5 mg daily-2 weeks + 125 mg twice daily-2 weeks (in absence of side effects) | No changes in urine ACR ratio, blood pressure and blood glucose Increase in ET-1 plasma levels | One patient with edema (discontinued intervention) |
| Avosentan | ETA | Wenzel et al., 2009 [93] | Randomized, double-blind, placebo-controlled, dosage-range, parallel-group phase 2 study | 252 | Type 1 and 2 | 5, 10, 25 and 50 mg (12 weeks) | Decrease in urinary albumin excretion rate (−20.9% to −29.9%) Reduction in urinary protein excretion | Dosage-dependent fluid retention (32.1% of patients in 50 mg dosage) |
| Mann et al., 2010 [94] | International, multicenter, randomized, double-blind phase 3 clinical trial | 1392 | Type 2 | 25 and 50 mg (prematurely terminated) | ACR declined in a range of 40%–50% in avosentan groups No changes in blood pressure | Increased early mortality mainly due to fluid overload and congestive heart failure. Prematurely terminated. | ||
| Atrasentan | ETA | Kohan et al., 2011 [95] | Randomized, double-blind, placebo-controlled phase 2a clinical trial | 81 | Type 2 | 0.25, 0.75 and 1.75 mg (8 weeks) | Up to 42% ACR reduction in atrasentan groups | Dose-dependent peripheral edema One patient with serious adverse effect (elevated baseline NT-pro BNP) |
| Andress et al., 2012 [96] | Randomized, double-blind, placebo-controlled phase 2a clinical trial | 89 | Type 2 | 0.25, 0.75 and 1.75 mg (8 weeks) | Up to 40% ACR reduction in atrasentan groups | Associated with 1.75 mg treatment group and baseline urinary ACR | ||
| de Zeeuw et al., 2014 [97] | Data pooled from two phase2b studies | 183 | Type 2 | 0.75 and 1.25 mg/day (12 weeks) | Up to 39% ACR reduction in atrasentan groups | Higher number of patients discontinued due to fluid retention-related events in 1.75 mg vs. 0.75 mg group | ||
| SONAR (actively enrolling) | Phase 3 clinical trial | 4148 (estimated enrolling) | Type 2 | Low dose (48 months) | Ongoing | Ongoing |
4.1.4. Darusentan
4.1.5. Avosentan
4.1.6. Sitaxsentan
4.1.7. Atrasentan
4.2. Human Studies and Clinical Trials
4.2.1. Bosentan
4.2.2. Avosentan
4.2.3. Atrasentan
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- KDOQI. Clinical Practice Guideline for Diabetes and CKD: 2012 Update. Am. J. Kidney Dis. 2012, 60, 850–886. [Google Scholar] [CrossRef] [PubMed]
- Hickey, K.A.; Rubanyi, G.; Paul, R.J.; Highsmith, R.F. Characterization of a coronary vasoconstrictor produced by cultured endothelial cells. Am. J. Physiol. 1985, 248, C550–C556. [Google Scholar] [PubMed]
- Inoue, A.; Yanagisawa, M.; Kimura, S.; Kasuya, Y.; Miyauchi, T.; Goto, K.; Masaki, T. The human endothelin family: Three structurally and pharmacologically distinct isopeptides predicted by three separate genes. Proc. Natl. Acad. Sci. USA 1989, 86, 2863–2867. [Google Scholar] [CrossRef] [PubMed]
- Karet, F.E.; Davenport, A.P. Localization of endothelin peptides in human kidney. Kidney Int. 1996, 49, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Morita, S.; Kitamura, K.; Yamamoto, Y.; Eto, T.; Osada, Y.; Sumiyoshi, A.; Koono, M.; Tanaka, K. Immunoreactive endothelin in human kidney. Ann. Clin. Biochem. 1991, 28, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Kohan, D.E. Endothelins in the normal and diseased kidney. Am. J. Kidney Dis. 1997, 29, 2–26. [Google Scholar] [CrossRef] [PubMed]
- Kuc, R.; Davenport, A.P. Comparison of endothelin-A and endothelin-B receptor distribution visualized by radioligand binding versus immunocytochemical localization using subtype selective antisera. J. Cardiovasc. Pharmacol. 2004, 44, S224–S226. [Google Scholar] [CrossRef] [PubMed]
- Saleh, M.A.; Boesen, E.I.; Pollock, J.S.; Savin, V.J.; Pollock, D.M. Endothelin-1 increases glomerular permeability and inflammation independent of blood pressure in the rat. Hypertension 2010, 56, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Elmarakby, A.A.; Loomis, E.D.; Pollock, J.S.; Pollock, D.M. NADPH oxidase inhibition attenuates oxidative stress but not hypertension produced by chronic ET-1. Hypertension 2005, 45, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Kohan, D.E.; Barton, M. Endothelin and endothelin antagonists in chronic kidney disease. Kidney Int. 2014, 86, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Kohan, D.E.; Rossi, N.F.; Inscho, E.W.; Pollock, D.M. Regulation of blood pressure and salt homeostasis by endothelin. Physiol. Rev. 2011, 91, 1–77. [Google Scholar] [CrossRef] [PubMed]
- Barton, M. Reversal of proteinuric renal disease and the emerging role of endothelin. Nat. Clin. Pract. Nephrol. 2008, 4, 490–501. [Google Scholar] [CrossRef] [PubMed]
- Pernow, J.; Franco-Cereceda, A.; Matran, R.; Lundberg, J.M. Effect of endothelin-1 on regional vascular resistances in the pig. J. Cardiovasc. Pharmacol. 1989, 13, S205–S206. [Google Scholar] [CrossRef] [PubMed]
- Nambi, P.; Pullen, M.; Wu, H.L.; Aiyar, N.; Ohlstein, E.H.; Edwards, R.M. Identification of endothelin receptor subtypes in human renal cortex and medulla using subtype-selective ligands. Endocrinology 1992, 131, 1081–1086. [Google Scholar] [PubMed]
- Furuya, S.; Naruse, S.; Nakayama, T.; Nokihara, K. Effect and distribution of intravenously injected 125I-endothelin-1 in rat kidney and lung examined by electron microscopic radioautography. Anat. Embryol. (Berl) 1992, 185, 87–96. [Google Scholar] [CrossRef]
- Frank, K.; Zeier, M.; Gross, M.L.; Waldherr, R.; Ritz, E.; Amann, K. Comprehensive immunohistological analysis of the endothelin system in human kidney grafts. Nephrol. Dial. Transplant. 2006, 21, 1365–1372. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Berrou, J.; Nguyen, G.; Sraer, J.D.; Rondeau, E. Endothelin-1 induces rapid and long lasting internalization of the thrombin receptor in human glomerular epithelial cells. Biochem. Biophys. Res. Commun. 1995, 217, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Hirohama, T.; Uemura, H. Endothelin B receptor-like immunoreactivity in podocytes of the rat kidney. Arch. Histol. Cytol. 2002, 65, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Wendel, M.; Knels, L.; Kummer, W.; Koch, T. Distribution of endothelin receptor subtypes ETA and ETB in the rat kidney. J. Histochem. Cytochem. 2006, 54, 1193–1203. [Google Scholar] [CrossRef] [PubMed]
- Herman, W.H.; Emancipator, S.N.; Rhoten, R.L.; Simonson, M.S. Vascular and glomerular expression of endothelin-1 in normal human kidney. Am. J. Physiol. 1998, 275, F8–F17. [Google Scholar] [PubMed]
- Orth, S.R.; Amann, K.; Gehlen, F.; Unger, L.; Wagner, J.; Raschack, M.; Ritz, E. Adult human mesangial cells (HMCs) express endothelin-B-receptors which mediate endothelin-1-induced cell growth. J. Cardiovasc. Pharmacol. 2000, 36, S232–S237. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, F.; Uchida, S.; Ogata, E.; Kurokawa, K. Endothelin-1 and endothelin-3 binding to rat nephrons. Am. J. Physiol. 1993, 264, F827–F832. [Google Scholar] [PubMed]
- Uchida, S.; Takemoto, F.; Ogata, E.; Kurokawa, K. Endothelin-1 and -3 binding to ETB receptors in rat renal tubules. J. Cardiovasc. Pharmacol. 1993, 22, S177–S180. [Google Scholar] [CrossRef] [PubMed]
- Terada, Y.; Tomita, K.; Nonoguchi, H.; Marumo, F. Different localization of two types of endothelin receptor mRNA in microdissected rat nephron segments using reverse transcription and polymerase chain reaction assay. J. Clin. Invest. 1992, 90, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Uemura, H. Distribution of endothelin-B receptor-like immunoreactivity in rat brain, kidney, and pancreas. J. Cardiovasc. Pharmacol. 1998, 31, S207–S211. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Suzuki, H.; Kubo, Y.; Matsumoto, A.; Uemura, H. Endothelin A receptor-like immunoreactivity on the basal infoldings of rat renal tubules and collecting ducts. Arch. Histol. Cytol. 2008, 71, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Moridaira, K.; Nodera, M.; Sato, G.; Yanagisawa, H. Detection of prepro-ET-1 mRNA in normal rat kidney by in situ RT-PCR. Nephron Exp. Nephrol. 2003, 95, e55–e61. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.A.; Haton, C.; Orea, V.; Sassard, J.; Bailly, C.; Unwin, R.J.; Imbert-Teboul, M. ETA receptor-mediated Ca2+ signaling in thin descending limbs of Henle’s loop: Impairment in genetic hypertension. Kidney Int. 2003, 63, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Dean, R.; Zhuo, J.; Alcorn, D.; Casley, D.; Mendelsohn, F.A. Cellular distribution of 125I-endothelin-1 binding in rat kidney following in vivo labeling. Am. J. Physiol. 1994, 267, F845–F852. [Google Scholar] [PubMed]
- Francis, B.N.; Abassi, Z.; Heyman, S.; Winaver, J.; Hoffman, A. Differential regulation of ET(A) and ET(B) in the renal tissue of rats with compensated and decompensated heart failure. J. Cardiovasc. Pharmacol. 2004, 44, S362–S365. [Google Scholar] [CrossRef] [PubMed]
- Ong, A.C.; Jowett, T.P.; Firth, J.D.; Burton, S.; Karet, F.E.; Fine, L.G. An endothelin-1 mediated autocrine growth loop involved in human renal tubular regeneration. Kidney Int. 1995, 48, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, J.M.; Ahlborg, G.; Hemsen, A.; Nisell, H.; Lunell, N.O.; Pernow, J.; Rudehill, A.; Weitzberg, E. Evidence for release of endothelin-1 in pigs and humans. J. Cardiovasc. Pharmacol. 1991, 17, S350–S353. [Google Scholar] [CrossRef] [PubMed]
- Backer, A.; Bokemeyer, D.; Kramer, H.J. Endothelin synthesis and receptors in porcine kidney. Acta Physiol. Scand. 2001, 171, 105–112. [Google Scholar] [PubMed]
- Karet, F.E.; Kuc, R.E.; Davenport, A.P. Novel ligands BQ123 and BQ3020 characterize endothelin receptor subtypes ETA and ETB in human kidney. Kidney Int. 1993, 44, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Fligny, C.; Barton, M.; Tharaux, P.L. Endothelin and podocyte injury in chronic kidney disease. Contrib. Nephrol. 2011, 172, 120–138. [Google Scholar] [PubMed]
- Morigi, M.; Buelli, S.; Angioletti, S.; Zanchi, C.; Longaretti, L.; Zoja, C.; Galbusera, M.; Gastoldi, S.; Mundel, P.; Remuzzi, G.; et al. In response to protein load podocytes reorganize cytoskeleton and modulate endothelin-1 gene: Implication for permselective dysfunction of chronic nephropathies. Am. J. Pathol. 2005, 166, 1309–1320. [Google Scholar] [CrossRef] [PubMed]
- Saleh, M.A.; Boesen, E.I.; Pollock, J.S.; Savin, V.J.; Pollock, D.M. Endothelin receptor A-specific stimulation of glomerular inflammation and injury in a streptozotocin-induced rat model of diabetes. Diabetologia 2011, 54, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, A.; Kohan, D.E. Physiology and pathology of endothelin-1 in renal mesangium. Am. J. Physiol. Renal Physiol. 2003, 285, F579–F589. [Google Scholar] [CrossRef] [PubMed]
- Badr, K.F.; Murray, J.J.; Breyer, M.D.; Takahashi, K.; Inagami, T.; Harris, R.C. Mesangial cell, glomerular and renal vascular responses to endothelin in the rat kidney. Elucidation of signal transduction pathways. J. Clin. Invest. 1989, 83, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Simonson, M.S.; Dunn, M.J. Endothelin-1 stimulates contraction of rat glomerular mesangial cells and potentiates beta-adrenergic-mediated cyclic adenosine monophosphate accumulation. J. Clin. Invest. 1990, 85, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Yokokawa, K.; Kohno, M.; Johchi, M.; Horio, T.; Murakawa, K.; Yasunari, K.; Yanagisawa, M.; Takeda, T. Effect of endothelin receptor antagonist, BQ-123, on Ca2+ signaling in cultured rat mesangial cells. Life Sci. 1993, 53, 1631–1641. [Google Scholar] [CrossRef] [PubMed]
- Inscho, E.W.; Imig, J.D.; Cook, A.K.; Pollock, D.M. ETA and ETB receptors differentially modulate afferent and efferent arteriolar responses to endothelin. Br. J. Pharmacol. 2005, 146, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.G.; Madden, A.C.; Oliver, J.J.; Lewis, T.V. Effects of ET(A)- and ET(B)-receptor antagonists on regional kidney blood flow, and responses to intravenous endothelin-1, in anaesthetized rabbits. J. Hypertens. 2001, 19, 1789–1799. [Google Scholar] [CrossRef] [PubMed]
- Dhaun, N.; Ferro, C.J.; Davenport, A.P.; Haynes, W.G.; Goddard, J.; Webb, D.J. Haemodynamic and renal effects of endothelin receptor antagonism in patients with chronic kidney disease. Nephrol. Dial. Transplant. 2007, 22, 3228–3234. [Google Scholar] [CrossRef] [PubMed]
- Denton, K.M.; Shweta, A.; Finkelstein, L.; Flower, R.L.; Evans, R.G. Effect of endothelin-1 on regional kidney blood flow and renal arteriole calibre in rabbits. Clin. Exp. Pharmacol. Physiol. 2004, 31, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Goddard, J.; Johnston, N.R.; Hand, M.F.; Cumming, A.D.; Rabelink, T.J.; Rankin, A.J.; Webb, D.J. Endothelin-A receptor antagonism reduces blood pressure and increases renal blood flow in hypertensive patients with chronic renal failure: A comparison of selective and combined endothelin receptor blockade. Circulation 2004, 109, 1186–1193. [Google Scholar] [CrossRef] [PubMed]
- Schildroth, J.; Rettig-Zimmermann, J.; Kalk, P.; Steege, A.; Fahling, M.; Sendeski, M.; Paliege, A.; Lai, E.Y.; Bachmann, S.; Persson, P.B.; et al. Endothelin type A and B receptors in the control of afferent and efferent arterioles in mice. Nephrol. Dial. Transplant. 2011, 26, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Samsell, L.; Baylis, C. Actions of endogenous endothelin on glomerular hemodynamics in the rat. Am. J. Physiol. 1995, 269, R469–R473. [Google Scholar] [PubMed]
- Yu, C.; Yang, Z.; Ren, H.; Zhang, Y.; Han, Y.; He, D.; Lu, Q.; Wang, X.; Yang, C.; Asico, L.D.; et al. D3 dopamine receptor regulation of ETB receptors in renal proximal tubule cells from WKY and SHRs. Am. J. Hypertens. 2009, 22, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, J.; Ren, H.; He, D.; Pascua, A.; Armando, M.I.; Yang, C.; Zhou, L.; Felder, R.A.; Jose, P.A.; et al. Inhibitory effect of ETB receptor on Na(+)-K(+) ATPase activity by extracellular Ca(2+) entry and Ca(2+) release from the endoplasmic reticulum in renal proximal tubule cells. Hypertens. Res. 2009, 32, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Laghmani, K.; Preisig, P.A.; Moe, O.W.; Yanagisawa, M.; Alpern, R.J. Endothelin-1/endothelin-B receptor-mediated increases in NHE3 activity in chronic metabolic acidosis. J. Clin. Invest. 2001, 107, 1563–1569. [Google Scholar] [CrossRef] [PubMed]
- Chu, T.S.; Peng, Y.; Cano, A.; Yanagisawa, M.; Alpern, R.J. Endothelin(B) receptor activates NHE-3 by a Ca2+-dependent pathway in OKP cells. J. Clin. Invest. 1996, 97, 1454–1462. [Google Scholar] [CrossRef] [PubMed]
- Herrera, M.; Hong, N.J.; Ortiz, P.A.; Garvin, J.L. Endothelin-1 inhibits thick ascending limb transport via Akt-stimulated nitric oxide production. J. Biol. Chem. 2009, 284, 1454–1460. [Google Scholar] [CrossRef] [PubMed]
- Plato, C.F.; Pollock, D.M.; Garvin, J.L. Endothelin inhibits thick ascending limb chloride flux via ET(B) receptor-mediated NO release. Am. J. Physiol. Renal Physiol. 2000, 279, F326–F333. [Google Scholar] [PubMed]
- Bailly, C. Effect of luminal atrial natriuretic peptide on chloride reabsorption in mouse cortical thick ascending limb: inhibition by endothelin. J. Am. Soc. Nephrol. 2000, 11, 1791–1797. [Google Scholar] [PubMed]
- Gerstung, M.; Roth, T.; Dienes, H.P.; Licht, C.; Fries, J.W. Endothelin-1 induces NF-kappaB via two independent pathways in human renal tubular epithelial cells. Am. J. Nephrol. 2007, 27, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Zeidel, M.L.; Brady, H.R.; Kone, B.C.; Gullans, S.R.; Brenner, B.M. Endothelin, a peptide inhibitor of Na(+)-K(+)-ATPase in intact renaltubular epithelial cells. Am. J. Physiol. 1989, 257, C1101–C1107. [Google Scholar] [PubMed]
- Star, R.A.; Nonoguchi, H.; Balaban, R.; Knepper, M.A. Calcium and cyclic adenosine monophosphate as second messengers for vasopressin in the rat inner medullary collecting duct. J. Clin. Invest. 1988, 81, 1879–1888. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Nonoguchi, H.; Marumo, F. Effects of endothelin on peptide-dependent cyclic adenosine monophosphate accumulation along the nephron segments of the rat. J. Clin. Invest. 1990, 85, 2014–2018. [Google Scholar] [CrossRef] [PubMed]
- Ahn, D.; Ge, Y.; Stricklett, P.K.; Gill, P.; Taylor, D.; Hughes, A.K.; Yanagisawa, M.; Miller, L.; Nelson, R.D.; Kohan, D.E. Collecting duct-specific knockout of endothelin-1 causes hypertension and sodium retention. J. Clin. Invest. 2004, 114, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, T.S.; Chahdi, A.; Ilatovskaya, D.V.; Levchenko, V.; Vandewalle, A.; Pochynyuk, O.; Sorokin, A.; Staruschenko, A. Endothelin-1 inhibits the epithelial Na+ channel through betaPix/14–3-3/Nedd4–2. J. Am. Soc. Nephrol. 2010, 21, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.P.; Ge, Y.; Pollock, D.M.; Pollock, J.S.; Kohan, D.E. Collecting duct-derived endothelin regulates arterial pressure and Na excretion via nitric oxide. Hypertension 2008, 51, 1605–1610. [Google Scholar] [CrossRef] [PubMed]
- Stricklett, P.K.; Hughes, A.K.; Kohan, D.E. Endothelin-1 stimulates NO production and inhibits cAMP accumulation in rat inner medullary collecting duct through independent pathways. Am. J. Physiol. Renal Physiol. 2006, 290, F1315–F1319. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Bagnall, A.; Stricklett, P.K.; Strait, K.; Webb, D.J.; Kotelevtsev, Y.; Kohan, D.E. Collecting duct-specific knockout of the endothelin B receptor causes hypertension and sodium retention. Am. J. Physiol. Renal Physiol. 2006, 291, F1274–F1280. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Ohnaka, K.; Takayanagi, R.; Umeda, F.; Nawata, H. Enhanced secretion of endothelin-1 by elevated glucose levels from cultured bovine aortic endothelial cells. FEBS Lett. 1990, 267, 16–18. [Google Scholar] [CrossRef] [PubMed]
- Hocher, B.; Thone-Reineke, C.; Rohmeiss, P.; Schmager, F.; Slowinski, T.; Burst, V.; Siegmund, F.; Quertermous, T.; Bauer, C.; Neumayer, H.H.; et al. Endothelin-1 transgenic mice develop glomerulosclerosis, interstitial fibrosis, and renal cysts but not hypertension. J. Clin. Invest. 1997, 99, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- King, A.J.; Brenner, B.M.; Anderson, S. Endothelin: A potent renal and systemic vasoconstrictor peptide. Am. J. Physiol. 1989, 256, F1051–F1058. [Google Scholar] [PubMed]
- Kawaguchi, H.; Sawa, H.; Yasuda, H. Endothelin stimulates angiotensin I to angiotensin II conversion in cultured pulmonary artery endothelial cells. J. Mol. Cell. Cardiol. 1990, 22, 839–842. [Google Scholar] [CrossRef] [PubMed]
- Barton, M.; Shaw, S.; d’Uscio, L.V.; Moreau, P.; Luscher, T.F. Angiotensin II increases vascular and renal endothelin-1 and functional endothelin converting enzyme activity in vivo: Role of ETA receptors for endothelin regulation. Biochem. Biophys. Res. Commun. 1997, 238, 861–865. [Google Scholar] [CrossRef] [PubMed]
- Lenoir, O.; Milon, M.; Virsolvy, A.; Henique, C.; Schmitt, A.; Masse, J.M.; Kotelevtsev, Y.; Yanagisawa, M.; Webb, D.J.; Richard, S.; et al. Direct action of endothelin-1 on podocytes promotes diabetic glomerulosclerosis. J. Am. Soc. Nephrol. 2014, 25, 1050–1062. [Google Scholar] [CrossRef] [PubMed]
- Morigi, M.; Buelli, S.; Zanchi, C.; Longaretti, L.; Macconi, D.; Benigni, A.; Moioli, D.; Remuzzi, G.; Zoja, C. Shigatoxin-induced endothelin-1 expression in cultured podocytes autocrinally mediates actin remodeling. Am. J. Pathol. 2006, 169, 1965–1975. [Google Scholar] [CrossRef] [PubMed]
- Granstam, S.O.; Granstam, E. Endothelin-induced changes in blood flow in STZ-diabetic and non-diabetic rats: Relation to nitric oxide synthase and cyclooxygenase inhibition. J. Physiol. Sci. 2011, 61, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Li, H.; Gou, R.; Cheng, G.; Guo, Y.; Fang, Y.; Chen, F. Endothelin-1 mediated high glucose-induced epithelial-mesenchymal transition in renal tubular cells. Diabetes Res. Clin. Pract. 2014, 104, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Clozel, M.; Breu, V.; Gray, G.A.; Kalina, B.; Loffler, B.M.; Burri, K.; Cassal, J.M.; Hirth, G.; Muller, M.; Neidhart, W.; et al. Pharmacological characterization of bosentan, a new potent orally active nonpeptide endothelin receptor antagonist. J. Pharmacol. Exp. Ther. 1994, 270, 228–235. [Google Scholar] [PubMed]
- Kelly, D.J.; Skinner, S.L.; Gilbert, R.E.; Cox, A.J.; Cooper, M.E.; Wilkinson-Berka, J.L. Effects of endothelin or angiotensin II receptor blockade on diabetes in the transgenic (mRen-2)27 rat. Kidney Int. 2000, 57, 1882–1894. [Google Scholar] [CrossRef] [PubMed]
- Cosenzi, A.; Bernobich, E.; Trevisan, R.; Milutinovic, N.; Borri, A.; Bellini, G. Nephroprotective effect of bosentan in diabetic rats. J. Cardiovasc. Pharmacol. 2003, 42, 752–756. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.S.; Qiu, C.; Hess, P.; Xi, J.F.; Zheng, N.; Clozel, M. Chronic endothelin receptor blockade prevents both early hyperfiltration and late overt diabetic nephropathy in the rat. J. Cardiovasc. Pharmacol. 2003, 42, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Gu, Y.; Lin, F.; Yang, H.; Zhu, W.; Ma, J.; Lin, S. Endothelin receptor antagonist combined with a calcium channel blocker attenuates renal injury in spontaneous hypertensive rats with diabetes. Chin. Med. J. (Engl) 2002, 115, 972–978. [Google Scholar]
- Chen, S.; Khan, Z.A.; Cukiernik, M.; Chakrabarti, S. Differential activation of NF-kappa B and AP-1 in increased fibronectin synthesis in target organs of diabetic complications. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E1089–E1097. [Google Scholar] [CrossRef] [PubMed]
- Hocher, B.; Lun, A.; Priem, F.; Neumayer, H.H.; Raschack, M. Renal endothelin system in diabetes: comparison of angiotensin-converting enzyme inhibition and endothelin-A antagonism. J. Cardiovasc. Pharmacol. 1998, 31, S492–S495. [Google Scholar] [CrossRef] [PubMed]
- Dhein, S.; Hochreuther, S.; Aus Dem Spring, C.; Bollig, K.; Hufnagel, C.; Raschack, M. Long-term effects of the endothelin(A) receptor antagonist LU 135252 and the angiotensin-converting enzyme inhibitor trandolapril on diabetic angiopathy and nephropathy in a chronic type I diabetes mellitus rat model. J. Pharmacol. Exp. Ther. 2000, 293, 351–359. [Google Scholar] [PubMed]
- Gross, M.L.; Ritz, E.; Schoof, A.; Helmke, B.; Parkman, A.; Tulp, O.; Munter, K.; Amann, K. Renal damage in the SHR/N-cp type 2 diabetes model: comparison of an angiotensin-converting enzyme inhibitor and endothelin receptor blocker. Lab. Invest. 2003, 83, 1267–1277. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.L.; El-Shakmak, A.; Szabo, A.; Koch, A.; Kuhlmann, A.; Munter, K.; Ritz, E.; Amann, K. ACE-inhibitors but not endothelin receptor blockers prevent podocyte loss in early diabetic nephropathy. Diabetologia 2003, 46, 856–868. [Google Scholar] [CrossRef] [PubMed]
- Gagliardini, E.; Corna, D.; Zoja, C.; Sangalli, F.; Carrara, F.; Rossi, M.; Conti, S.; Rottoli, D.; Longaretti, L.; Remuzzi, A.; et al. Unlike each drug alone, lisinopril if combined with avosentan promotes regression of renal lesions in experimental diabetes. Am. J. Physiol. Renal Physiol. 2009, 297, F1448–F1456. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.M.; Li, J.; Schumacher, C.; de Gasparo, M.; Feng, B.; Thomas, M.C.; Allen, T.J.; Cooper, M.E.; Jandeleit-Dahm, K.A. The endothelin receptor antagonist avosentan ameliorates nephropathy and atherosclerosis in diabetic apolipoprotein E knockout mice. Diabetologia 2010, 53, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Zoja, C.; Cattaneo, S.; Fiordaliso, F.; Lionetti, V.; Zambelli, V.; Salio, M.; Corna, D.; Pagani, C.; Rottoli, D.; Bisighini, C.; et al. Distinct cardiac and renal effects of ETA receptor antagonist and ACE inhibitor in experimental type 2 diabetes. Am. J. Physiol. Renal Physiol. 2011, 301, F1114–F1123. [Google Scholar] [CrossRef] [PubMed]
- Opgenorth, T.J.; Adler, A.L.; Calzadilla, S.V.; Chiou, W.J.; Dayton, B.D.; Dixon, D.B.; Gehrke, L.J.; Hernandez, L.; Magnuson, S.R.; Marsh, K.C.; et al. Pharmacological characterization of A-127722: An orally active and highly potent ETA-selective receptor antagonist. J. Pharmacol. Exp. Ther. 1996, 276, 473–481. [Google Scholar] [PubMed]
- Saleh, M.A.; Pollock, J.S.; Pollock, D.M. Distinct actions of endothelin A-selective versus combined endothelin A/B receptor antagonists in early diabetic kidney disease. J. Pharmacol. Exp. Ther. 2011, 338, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Sasser, J.M.; Sullivan, J.C.; Hobbs, J.L.; Yamamoto, T.; Pollock, D.M.; Carmines, P.K.; Pollock, J.S. Endothelin A receptor blockade reduces diabetic renal injury via an anti-inflammatory mechanism. J. Am. Soc. Nephrol. 2007, 18, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Song, G.G. Meta-analysis of randomized controlled trials of bosentan for treatment of pulmonary arterial hypertension. Korean J. Intern. Med. 2013, 28, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Bosentan: A review of its use in the management of digital ulcers associated with systemic sclerosis. Drugs 2009, 69, 2005–2024. [Google Scholar] [CrossRef] [PubMed]
- Rafnsson, A.; Bohm, F.; Settergren, M.; Gonon, A.; Brismar, K.; Pernow, J. The endothelin receptor antagonist bosentan improves peripheral endothelial function in patients with type 2 diabetes mellitus and microalbuminuria: A randomised trial. Diabetologia 2012, 55, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, R.R.; Littke, T.; Kuranoff, S.; Jurgens, C.; Bruck, H.; Ritz, E.; Philipp, T.; Mitchell, A. Avosentan reduces albumin excretion in diabetics with macroalbuminuria. J. Am. Soc. Nephrol. 2009, 20, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.F.; Green, D.; Jamerson, K.; Ruilope, L.M.; Kuranoff, S.J.; Littke, T.; Viberti, G. Avosentan for overt diabetic nephropathy. J. Am. Soc. Nephrol. 2010, 21, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Kohan, D.E.; Pritchett, Y.; Molitch, M.; Wen, S.; Garimella, T.; Audhya, P.; Andress, D.L. Addition of atrasentan to renin-angiotensin system blockade reduces albuminuria in diabetic nephropathy. J. Am. Soc. Nephrol. 2011, 22, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Andress, D.L.; Coll, B.; Pritchett, Y.; Brennan, J.; Molitch, M.; Kohan, D.E. Clinical efficacy of the selective endothelin A receptor antagonist, atrasentan, in patients with diabetes and chronic kidney disease (CKD). Life Sci. 2012, 91, 739–742. [Google Scholar] [CrossRef] [PubMed]
- De Zeeuw, D.; Coll, B.; Andress, D.; Brennan, J.J.; Tang, H.; Houser, M.; Correa-Rotter, R.; Kohan, D.; Lambers Heerspink, H.J.; Makino, H.; et al. The endothelin antagonist atrasentan lowers residual albuminuria in patients with type 2 diabetic nephropathy. J. Am. Soc. Nephrol. 2014, 25, 1083–1093. [Google Scholar] [CrossRef] [PubMed]
- Barst, R.J.; Rich, S.; Widlitz, A.; Horn, E.M.; McLaughlin, V.; McFarlin, J. Clinical efficacy of sitaxsentan, an endothelin-A receptor antagonist, in patients with pulmonary arterial hypertension: Open-label pilot study. Chest 2002, 121, 1860–1868. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Olsson, K.M.; Schneider, A.; Golpon, H. Severe hepatitis associated with sitaxentan and response to glucocorticoid therapy. Eur. Respir. J. 2009, 33, 1518–1519. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.T.; Kirkham, N.; Johnson, M.K.; Lordan, J.L.; Fisher, A.J.; Peacock, A.J. Sitaxentan-related acute liver failure in a patient with pulmonary arterial hypertension. Eur. Respir. J. 2011, 37, 472–474. [Google Scholar] [CrossRef] [PubMed]
- Lavelle, A.; Sugrue, R.; Lawler, G.; Mulligan, N.; Kelleher, B.; Murphy, D.M.; Gaine, S.P. Sitaxentan-induced hepatic failure in two patients with pulmonary arterial hypertension. Eur. Respir. J. 2009, 34, 770–771. [Google Scholar] [CrossRef] [PubMed]
- Galie, N.; Olschewski, H.; Oudiz, R.J.; Torres, F.; Frost, A.; Ghofrani, H.A.; Badesch, D.B.; McGoon, M.D.; McLaughlin, V.V.; Roecker, E.B.; et al. Ambrisentan for the treatment of pulmonary arterial hypertension: Results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation 2008, 117, 3010–3019. [Google Scholar] [CrossRef] [PubMed]
- McGoon, M.D.; Frost, A.E.; Oudiz, R.J.; Badesch, D.B.; Galie, N.; Olschewski, H.; McLaughlin, V.V.; Gerber, M.J.; Dufton, C.; Despain, D.J.; et al. Ambrisentan therapy in patients with pulmonary arterial hypertension who discontinued bosentan or sitaxsentan due to liver function test abnormalities. Chest 2009, 135, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Tikkanen, I.; Tikkanen, T.; Cao, Z.; Allen, T.J.; Davis, B.J.; Lassila, M.; Casley, D.; Johnston, C.I.; Burrell, L.M.; Cooper, M.E. Combined inhibition of neutral endopeptidase with angiotensin converting enzyme or endothelin converting enzyme in experimental diabetes. J. Hypertens. 2002, 20, 707–714. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anguiano, L.; Riera, M.; Pascual, J.; Soler, M.J. Endothelin Blockade in Diabetic Kidney Disease. J. Clin. Med. 2015, 4, 1171-1192. https://doi.org/10.3390/jcm4061171
Anguiano L, Riera M, Pascual J, Soler MJ. Endothelin Blockade in Diabetic Kidney Disease. Journal of Clinical Medicine. 2015; 4(6):1171-1192. https://doi.org/10.3390/jcm4061171
Chicago/Turabian StyleAnguiano, Lidia, Marta Riera, Julio Pascual, and María José Soler. 2015. "Endothelin Blockade in Diabetic Kidney Disease" Journal of Clinical Medicine 4, no. 6: 1171-1192. https://doi.org/10.3390/jcm4061171
APA StyleAnguiano, L., Riera, M., Pascual, J., & Soler, M. J. (2015). Endothelin Blockade in Diabetic Kidney Disease. Journal of Clinical Medicine, 4(6), 1171-1192. https://doi.org/10.3390/jcm4061171