
Using iPS Cells toward the Understanding of Parkinson’s Disease

Abstract
:1. Parkinson’s Disease
2. Models of Parkinson’s Disease
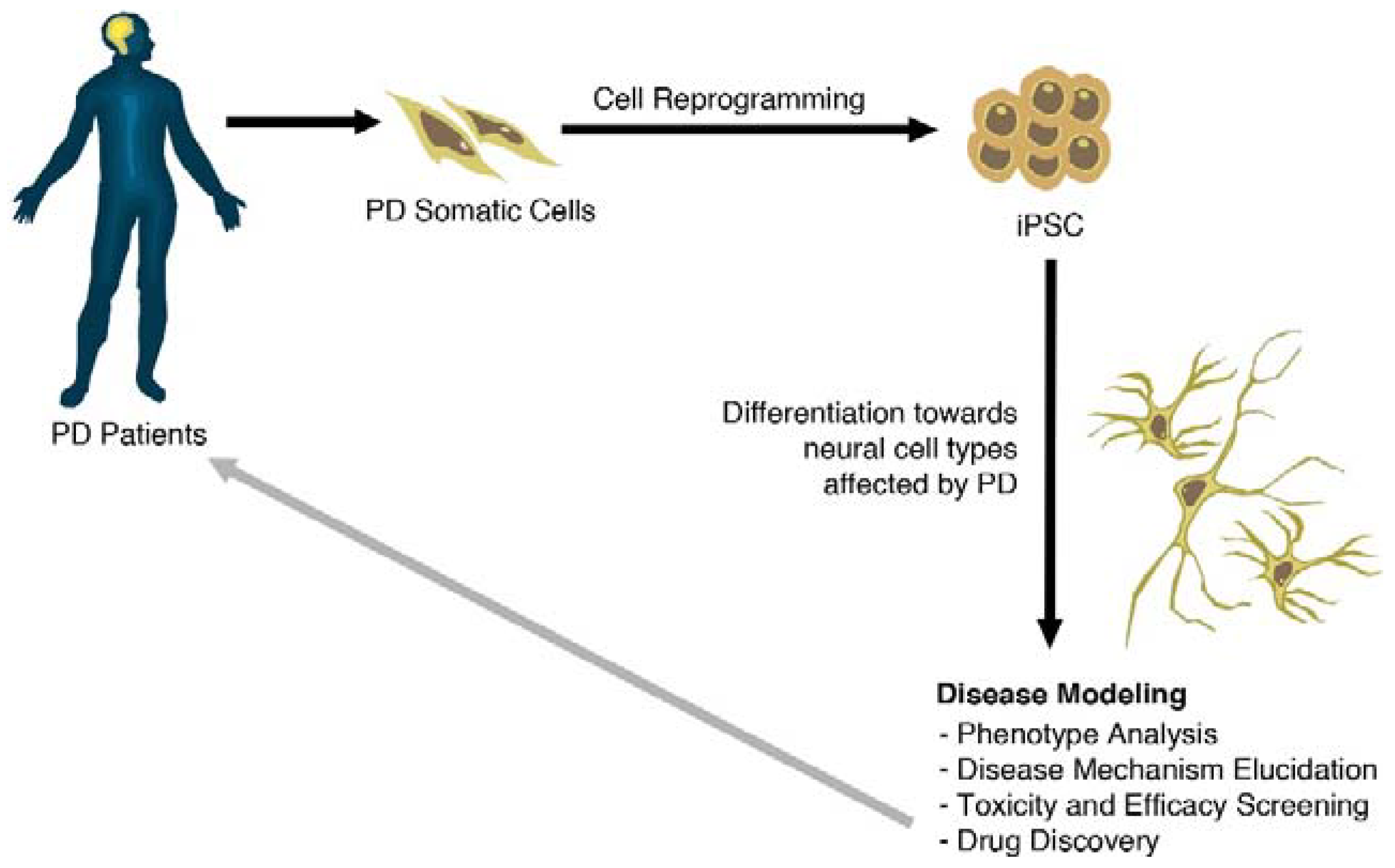
3. Generation of PD-Specific iPSCs
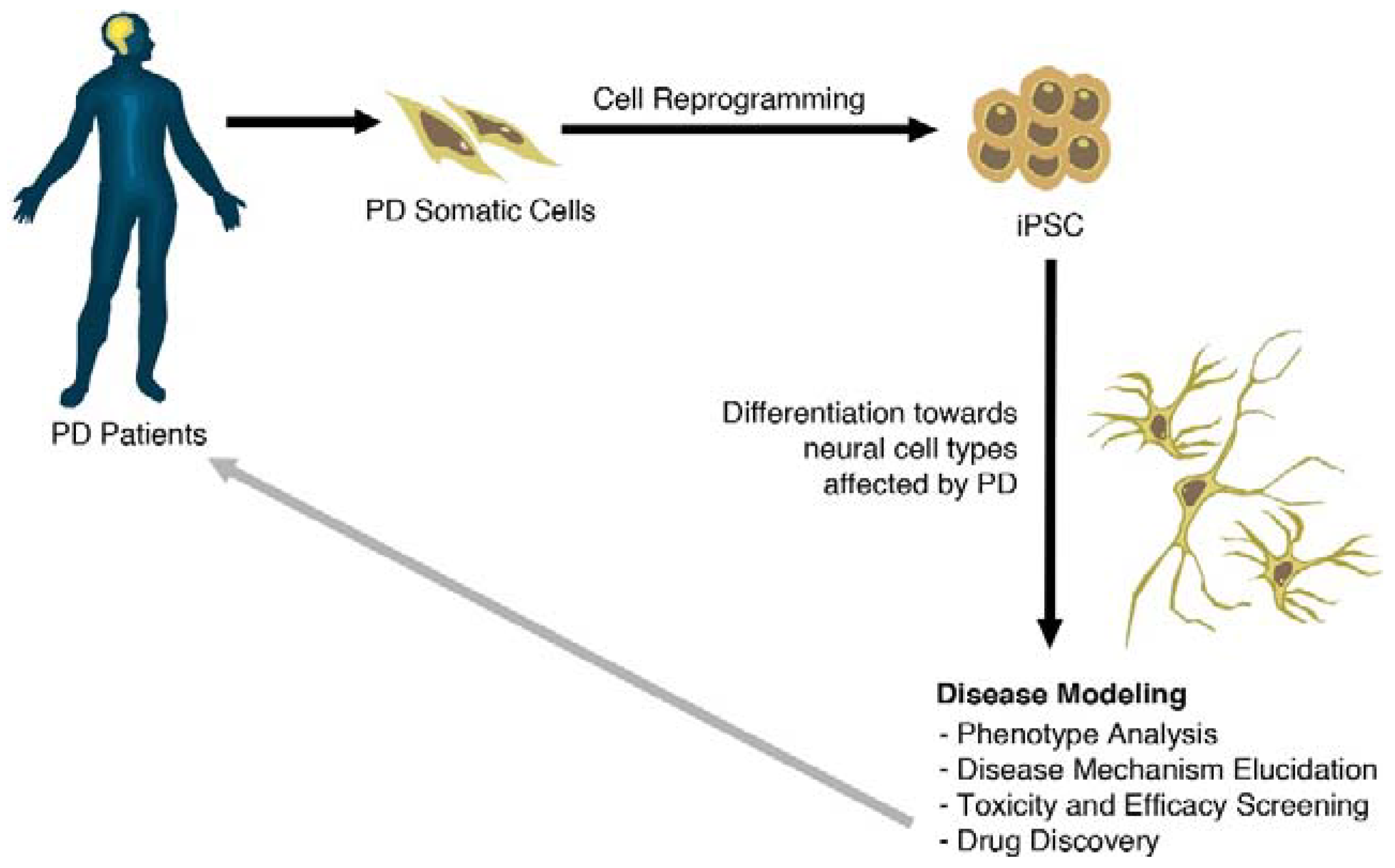
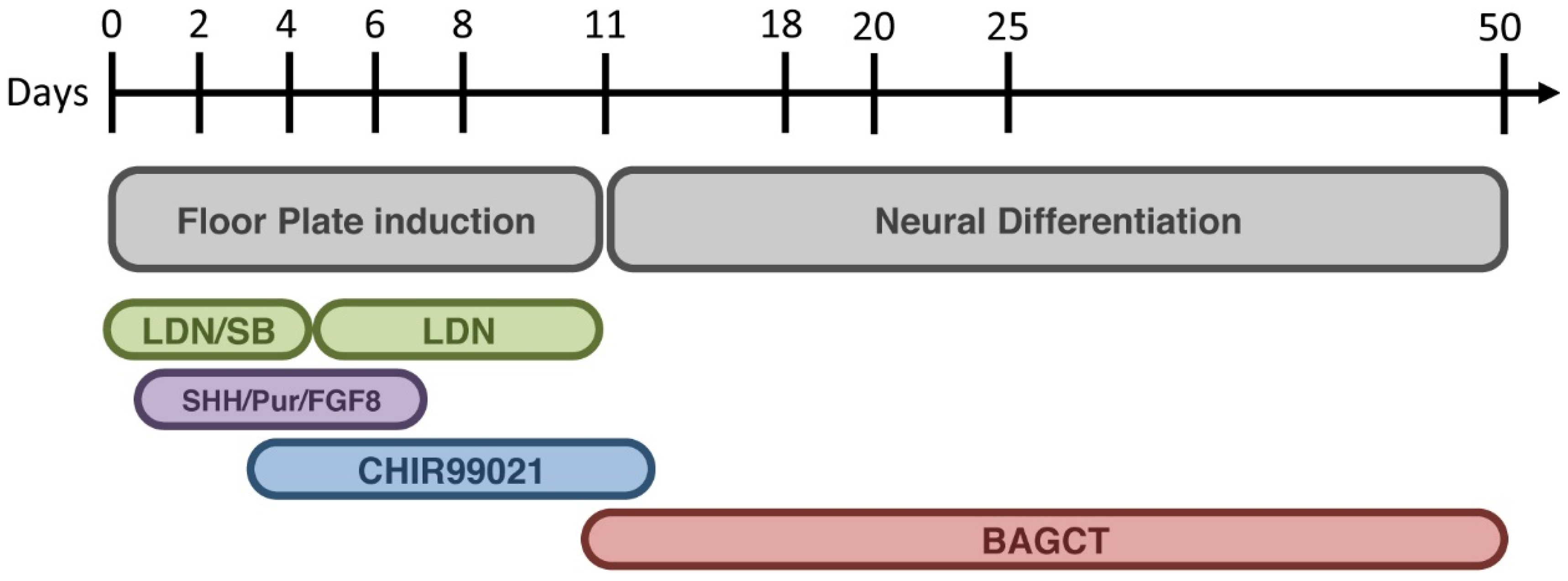
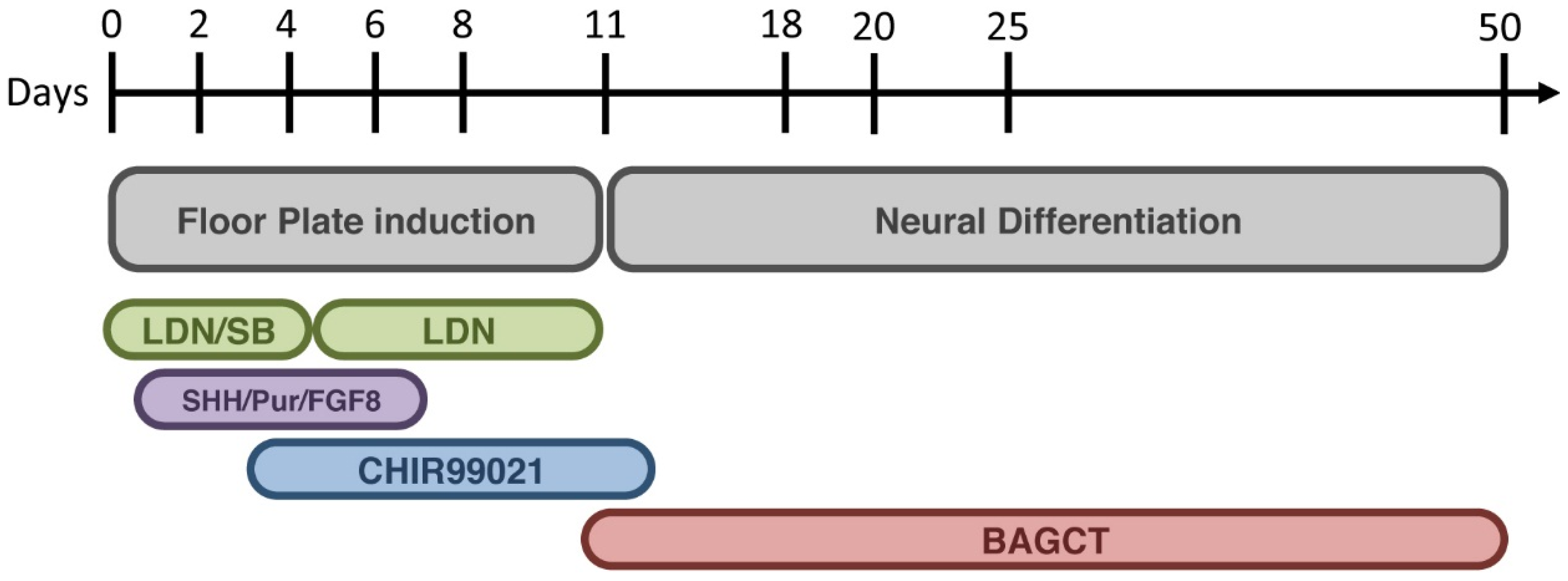
4. Modeling Sporadic and Familial PD Using iPSC

{kind=link}
{kind=link}
| Gene | Publication | Mutation | Number of patients | Isogenic Controls | Cell Type Differentiation | Findings |
|---|---|---|---|---|---|---|
| SNCA | Devine et al., 2011 [30] | Triplication | 1 | NO | Floor-plate DAn differentiation (21–30 days): 28%–37% TH+/TUJ1+ | mRNA doubled expression of SNCA |
| Byers et al., 2011 [31] | Triplication | 1 | NO | DAn differentiation (50 days): 6%–11% TH+ | Double expression of SNCA, increased susceptibility to OS | |
| Chung et al., 2013 [32] | A53T | 2 | YES | Neuronal differentiation (56–84 days): DAn yield not specified. | Increased nitrosative stress, and ER stress, reversed by adding NAB2. | |
| Ryan et al., 2013 [25] | A53T | 1 | YES | Kriks’s Floor-plate DAn differentiation: ~80% A9 DAn of total neurons. | Diminished spare respiration mitochondrial capacity; increased ROS/RNS and attenuation of MEF2/PGC1α neuroprotective pathway | |
| GBA1 | Mazzulli et al., 2011 [33] | N370S/84GG insertion | 1 | NO | DAn diff. (30 days): 80% TUJ1+, 10% TH+/TUJ1+ | Formation of soluble α-syn oligomers, correlated with a decline of lysosomal proteolysis. |
| Schöndorf et al., 2014 [34] | GBA1 (RecNcil/wt) GD (N370S; L444P) | 4 GBA1 4 GD | YES | Kriks’s Floor-plate DAn differentiation: 15%–20% TH+/GIRK2+/FOXA2+/VMAT2+ There is also further purification of DAn by FACS | Causal relation of GBA1 mutations with increased a-syn and LB inclusions, correlated with autophagic/lysosomal system impairment | |
| PARK2 | Jiang et al., 2012 [35] | Exon 3/5 deletion | 2 | NO | DAn differentiation (70 days): yield not specified | Loss of Parkin function; decreased DA uptake and incorrectly folded DAT protein, with increased OS susceptibility. Transduction of WT PARK2 reversed OS sensitiveness. |
| Imaizumi et al., 2012 [36] | Exons 2–4 and 6,7 homozygous deletion | 2 | NO | DAn differentiation (10 days): yield not specified | Abnormal mitochondrial morphology and impaired mitochondrial homeostasis. | |
| PARK2 PINK1 | Miller et al., 2013 [26] | PINK1 (Q456X) Parkin (V324a) | 1 1 | NO | Kriks’s Floor-plate DAn differentiation yield not specified | Loss of dendrite lenght and decreased neuronal survival, as seen by decreased p-ATK values, when exposing mDA neurons to progerin. |
| PINK1 | Seibler et al., 2013 [37] | C1366T, C509G | 3 | NO | Floor-plate DAn differentiation: 11%–16% TH+/TUJ1+ | Endogenous mutant PINK1 diminished Parkin recruitment to the mitochondrial membrane under the presence of valynomycin. WT PINK1 rescued Parkin recruitment. |
| (PINK1) | Cooper et al., 2012 [38] | Q456X | 2 | NO | DAn differentiation (22 days): 35% TUJ1+; 10% TH+ | Increased vulnerability of neural cells to chemical stressors, with common defects to protect against OS. |
| LRRK2 | Nguyen et al., 2011 [39] | G2019S, R1441C | 2 | NO | Floor-plate DAn differentiation (30–35 days): 3.6%–5% TH+ | α-syn accumulation, increased OS genes, and increased susceptibility to hydrogen peroxide. |
| Sánchez-Danes et al., 2012 [40] | G2019S | 7 Sporadic 4 LRRK2 (G2019S) | NO | DAn diff (Lentiviral-mediated forced expression LMX1A in neural precursors) (75 days): 55% TH+/TUJ1+ (Majority TH+GIRK2+) | Reduced neurite lenght and number. Accumulation of α-syn in LRRK2 DAn. Reduction of autophagic flux and accumulation of early autophagosomes. | |
| Orenstein et al., 2013 [41] | G2019S | 4 LRRK2 (G2019S) | NO | As described in [40] | Blockage of the CMA degradation pathway due to accumulated α-syn with correlated increased expression of LAMP-2A. | |
| Reinhardt et al., 2013 [42] | G2019S | 2 | YES | Floor-plate DAn differentiation (30–35 days): 20% TH/TUJ1/DAPI | Decreased neurite lenght levels. Increased ERK activation levels, and discover of novel genes dysregulated in LRRK2 DAn. |
5. Patient-Derived Stem Cells Could Improve Drug Research for PD
6. Limitations of Using iPSC in Disease Modeling: From Overall Neurodegeneration to the Detailed Mechanisms Involved
6.1. Reprogramming and Epigenetic Signatures
6.2. Reliable Control Lines and Gene-Editing
6.3. Cell Differentiation and Sorting
7. Conclusions and Challenges
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lees, A.J.; Hardy, J.; Revesz, T. Parkinson’s disease. Lancet 2009, 373, 2055–2066. [Google Scholar] [PubMed]
- Obeso, J.A.; Rodriguez-Oroz, M.C.; Goetz, C.G.; Marin, C.; Kordower, J.H.; Rodriguez, M.; Hirsch, E.C.; Farrer, M.; Schapira, A.H.; Halliday, G. Missing pieces in the Parkinson’s disease puzzle. Nat. Med. 2010, 16, 653–661. [Google Scholar] [PubMed]
- Schapira, A.H.; Tolosa, E. Molecular and clinical prodrome of Parkinson disease: Implications for treatment. Nat. Rev. Neurol. 2010, 6, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–923. [Google Scholar] [CrossRef] [PubMed]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron. 2003, 39, 889–909. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Byun, J.W.; Choi, I.; Kim, B.; Jeong, H.K.; Jou, I.; Joe, E. PINK1 Deficiency Enhances Inflammatory Cytokine Release from Acutely Prepared Brain Slices. Exp. Neurobiol. 2013, 22, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Melrose, H.; Lincoln, S.; Tyndall, G.; Dickson, D.; Farrer, M. Anatomical localization of leucine-rich repeat kinase 2 in mouse brain. Neuroscience 2006, 139, 791–794. [Google Scholar] [CrossRef] [PubMed]
- Dawson, T.M.; Ko, H.S.; Dawson, V.L. Genetic animal models of Parkinson’s disease. Neuron 2010, 66, 646–661. [Google Scholar] [CrossRef] [PubMed]
- Gispert, S.; del Turco, D.; Garrett, L.; Chen, A.; Bernard, D.J.; Hamm-Clement, J.; Korf, H.W.; Deller, T.; Braak, H.; Auburger, G.; et al. Transgenic mice expressing mutant A53T human alpha-synuclein show neuronal dysfunction in the absence of aggregate formation. Mol. Cell Neurosci. 2003, 24, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Gispert, S.; Ricciardi, F.; Kurz, A.; Azizov, M.; Hoepken, H.H.; Becker, D.; Voos, W.; Leuner, K.; Müller, W.E.; Kudin, A.P.; et al. Parkinson phenotype in aged PINK1-deficient mice is accompanied by progressive mitochondrial dysfunction in absence of neurodegeneration. PLoS ONE 2009, 4, e5777. [Google Scholar] [CrossRef] [PubMed]
- Chesselet, M.F.; Richter, F. Modelling of Parkinson’s disease in mice. Lancet Neurol. 2011, 10, 1108–1118. [Google Scholar] [CrossRef] [PubMed]
- Magen, I.; Chesselet, M.F. Genetic mouse models of Parkinson’s disease. The state of the art. Prog Brain Res. 2010, 184, 53–87. [Google Scholar]
- Tieu, K. A guide to neurotoxic animal models of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2011. [Google Scholar] [CrossRef]
- Kume, T.; Kawato, Y.; Osakada, F.; Izumi, Y.; Katsuki, H.; Nakagawa, T.; Kaneko, S.; Niidome, T.; Takada-Takatori, Y.; Akaike, A. Dibutyryl cyclic AMP induces differentiation of human neuroblastoma SH-SY5Y cells into a noradrenergic phenotype. Neurosci. Lett. 2008, 443, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Pizà, I.; Richaud-Patin, Y.; Vassena, R.; González, F.; Barrero, M.J.; Veiga, A.; Raya, A.; Izpisúa Belmonte, J.C. Reprogramming of human fibroblasts to induced pluripotent stem cells under xeno-free conditions. Stem Cells 2010, 28, 36–44. [Google Scholar] [PubMed]
- Ebert, A.D.; Yu, J.; Rose, F.F., Jr.; Mattis, V.B.; Lorson, C.L.; Thomson, J.A.; Svendsen, C.N. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 2009, 457, 277–280. [Google Scholar] [CrossRef]
- Lee, G.; Papapetrou, E.P.; Kim, H.; Chambers, S.M.; Tomishima, M.J.; Fasano, C.A.; Ganat, Y.M.; Menon, J.; Shimizu, F.; Viale, A.; et al. Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature 2009, 461, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Raya, A.; Rodriguez-Piza, I.; Guenechea, G.; Vassena, R.; Navarro, S.; Barrero, M.J.; Consiglio, A.; Castella, M.; Rio, P.; Sleep, E.; et al. Disease-corrected haematopoietic progenitors from Fanconi anaemia induced pluripotent stem cells. Nature 2009, 460, 53–55. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-Vergara, X.; Sevilla, A.; D’Souza, S.L.; Ang, Y.S.; Schaniel, C.; Lee, D.F.; Yang, L.; Kaplan, A.D.; Adler, E.D.; Rozov, R.; et al. Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature 2010, 465, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Ku, S.; Soragni, E.; Campau, E.; Thomas, E.A.; Altun, G.; Laurent, L.C.; Loring, J.F.; Napierala, M.; Gottesfeld, J.M. Friedreich’s ataxia induced pluripotent stem cells model intergenerational GAATTC triplet repeat instability. Cell Stem Cell 2010, 7, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Moretti, A.; Bellin, M.; Welling, A.; Jung, C.B.; Lam, J.T.; Bott-Flugel, L.; Dorn, T.; Goedel, A.; Hohnke, C.; Hofmann, F.; et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N. Engl. J. Med. 2010, 363, 1397–1409. [Google Scholar] [CrossRef]
- Rashid, S.T.; Corbineau, S.; Hannan, N.; Marciniak, S.J.; Miranda, E.; Alexander, G.; Huang-Doran, I.; Griffin, J.; Ahrlund-Richter, L.; Skepper, J.; et al. Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells. J. Clin. Invest. 2010, 120, 3127–3136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lian, Q.; Zhu, G.; Zhou, F.; Sui, L.; Tan, C.; Mutalif, R.A.; Navasankari, R.; Zhang, Y.; Tse, H.F.; et al. A human iPSC model of Hutchinson Gilford Progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell 2011, 8, 31–45. [Google Scholar] [CrossRef]
- Ryan, S.D.; Dolatabadi, N.; Chan, S.F.; Zhang, X.; Akhtar, M.W.; Parker, J.; Soldner, F.; Sunico, C.R.; Nagar, S.; Talantova, M.; et al. Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in mef2-pgc1alpha transcription. Cell 2013, 155, 1351–1364. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.D.; Ganat, Y.M.; Kishinevsky, S.; Bowman, R.L.; Liu, B.; Tu, E.Y.; Mandal, P.K.; Vera, E.; Shim, J.W.; Kriks, S.; et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 2013, 13, 691–705. [Google Scholar] [CrossRef]
- Park, I.H.; Arora, N.; Huo, H.; Maherali, N.; Ahfeldt, T.; Shimamura, A.; Lensch, M.W.; Cowan, C.; Hochedlinger, K.; Daley, G.Q. Disease-specific induced pluripotent stem cells. Cell 2008, 134, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Soldner, F.; Hockemeyer, D.; Beard, C.; Gao, Q.; Bell, G.W.; Cook, E.G.; Hargus, G.; Blak, A.; Cooper, O.; Mitalipova, M.; et al. Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell 2009, 136, 964–977. [Google Scholar] [CrossRef] [PubMed]
- Hargus, G.; Cooper, O.; Deleidi, M.; Levy, A.; Lee, K.; Marlow, E.; Yow, A.; Soldner, F.; Hockemeyer, D.; Hallett, P.J.; et al. Differentiated Parkinson patient-derived induced pluripotent stem cells grow in the adult rodent brain and reduce motor asymmetry in Parkinsonian rats. Proc. Natl. Acad. Sci. USA 2010, 107, 15921–15926. [Google Scholar] [CrossRef]
- Devine, M.J.; Ryten, M.; Vodicka, P.; Thomson, A.J.; Burdon, T.; Houlden, H.; Cavaleri, F.; Nagano, M.; Drummond, N.J.; Taanman, J.W.; et al. Parkinson’s disease induced pluripotent stem cells with triplication of the alpha-synuclein locus. Nat. Commun. 2011, 2. [Google Scholar] [CrossRef]
- Byers, B.; Cord, B.; Nguyen, H.N.; Schüle, B.; Fenno, L.; Lee, P.C.; Deisseroth, K.; Langston, J.W.; Pera, R.R.; Palmer, T.D. SNCA triplication Parkinson’s patient’s iPSC-derived DA neurons accumulate α-synuclein and are susceptible to oxidative stress. PLoS ONE 2011, 6, e26159. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.Y.; Khurana, V.; Auluck, P.K.; Tardiff, D.F.; Mazzulli, J.R.; Soldner, F.; Baru, V.; Lou, Y.; Freyzon, Y.; Cho, S.; et al. Identification and rescue of α-synuclein toxicity in Parkinson patient-derived neurons. Science 2013, 342, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Mazzulli, J.R.; Xu, Y.H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Schöndorf, D.C.; Aureli, M.; McAllister, F.E.; Hindley, C.J.; Mayer, F.; Schmid, B.; Sardi, S.P.; Valsecchi, M.; Hoffmann, S.; Schwarz, L.K.; et al. iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Ren, Y.; Yuen, E.Y.; Zhong, P.; Ghaedi, M.; Hu, Z.; Azabdaftari, G.; Nakaso, K.; Yan, Z.; Feng, J. Parkin controls dopamine utilization in human midbrain dopaminergic neurons derived from induced pluripotent stem cells. Nat. Commun. 2012, 3. [Google Scholar] [CrossRef]
- Imaizumi, Y.; Okada, Y.; Akamatsu, W.; Koike, M.; Kuzumaki, N.; Hayakawa, H.; Nihira, T.; Kobayashi, T.; Ohyama, M.; Sato, S.; et al. Mitochondrial dysfunction associated with increased oxidative stress and α-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue. Mol. Brain. 2012, 5. [Google Scholar] [CrossRef]
- Seibler, P.; Graziotto, J.; Jeong, H.; Simunovic, F.; Klein, C.; Krainc, D. Mitochondrial Parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. J. Neurosci. 2011, 31, 5970–5976. [Google Scholar] [CrossRef] [PubMed]
- Cooper, O.; Seo, H.; Andrabi, S.; Guardia-Laguarta, C.; Graziotto, J.; Sundberg, M.; McLean, J.R.; Carrillo-Reid, L.; Xie, Z.; Osborn, T.; et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci. Transl. Med. 2012, 4. [Google Scholar] [CrossRef]
- Nguyen, H.N.; Byers, B.; Cord, B.; Shcheglovitov, A.; Byrne, J.; Gujar, P.; Kee, K.; Schule, B.; Dolmetsch, R.E.; Langston, W.; et al. LRRK2 Mutant iPSC-Derived DA Neurons Demonstrate Increased Susceptibility to Oxidative Stress. Cell Stem Cell 2011, 8, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Danes, A.; Richaud-Patin, Y.; Carballo-Carbajal, I.; Jimenez-Delgado, S.; Caig, C.; Mora, S.; Di Guglielmo, C.; Ezquerra, M.; Patel, B.; Giralt, A.; et al. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol. Med. 2012, 4, 380–395. [Google Scholar] [CrossRef] [PubMed]
- Orenstein, S.J.; Kuo, S.-H.; Tasset, I.; Arias, E.; Koga, H.; Fernandez Carasa, I.; Cortes, E.; Honig, L.S.; Dauer, W.; Consiglio, A.; et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci. 2013, 16, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, P.; Schmid, B.; Burbulla, L.F.; Schöndorf, D.C.; Wagner, L.; Glatza, M.; Höing, S.; Hargus, G.; Heck, S.A.; Dhingra, A.; et al. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 2013, 12, 354–367. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.; Simon, D.K. Transcribe to survive: Transcriptional control of antioxidant defense programs for neuroprotection in Parkinson’s disease. Antiox. Redox Signal. 2009, 11, 509–528. [Google Scholar] [CrossRef]
- Chan, P.; Jiang, X.; Forno, L.S.; Di Monte, D.A.; Tanner, C.M.; Langston, J.W. Absence of mutations in the coding region of the alpha-synuclein gene in pathologically proven Parkinson’s disease. Neurology 1998, 50, 1136–1137. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Djarmati, A.; Hedrich, K.; Schäfer, N.; Scaglione, C.; Marchese, R.; Kock, N.; Schüle, B.; Hiller, A.; Lohnau, T.; et al. PINK1, Parkin, and DJ-1 mutations in Italian patients with early-onset parkinsonism. Eur. J. Hum. Genet. 2005, 13, 1086–1093. [Google Scholar] [CrossRef] [PubMed]
- Singleton, A.B.; Farrer, M.J.; Bonifati, V. The genetics of Parkinson’s disease: Progress and therapeutic implications. Mov. Disord. 2013, 28, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.M.M.; Dawson, T.M. Parkin blushed by PINK1. Neuron 2006, 50, 527–529. [Google Scholar] [CrossRef] [PubMed]
- Okatsu, K.; Oka, T.; Iguchi, M.; Imamura, K.; Kosako, H.; Tani, N.; Kimura, M.; Go, E.; Koyano, F.; Funayama, M.; et al. PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat. Commun. 2012, 3. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed]
- Haugarvoll, K.; Rademakers, R.; Kachergus, J.M.; Nuytemans, K.; Ross, O.A.; Gibson, J.M.; Tan, E.K.; Gaig, C.; Tolosa, E.; Goldwurm, S.; et al. LRRK2 R1441C parkinsonism is clinically similar to sporadic Parkinson disease. Neurology 2008, 70, 1456–1460. [Google Scholar] [CrossRef] [PubMed]
- Yahata, N.; Asai, M.; Kitaoka, S.; Takahashi, K.; Asaka, I.; Hioki, H.; Kaneko, T.; Maruyama, K.; Saido, T.C.; Nakahata, T.; et al. Anti-Aβ drug screening platform using human iPS cell-derived neurons for the treatment of Alzheimer’s disease. PLoS ONE 2011, 6, e25788. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D.; Chesi, A.; Geddie, M.L.; Strathearn, K.E.; Hamamichi, S.; Hill, K.J.; Caldwell, K.A.; Caldwell, G.A.; Cooper, A.A.; Rochet, J.S.; et al. α-Synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat. Genet. 2009, 41, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Tardiff, D.F.; Jui, N.T.; Khurana, V.; Tambe, M.A.; Thompson, M.L.; Chung, C.Y.; Kamadurai, H.B.; Kim, H.T.; Lancaster, A.K.; Caldwell, K.A.; et al. Yeast Reveal a “Druggable” Rsp5/Nedd4 Network that Ameliorates α-synuclein Toxicity in Neurons. Science 2013, 343, 979–983. [Google Scholar] [CrossRef]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. α-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Gore, A.; Li, Z.; Fung, H.L.; Young, J.E.; Agarwal, S.; Antosiewicz-Bourget, J.; Canto, I.; Giorgetti, A.; Israel, M.A.; Kiskinis, E.; et al. Somatic coding mutations in human induced pluripotent stem cells. Nature 2011, 471, 63–67. [Google Scholar] [CrossRef]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Marchetto, M.C.; Yeo, G.W.; Kainohana, O.; Marsala, M.; Gage, F.H.; Muotri, A.R. Transcriptional signature and memory retention of human-induced pluripotent stem cells. PLoS ONE 2009, 4, e7076. [Google Scholar] [CrossRef] [PubMed]
- Hussein, S.M.; Batada, N.N.; Vuoristo, S.; Ching, R.W.; Autio, R.; Närvä, E.; Ng, S.; Sourour, M.; Hämäläinen, R.; Olsson, C.; et al. Copy number variation and selection during reprogramming to pluripotency. Nature 2011, 471, 58–62. [Google Scholar] [CrossRef]
- Lister, R.; Pelizzola, M.; Kida, Y.S.; Hawkins, R.D.; Nery, J.R.; Hon, G.; Antosiewicz-Bourget, J.; O’Malley, R.; Castanon, R.; Klugman, S.; et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature 2011, 471, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Laurent, L.C.; Ulitsky, I.; Slavin, I.; Tran, H.; Schork, A.; Morey, R.; Lynch, C.; Harness, J.V.; Lee, S.; Barrero, M.J.; et al. Dynamic changes in the copy number of pluripotency and cell proliferation genes in human escs and ipscs during reprogramming and time in culture. Cell Stem Cell 2011, 8, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Morey, R.; O’Neil, R.C.; He, Y.; Daughtry, B.; Schultz, M.D.; Hariharan, M.; Nery, J.R.; Castanon, R.; Sabatini, K.; et al. Abnormalities in human pluripotent cells due to reprogramming mechanisms. Nature 2014, 511, 177–183. [Google Scholar] [CrossRef]
- Ban, H.; Nishishita, N.; Fusaki, N.; Tabata, T.; Saeki, K.; Shikamura, M.; Takada, N.; Inoue, M.; Hasegawa, M.; Kawamata, S.; et al. Efficient generation of transgene-free human induced plutipotent stem cells (iPSCs) by temperature-sensitive Sendai virus vectors. Proc. Natl. Acad. Sci. USA 2011, 108, 13234–14239. [Google Scholar] [CrossRef]
- Okita, K.; Matsumura, Y.; Sato, Y.; Okada, A.; Morizane, A.; Okamoto, S.; Hong, H.; Nakagawa, M.; Tanabe, K.; Tezuka, K.; et al. A more efficient method to generate integration-free human iPS cells. Nat. Methods 2011, 8, 409–412. [Google Scholar] [CrossRef]
- Kim, D.; Kim, C.H.; Moon, J.I.; Chung, Y.G.; Chang, M.Y.; Han, B.S.; Ko, S.; Yang, E.; Cha, K.Y.; Lanza, R.; et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell 2009, 4, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.H.; Li, H.; Lau, F.; Ebina, W.; MAndal, P.K.; Smith, Z.D.; Meissner, A.; et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef] [PubMed]
- International Parkinson Disease Genomics Consortium; Nalls, M.A.; Plagnol, V.; Hernandez, D.G.; Sharma, M.; Sheerin, U.M.; Saad, M.; Simón-Sánchez, J.; Schulte, C.; Lesage, S.; et al. Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet 2011, 377, 641–649. [Google Scholar]
- Liu, G.H.; Qu, J.; Suzuki, K.; Nivet, E.; Li, M.; Montserrat, N.; Yi, F.; Xu, X.; Ruiz, S.; Zhang, W.; et al. Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature 2012, 491, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Bernitz, J.; Lee, D.F.; Lemischka, I.R. Genomic editing tools to model human diseases with isogenic pluripotent stem cells. Stem Cells Dev. 2014, 23, 2673–2686. [Google Scholar] [CrossRef]
- Kriks, S.; Shim, J.W.; Piao, J.; Ganat, Y.M.; Wakeman, D.R.; Xie, Z.; Carrillo-Reid, L.; Auyeung, G.; Antonacci, C.; Buch, A.; et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 2011, 480, 547–551. [Google Scholar] [PubMed]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2001, 27, 275–280. [Google Scholar] [CrossRef]
- Muroyama, Y.; Fujihara, M.; Ikeya, M.; Kondoh, H.; Takada, S. Wnt signaling plays an essential role in neuronal specification of the dorsal spinal cord. Genes Dev. 2002, 16, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Joksimovic, M.; Yun, B.A.; Kittappa, R.; Anderegg, A.M.; Chang, W.W.; Taketo, M.M.; McKay, R.D.; Awatrami, R.B. Wnt antagonism of Shh facilitates midbrain floor plate neurogenesis. Nat. Neurosci. 2009, 12, 125–131. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torrent, R.; De Angelis Rigotti, F.; Dell'Era, P.; Memo, M.; Raya, A.; Consiglio, A. Using iPS Cells toward the Understanding of Parkinson’s Disease. J. Clin. Med. 2015, 4, 548-566. https://doi.org/10.3390/jcm4040548
Torrent R, De Angelis Rigotti F, Dell'Era P, Memo M, Raya A, Consiglio A. Using iPS Cells toward the Understanding of Parkinson’s Disease. Journal of Clinical Medicine. 2015; 4(4):548-566. https://doi.org/10.3390/jcm4040548
Chicago/Turabian StyleTorrent, Roger, Francesca De Angelis Rigotti, Patrizia Dell'Era, Maurizio Memo, Angel Raya, and Antonella Consiglio. 2015. "Using iPS Cells toward the Understanding of Parkinson’s Disease" Journal of Clinical Medicine 4, no. 4: 548-566. https://doi.org/10.3390/jcm4040548
APA StyleTorrent, R., De Angelis Rigotti, F., Dell'Era, P., Memo, M., Raya, A., & Consiglio, A. (2015). Using iPS Cells toward the Understanding of Parkinson’s Disease. Journal of Clinical Medicine, 4(4), 548-566. https://doi.org/10.3390/jcm4040548