
Recent Insights into the Control of Human Papillomavirus (HPV) Genome Stability, Loss, and Degradation

Abstract
:
1. Introduction
1.2. HPV Infection
1.3. Replicative Phases of the HPV Lifecycle
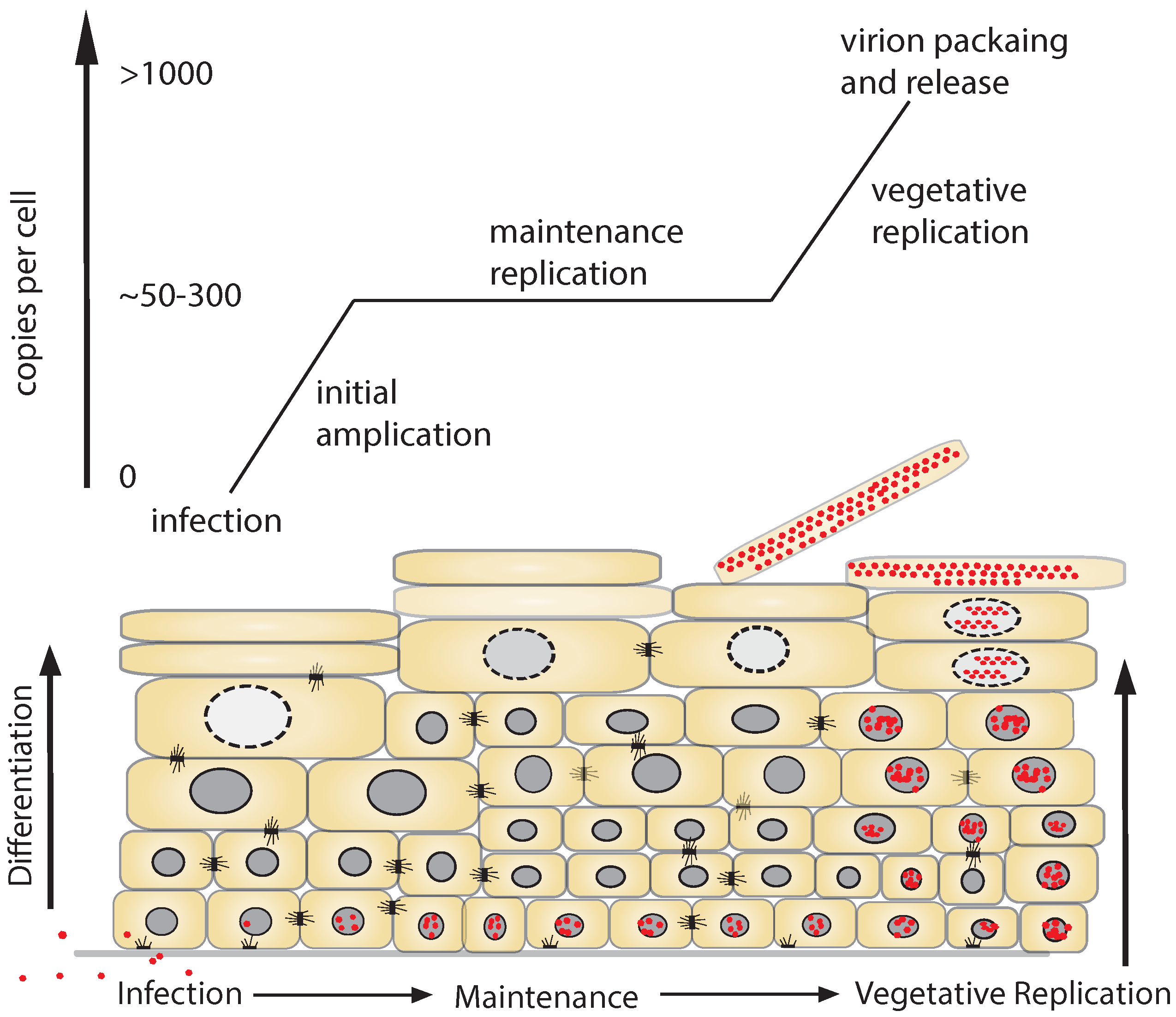
1.4. DNA Damage Response (DDR) and DNA Viruses
2. HPV Episome Stability and Degradation
2.1. Interferon
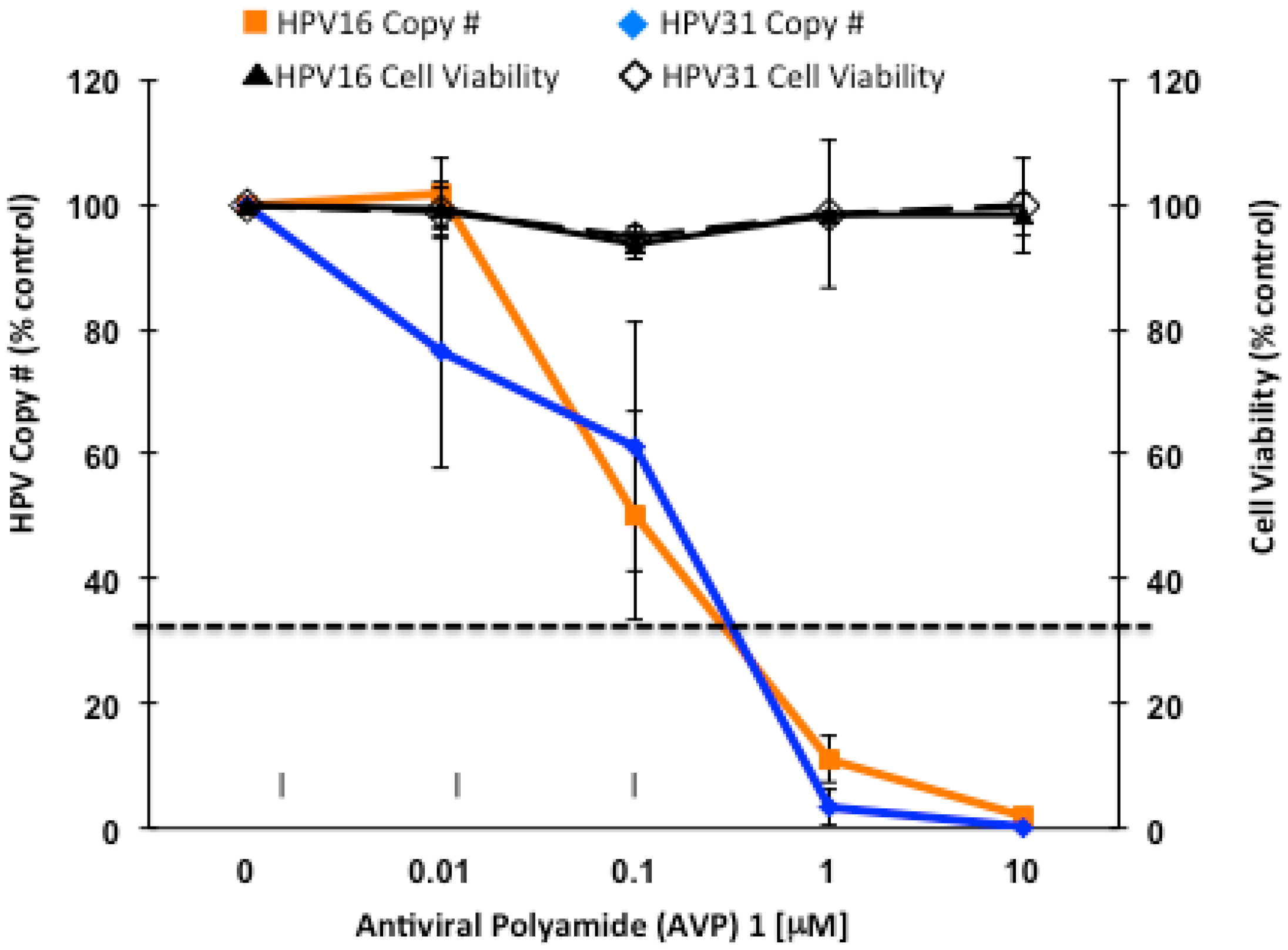
2.2. Antiviral Polyamides (AVPs) that Promote Massive HPV Episome Instability and Degradation
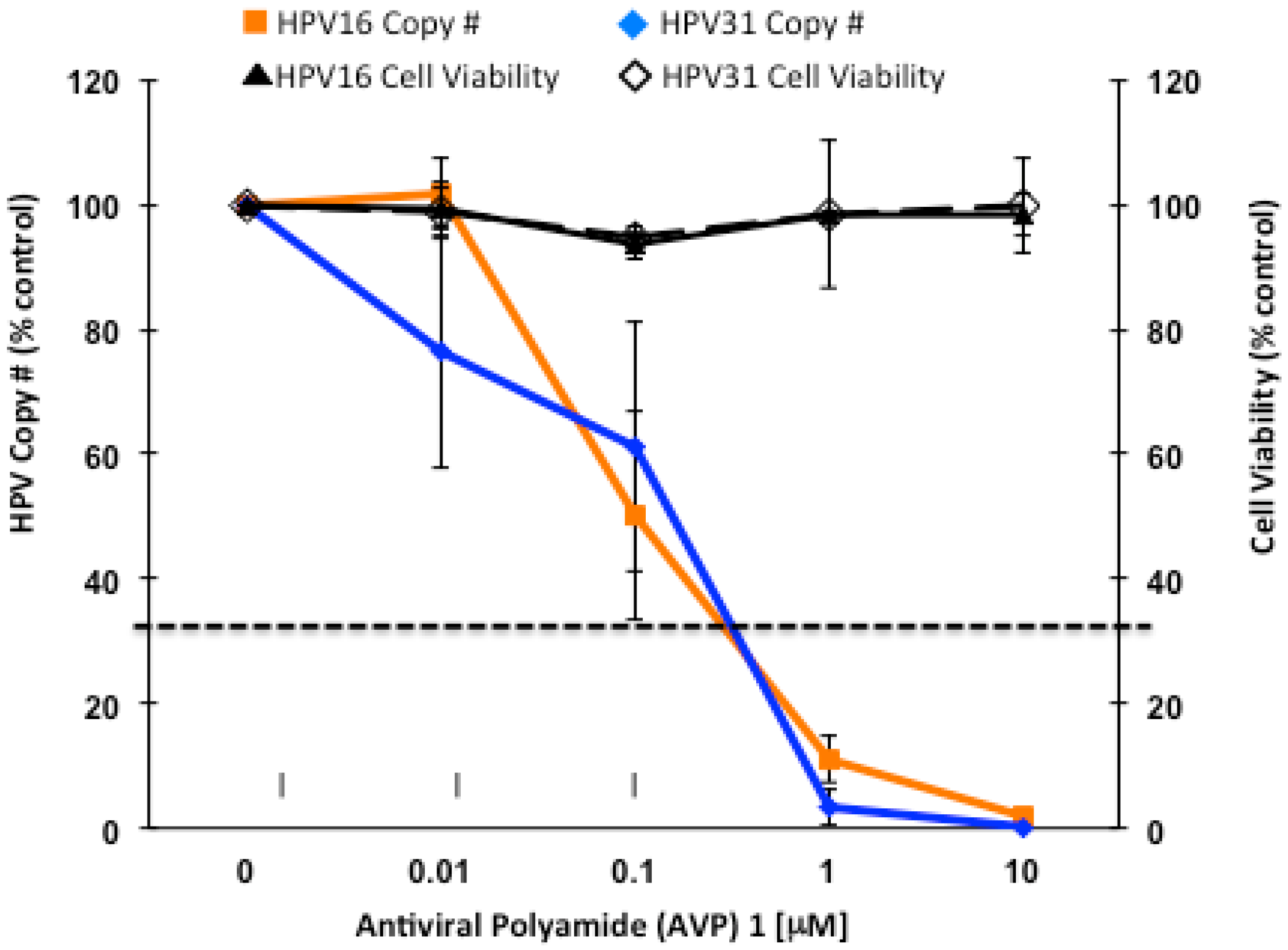
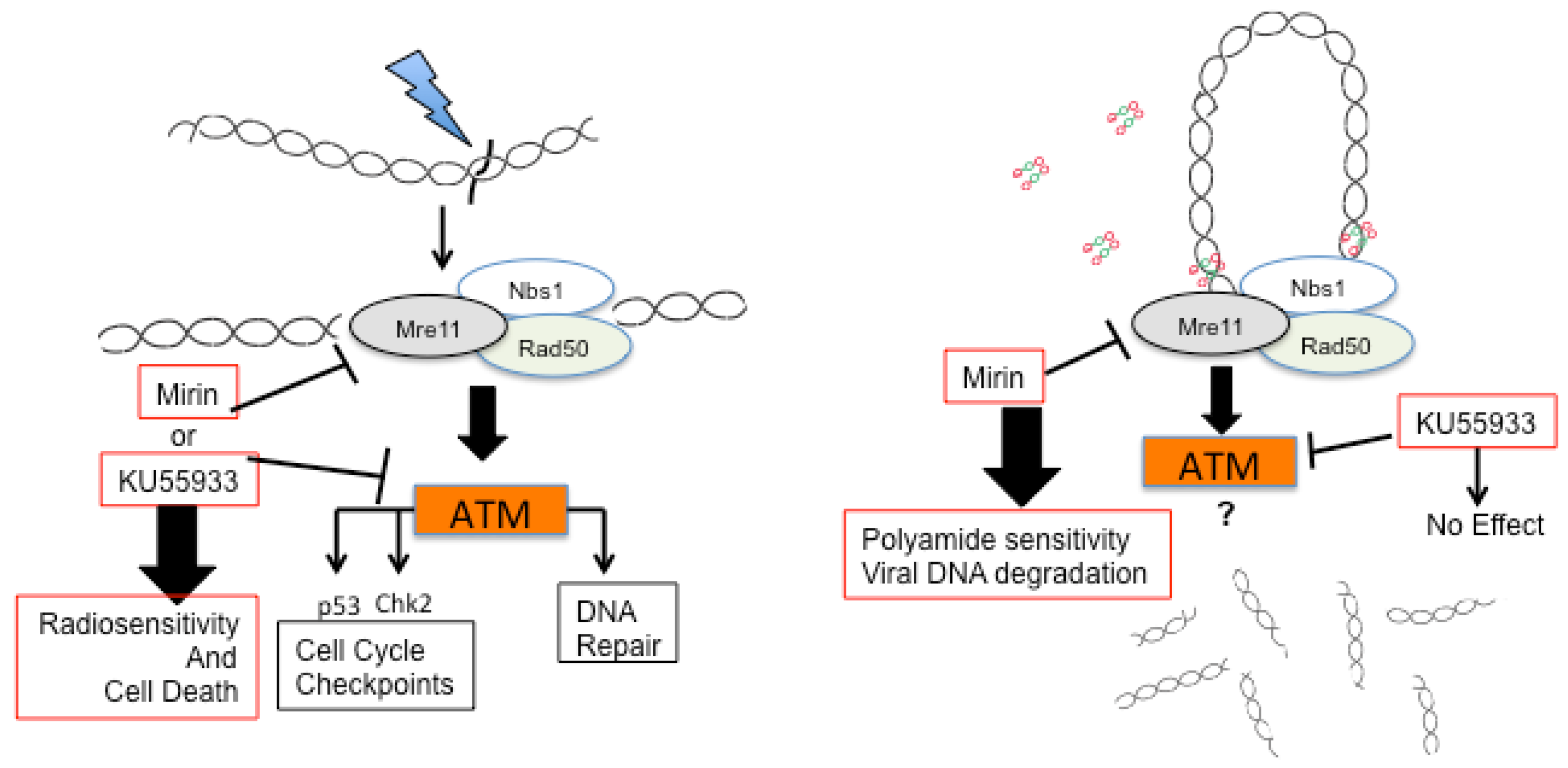
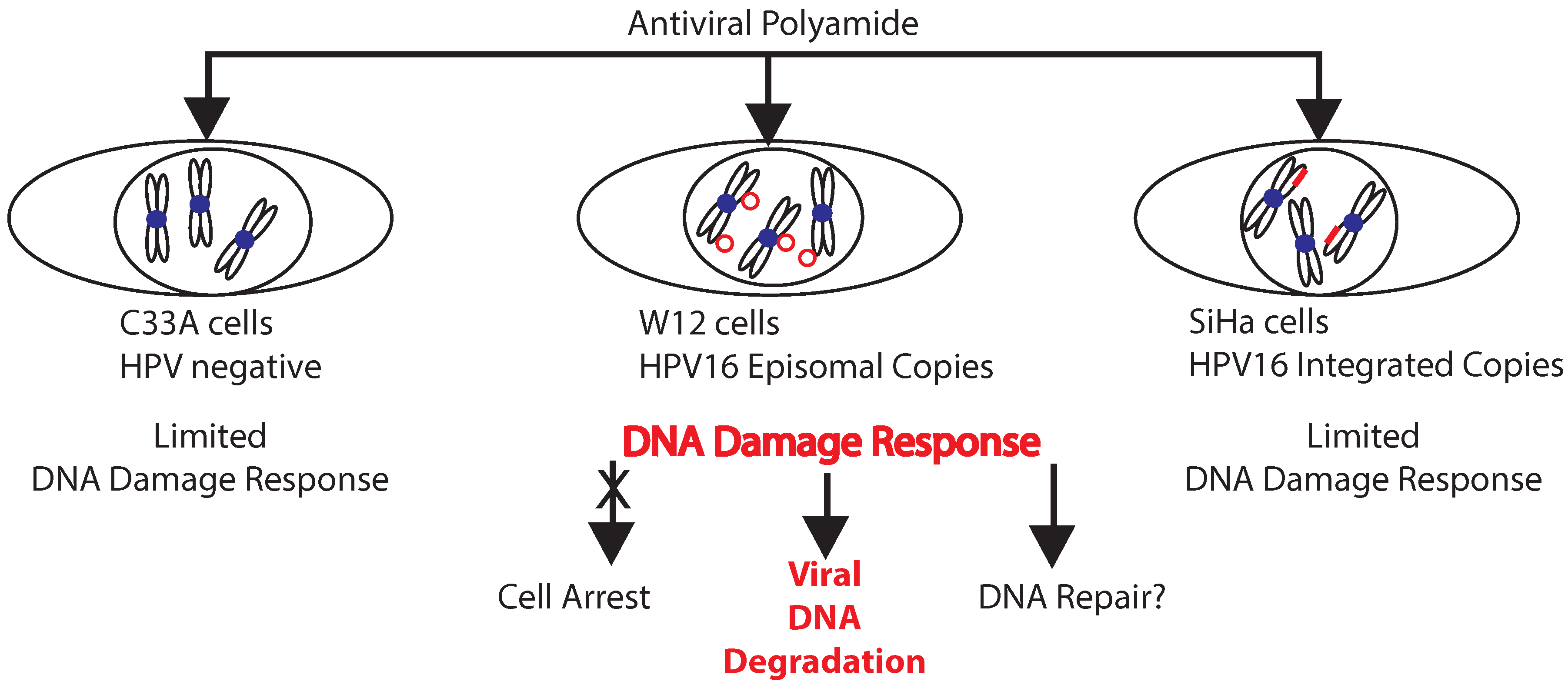
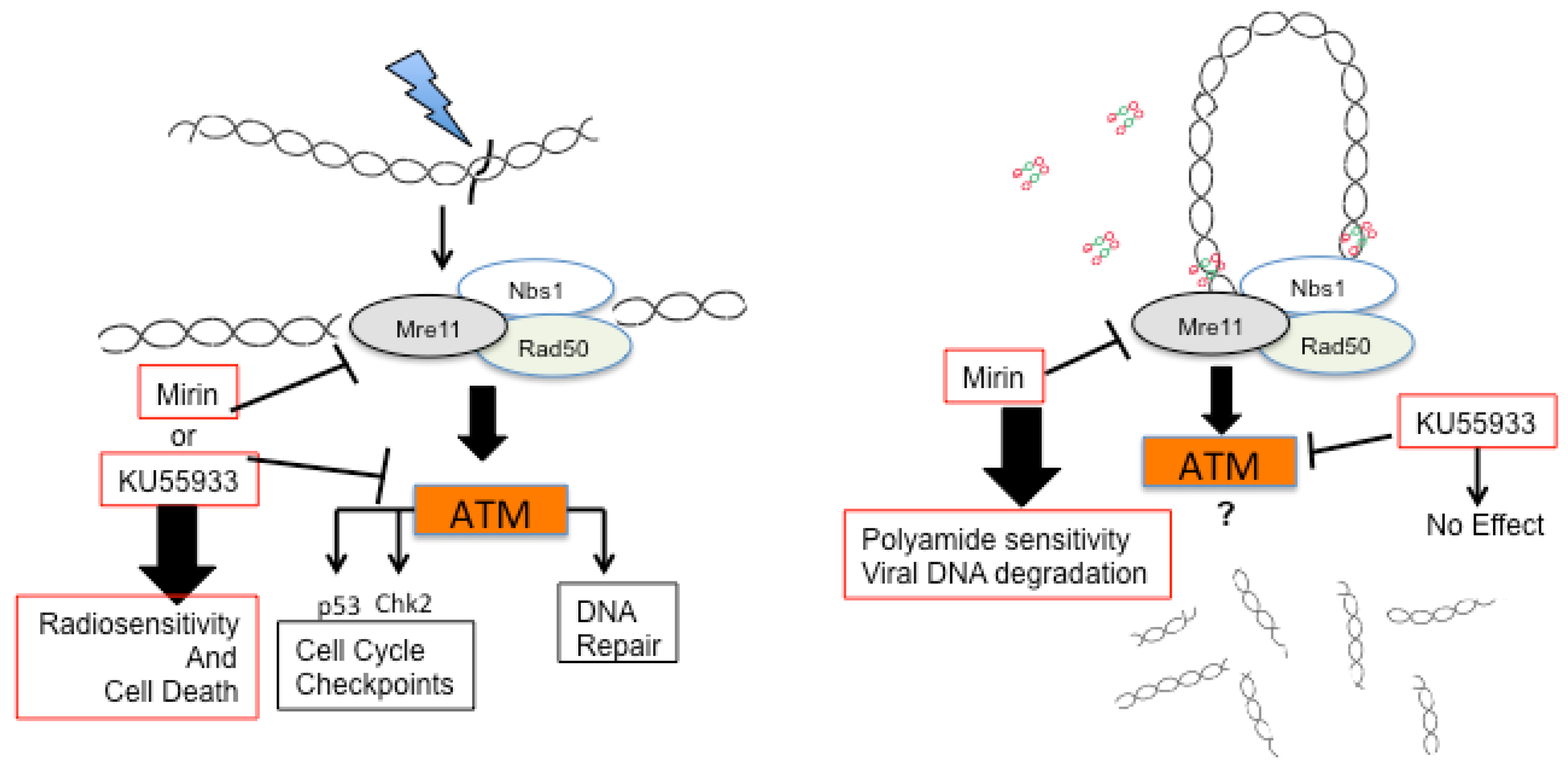
2.3. A Role for DNA Damage Response (DDR) Pathways in HPV Episome Stability, Loss, and Degradation
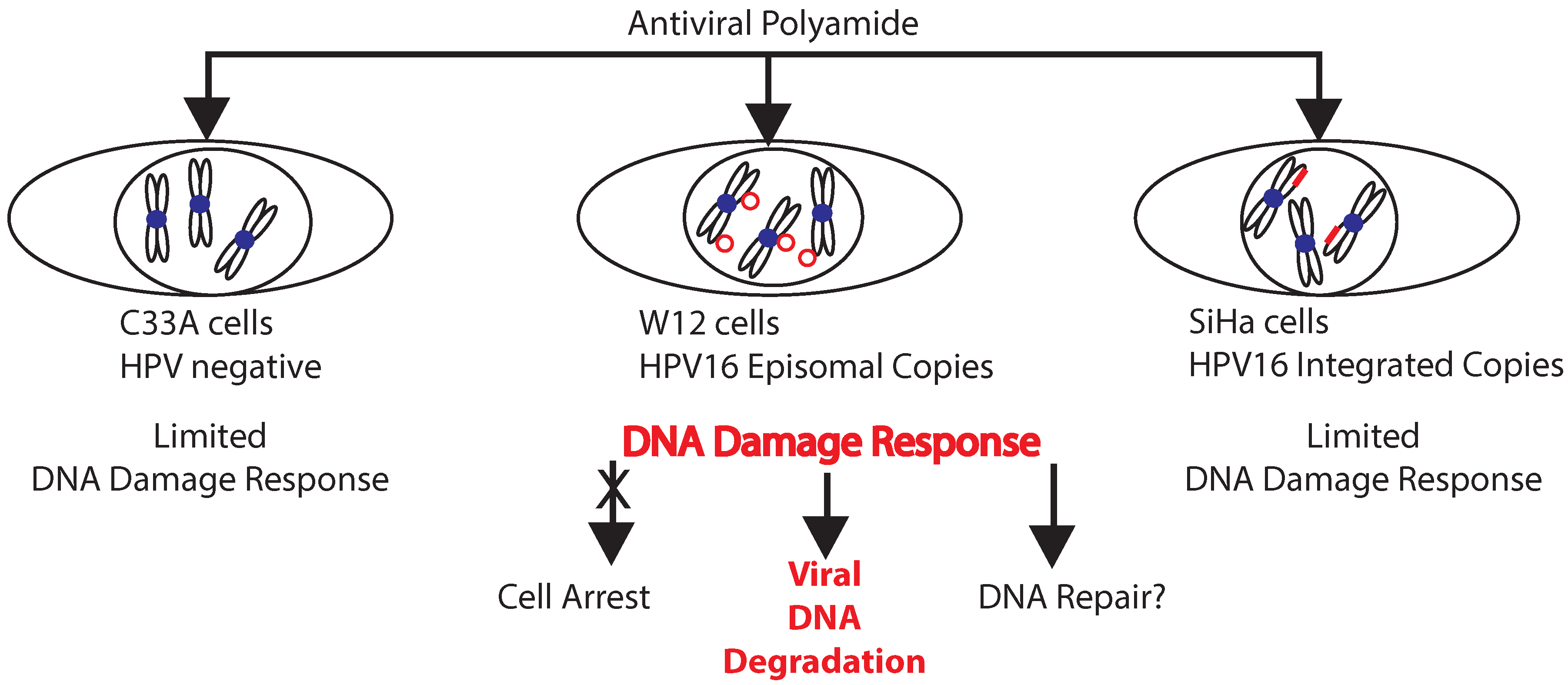
2.4. Enhancers and Repressors of AVP-Mediated HPV DNA Instability
2.5. Identification of an AVP Sensitizer
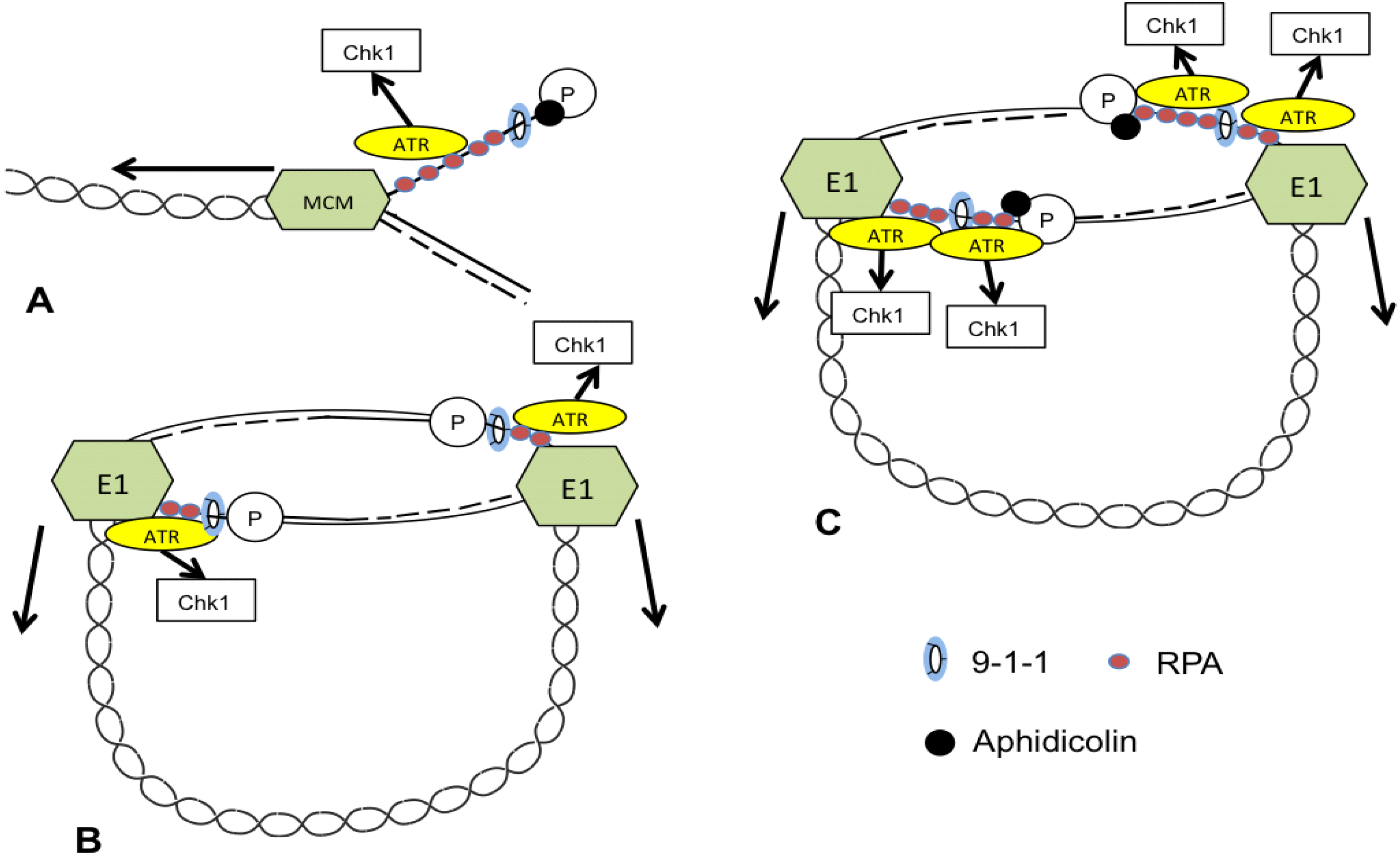
3. ATR and CHK1 Stabilize Episomes during the Maintenance Phase
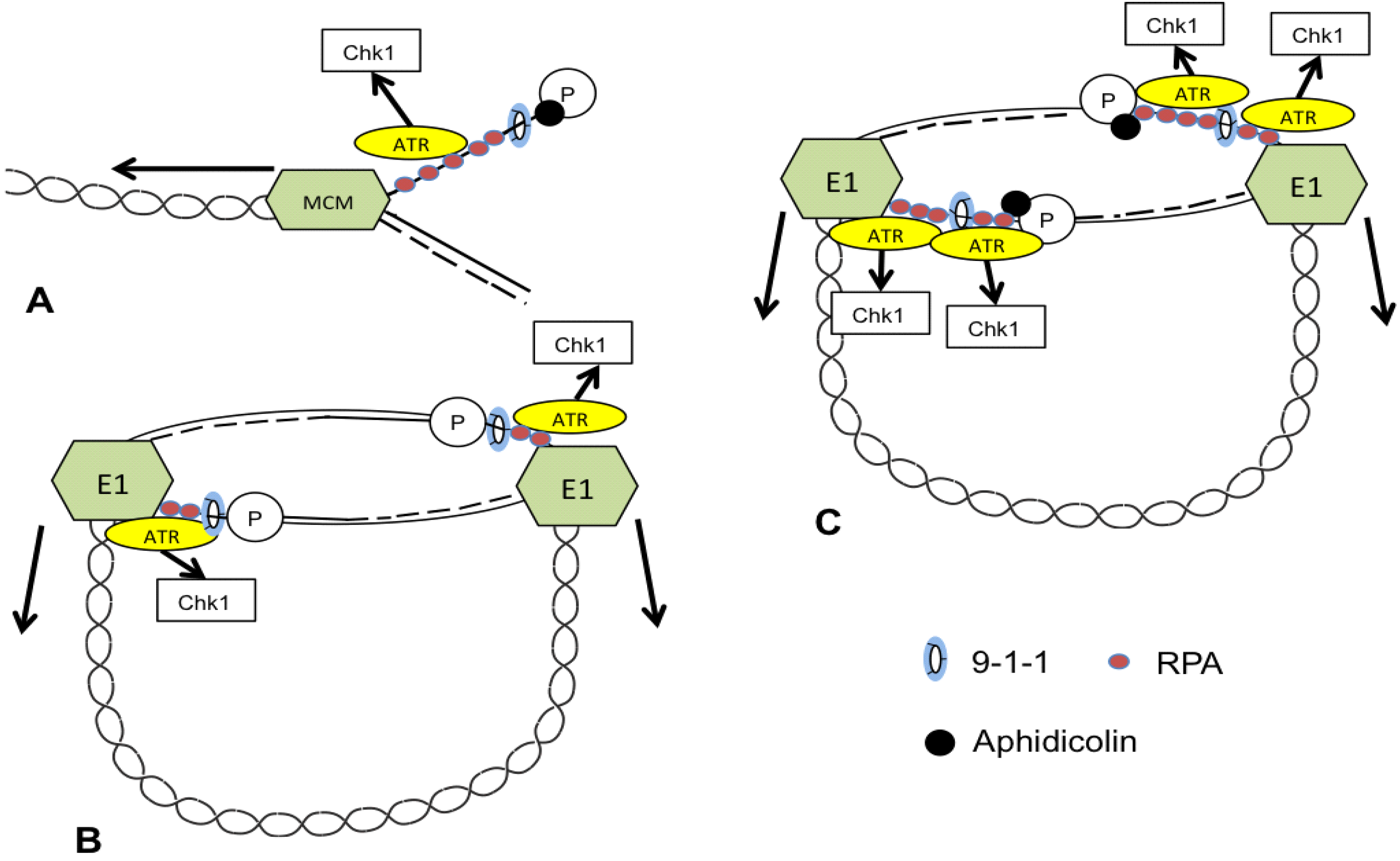
4. Additional Mediators of Episome Stability

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Effect on HPV Episomes [6]; Function | Other Known HPV Roles |
|---|---|---|
| MGMT | Repressor of AVPs; Base excision repair methyltransferase | Proteolytic Target of E6 [139] Promoter methylated in cervical cancer [140] |
| TP73 | Repressor of AVPs; Tumor suppressor; Transcription Factor (P53 family) | Promoter methylated in cervical cancer [140]; linked to cervical carcinogenesis [141] |
| MLH3 | Repressor of AVPs; knockdown decreases normal HPV episome levels; mutL homolog | Associated with risk of HPV infection and cervical carcinogenesis [142] |
| TYMS | Repressor of AVPs; Thymidylate synthetase | Associated with infection by high-risk HPV [143] |
| FAN1 | Repressor of AVPs; FANCD2-associated nuclease; Fanconi anemia pathway | Fanconi anemia pathways implicated in HPV infection [116,144] |
| FANCC | Knockdown decreases normal episome levels; Fanconi anemia pathway | Fanconi anemia pathways implicated in HPV infection [116,144] |
| FANCF | Knockdown increases normal episome levels; Fanconi anemia pathway | Fanconi anemia pathways implicated in HPV infection [116,144] |
| MTOR | Knockdown decreases normal episome levels; kinase, central regulator | MTOR activated by HPV E6 [145]; implicated in HPV infection/entry [146] |
5. TREX1: A Candidate Mediator of Episome Degradation
6. Caveats of Viral DNA Integration
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Chen, H.C.; Schiffman, M.; Lin, C.Y.; Pan, M.H.; You, S.L.; Chuang, L.C.; Hsieh, C.Y.; Liaw, K.L.; Hsing, A.W.; Chen, C.J. Persistence of type-specific human papillomavirus infection and increased long-term risk of cervical cancer. J. Natl. Cancer Inst. 2011, 103, 1387–1396. [Google Scholar] [CrossRef] [PubMed]
- Bodily, J.; Laimins, L.A. Persistence of human papillomavirus infection: Keys to malignant progression. Trends Microbiol. 2011, 19, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Woodman, C.B.; Collins, S.I.; Young, L.S. The natural history of cervical HPV infection: Unresolved issues. Nat. Rev. Cancer 2007, 7, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Edwards, T.G.; Helmus, M.J.; Koeller, K.; Bashkin, J.K.; Fisher, C. Human papillomavirus episome stability is reduced by aphidicolin and controlled by DNA damage response pathways. J. Virol. 2013, 87, 3979–3989. [Google Scholar] [CrossRef] [PubMed]
- Edwards, T.G.; Koeller, K.J.; Slomczynska, U.; Fok, K.; Helmus, M.; Bashkin, J.K.; Fisher, C. HPV episome levels are potently decreased by pyrrole-imidazole polyamides. Antivir. Res. 2011, 91, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Edwards, T.G.; Vidmar, T.J.; Koeller, K.; Bashkin, J.K.; Fisher, C. DNA damage repair genes controlling human papillomavirus (HPV) episome levels under conditions of stability and extreme instability. PLoS One 2013, 8, e75406. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S. DNA degradation in development and programmed cell death. Annu. Rev. Immunol. 2005, 23, 853–875. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.W.; Elledge, S.J. The DNA damage response: Ten years after. Mol. Cell 2007, 28, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Regalado-Magdos, A.D.; Stiggelbout, B.; Lee-Kirsch, M.A.; Lieberman, J. The cytosolic exonuclease trex1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat. Immunol. 2010, 11, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Laguette, N.; Bregnard, C.; Hue, P.; Basbous, J.; Yatim, A.; Larroque, M.; Kirchhoff, F.; Constantinou, A.; Sobhian, B.; Benkirane, M. Premature activation of the SLX4 complex by Vpr promotes G2/M arrest and escape from innate immune sensing. Cell 2014, 156, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, J.; Xia, Y.; Reisinger, F.; Zhang, K.; Stadler, D.; Cheng, X.; Sprinzl, M.F.; Koppensteiner, H.; Makowska, Z.; Volz, T.; et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccdna. Science 2014, 343, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Archambault, J.; Melendy, T. Targeting human papillomavirus genome replication for antiviral drug discovery. Antivir. Ther. 2013, 18, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Fradet-Turcotte, A.; Archambault, J. Recent advances in the search for antiviral agents against human papillomaviruses. Antivir. Ther. 2007, 12, 431–451. [Google Scholar] [PubMed]
- De Martel, C.; Ferlay, J.; Franceschi, S.; Vignat, J.; Bray, F.; Forman, D.; Plummer, M. Global burden of cancers attributable to infections in 2008: A review and synthetic analysis. Lancet Oncol. 2012, 13, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Arbyn, M.; de Sanjose, S.; Saraiya, M.; Sideri, M.; Palefsky, J.; Lacey, C.; Gillison, M.; Bruni, L.; Ronco, G.; Wentzensen, N.; et al. Eurogin 2011 roadmap on prevention and treatment of HPV-related disease. Int. J. Cancer 2012, 131, 1969–1982. [Google Scholar] [CrossRef] [PubMed]
- Schiller, J.T.; Lowy, D.R. Understanding and learning from the success of prophylactic human papillomavirus vaccines. Nat. Rev. Microbiol. 2012, 10, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Van Doorslaer, K. Evolution of the papillomaviridae. Virology 2013, 445, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Chouhy, D.; Bolatti, E.M.; Perez, G.R.; Giri, A.A. Analysis of the genetic diversity and phylogenetic relationships of putative human papillomavirus types. J. Gen. Virol. 2013, 94, 2480–2488. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.M.; Kines, R.C.; Roberts, J.N.; Lowy, D.R.; Schiller, J.T.; Day, P.M. Role of heparan sulfate in attachment to and infection of the murine female genital tract by human papillomavirus. J. Virol. 2009, 83, 2067–2074. [Google Scholar] [CrossRef] [PubMed]
- Laniosz, V.; Dabydeen, S.A.; Havens, M.A.; Meneses, P.I. Human papillomavirus type 16 infection of human keratinocytes requires clathrin and caveolin-1 and is brefeldin a sensitive. J. Virol. 2009, 83, 8221–8232. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Lowy, D.R.; Schiller, J.T. Papillomaviruses infect cells via a clathrin-dependent pathway. Virology 2003, 307, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Schelhaas, M.; Shah, B.; Holzer, M.; Blattmann, P.; Kuhling, L.; Day, P.M.; Schiller, J.T.; Helenius, A. Entry of human papillomavirus type 16 by actin-dependent, clathrin- and lipid raft-independent endocytosis. PLoS Pathog. 2012, 8, e1002657. [Google Scholar] [CrossRef] [PubMed]
- Klucevsek, K.; Daley, J.; Darshan, M.S.; Bordeaux, J.; Moroianu, J. Nuclear import strategies of high-risk HPV18 L2 minor capsid protein. Virology 2006, 352, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Florin, L.; Sapp, M.; Spoden, G.A. Host-cell factors involved in papillomavirus entry. Med. Microbiol. Immunol. 2012, 201, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Kadaja, M.; Silla, T.; Ustav, E.; Ustav, M. Papillomavirus DNA replication—From initiation to genomic instability. Virology 2009, 384, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Garner-Hamrick, P.A.; Fisher, C. HPV episomal copy number closely correlates with cell size in keratinocyte monolayer cultures. Virology 2002, 301, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Maglennon, G.A.; McIntosh, P.; Doorbar, J. Persistence of viral DNA in the epithelial basal layer suggests a model for papillomavirus latency following immune regression. Virology 2011, 414, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life-cycle of human papillomaviruses. Vaccine 2012, 30. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.M.; Ustav, M.; Stenlund, A.; Ho, T.F.; Broker, T.R.; Chow, L.T. Viral-E1 and viral-E2 proteins support replication of homologous and heterologous papillomaviral origins. Proc. Natl. Acad. Sci. USA 1992, 89, 5799–5803. [Google Scholar] [CrossRef] [PubMed]
- Frattini, M.G.; Laimins, L.A. The role of the E1 and E2 proteins in the replication of human papillomavirus type 31b. Virology 1994, 204, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Ozbun, M.A. Human papillomavirus type 31b infection of human keratinocytes and the onset of early transcription. J. Virol. 2002, 76, 11291–11300. [Google Scholar] [CrossRef] [PubMed]
- Stubenrauch, F.; Colbert, A.M.; Laimins, L.A. Transactivation by the E2 protein of oncogenic human papillomavirus type 31 is not essential for early and late viral functions. J. Virol. 1998, 72, 8115–8123. [Google Scholar] [PubMed]
- Ammermann, I.; Bruckner, M.; Matthes, F.; Iftner, T.; Stubenrauch, F. Inhibition of transcription and DNA replication by the papillomavirus E8(lambda)E2C protein is mediated by interaction with corepressor molecules. J. Virol. 2008, 82, 5127–5136. [Google Scholar] [CrossRef] [PubMed]
- Lace, M.J.; Anson, J.R.; Thomas, G.S.; Turek, L.P.; Haugen, T.H. The E8 boolean and E2 gene product of human papillomavirus type 16 represses early transcription and replication but is dispensable for viral plasmid persistence in keratinocytes. J. Virol. 2008, 82, 10841–10853. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, R.; Hirt, B.; Bechtold, V.; Beard, P.; Raj, K. Different modes of human papillomavirus DNA replication during maintenance. J. Virol. 2006, 80, 4431–4439. [Google Scholar] [CrossRef] [PubMed]
- Frattini, M.G.; Lim, H.B.; Laimins, L.A. In vitro synthesis of oncogenic human papillomaviruses requires episomal genomes for differentiation-dependent late expression. Proc. Natl. Acad. Sci. USA 1996, 93, 3062–3067. [Google Scholar] [CrossRef] [PubMed]
- Meyers, C.; Mayer, T.J.; Ozbun, M.A. Synthesis of infectious human papillomavirus type 18 in differentiating epithelium transfected with viral DNA. J. Virol. 1997, 71, 7381–7386. [Google Scholar] [PubMed]
- Stubenrauch, F.; Lim, H.B.; Laimins, L.A. Differential requirements for conserved E2 binding sites in the life cycle of oncogenic human papillomavirus type 31. J. Virol. 1998, 72, 1071–1077. [Google Scholar] [PubMed]
- Thomas, J.T.; Hubert, W.G.; Ruesch, M.N.; Laimins, L.A. Human papillomavirus type 31 oncoproteins E6 and E7 are required for the maintenance of episomes during the viral life cycle in normal human keratinocytes. Proc. Natl. Acad. Sci. USA 1999, 96, 8449–8454. [Google Scholar] [CrossRef] [PubMed]
- Park, R.B.; Androphy, E.J. Genetic analysis of high-risk E6 in episomal maintenance of human papillomavirus genomes in primary human keratinocytes. J. Virol. 2002, 76, 11359–11364. [Google Scholar] [CrossRef] [PubMed]
- Bergvall, M.; Melendy, T.; Archambault, J. The E1 proteins. Virology 2013, 445, 35–56. [Google Scholar] [CrossRef] [PubMed]
- Fradet-Turcotte, A.; Moody, C.; Laimins, L.A.; Archambault, J. Nuclear export of human papillomavirus type 31 E1 is regulated by CDK2 phosphorylation and required for viral genome maintenance. J. Virol. 2010, 84, 11747–11760. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.-H.; Lin, B.Y.; Deng, W.; Broker, T.R.; Chow, L.T. Mitogen-activated protein kinases activated the nuclear localization sequence of human papillomavirus type 11 E1 DNA helicase to promote efficient nuclear import. J. Virol. 2007, 81, 5066–5078. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Fradet-Turcotte, A.; Archambault, J.; Laimins, L.A. Human papillomaviruses activate caspases upon epithelial differentiation to induce viral genome amplification. Proc. Natl. Acad. Sci. USA 2007, 104, 19541–19546. [Google Scholar] [CrossRef] [PubMed]
- Egawa, N.; Nakahara, T.; Ohno, S.-I.; Narisawa-Saito, M.; Yugawa, T.; Fujita, M.; Yamato, K.; Natori, Y.; Kiyono, T. The E1 protein of human papillomavirus type 16 is dispensable for maintenance replication of the viral genome. J. Virol. 2012, 86, 3276–3283. [Google Scholar] [CrossRef] [PubMed]
- Pittayakhajonwut, D.; Angeletti, P.C. Viral trans-factor independent replication of human papillomavirus genomes. Virol. J. 2010, 7. [Google Scholar] [CrossRef] [PubMed]
- Steger, G.; Corbach, S. Dose-dependent regulation of the early promoter of human papillomavirus type 18 by the viral E2 protein. J. Virol. 1997, 71, 50–58. [Google Scholar] [PubMed]
- Zobel, T.; Iftner, T.; Stubenrauch, F. The papillomavirus E8 boolean and E2C protein represses DNA replication from extrachromosomal origins. Mol. Cell Biol. 2003, 23, 8352–8362. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A.; Sakakibara, N.; Stepp, W.H.; Jang, M.K. Hitchhiking on host chromatin: How papillomaviruses persist. Biochim. Biophys. Acta Gene Regul. Mech. 2012, 1819, 820–825. [Google Scholar] [CrossRef]
- McBride, A.A. The papillomavirus e2 proteins. Virology 2013, 445, 57–79. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.; Lambert, P.F. Integration of human papillomavirus type 16 DNA into the human genome leads to increased stability of E6 and E7 mRNAs: Implications for cervical carcinogenesis. Proc. Natl Acad. Sci. USA 1995, 92, 1654–1658. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.; Allen-Hoffmann, B.L.; Lambert, P.F. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J. Virol. 1995, 69, 2989–2997. [Google Scholar] [PubMed]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, E.C.; Yang, E.; Lee, C.J.; Lee, H.W.; DiMaio, D.; Hwang, E.S. Rapid induction of senescence in human cervical carcinoma cells. Proc. Natl. Acad. Sci. USA 2000, 97, 10978–10983. [Google Scholar] [CrossRef] [PubMed]
- Desaintes, C.; Demeret, C.; Goyat, S.; Yaniv, M.; Thierry, F. Expression of the papillomavirus E2 protein in hela cells leads to apoptosis. Embo J. 1997, 16, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Johung, K.; Goodwin, E.C.; DiMaio, D. Human papillomavirus E7 repression in cervical carcinoma cells initiates a transcriptional cascade driven by the retinoblastoma family, resulting in senescence. J. Virol. 2007, 81, 2102–2116. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Drubin, M.E.; Munger, K. The human papillomavirus E7 oncoprotein. Virology 2009, 384, 335–344. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Staples, D.; Smith, C.; Fisher, C. Direct activation of cyclin-dependent kinase 2 by human papillomavirus E7. J. Virol. 2003, 77, 10566–10574. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.L.; Munger, K. Direct association of the HPV16 E7 oncoprotein with cyclin A/CDK2 and cyclin E/CDK2 complexes. Virology 2008, 380, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Howie, H.L.; Katzenellenbogen, R.A.; Galloway, D.A. Papillomavirus E6 proteins. Virology 2009, 384, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Garner-Hamrick, P.A.; Fostel, J.M.; Chien, W.M.; Banerjee, N.S.; Chow, L.T.; Broker, T.R.; Fisher, C. Global effects of human papillomavirus type 18 E6/E7 in an organotypic keratinocyte culture system. J. Virol. 2004, 78, 9041–9050. [Google Scholar] [CrossRef] [PubMed]
- Hummel, M.; Hudson, J.B.; Laimins, L.A. Differentiation-induced and constitutive transcription of human papillomavirus type 31b in cell lines containing viral episomes. J. Virol. 1992, 66, 6070–6080. [Google Scholar] [PubMed]
- Ozbun, M.A.; Meyers, C. Characterization of late gene transcripts expressed during vegetative replication of human papillomavirus type 31b. J. Virol. 1997, 71, 5161–5172. [Google Scholar] [PubMed]
- Ozbun, M.A.; Meyers, C. Temporal usage of multiple promoters during the life cycle of human papillomavirus type 31b. J. Virol. 1998, 72, 2715–2722. [Google Scholar] [PubMed]
- Bedell, M.A.; Hudson, J.B.; Golub, T.R.; Turyk, M.E.; Hosken, M.; Wilbanks, G.D.; Laimins, L.A. Amplification of human papillomavirus genomes in vitro is dependent on epithelial differentiation. J. Virol. 1991, 65, 2254–2260. [Google Scholar] [PubMed]
- Frattini, M.G.; Lim, H.B.; Doorbar, J.; Laimins, L.A. Induction of human papillomavirus type 18 late gene expression and genomic amplification in organotypic cultures from transfected DNA templates. J. Virol. 1997, 71, 7068–7072. [Google Scholar] [PubMed]
- Meyers, C.; Frattini, M.G.; Hudson, J.B.; Laimins, L.A. Biosynthesis of human papillomavirus from a continuous cell line upon epithelial differentiation. Science 1992, 257, 971–973. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, N.S.; Wang, H.-K.; Broker, T.R.; Chow, L.T. Human papillomavirus (HPV) E7 induces prolonged g(2) following s phase reentry in differentiated human keratinocytes. J. Biol. Chem. 2011, 286, 15473–15482. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J. The papillomavirus life cycle. J. Clin. Virol. 2005, 32 (Suppl. S1), 7–15. [Google Scholar] [CrossRef]
- Hong, S.Y.; Laimins, L.A. Regulation of the life cycle of HPVs by differentiation and the DNA damage response. Futur. Microbiol. 2013, 8, 1547–1557. [Google Scholar] [CrossRef]
- Turnell, A.S.; Grand, R.J. DNA viruses and the cellular DNA-damage response. J. Gen. Virol. 2012, 93, 2076–2097. [Google Scholar] [CrossRef] [PubMed]
- McFadden, K.; Luftig, M.A. Interplay between DNA tumor viruses and the host DNA damage response. Curr. Top. Microbiol. Immunol. 2013, 371, 229–257. [Google Scholar] [PubMed]
- Luftig, M. Viruses and the DNA damage response: Activation and antagonism. Annu. Rev. Virol. 2014, 1, 605–625. [Google Scholar] [CrossRef]
- Stracker, T.H.; Usui, T.; Petrini, J.H. Taking the time to make important decisions: The checkpoint effector kinases CHK1 and CHK2 and the DNA damage response. DNA Repair (Amst) 2009, 8, 1047–1054. [Google Scholar] [CrossRef]
- Moody, C.A.; Laimins, L.A. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 2009, 5. [Google Scholar] [CrossRef] [PubMed]
- Fradet-Turcotte, A.; Bergeron-Labrecque, F.; Moody, C.A.; Lehoux, M.; Laimins, L.A.; Archambault, J. Nuclear accumulation of the papillomavirus E1 helicase blocks S-phase progression and triggers an atm-dependent DNA damage response. J. Virol. 2011, 85, 8996–9012. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, N.; Mitra, R.; McBride, A. The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. J. Virol. 2011, 85, 8981–8995. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, K.A.; Mehta, K.P.; Laimins, L.A.; Moody, C.A. Human papillomaviruses recruit cellular DNA repair and homologous recombination factors to viral replication centers. J. Virol. 2012, 86, 9520–9526. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.Y.; Laimins, L.A. The JAK-STAT transcriptional regulator, STAT-5, activates the ATM DNA damage pathway to induce HPV 31 genome amplification upon epithelial differentiation. PLoS Pathog 2013, 9. [Google Scholar] [CrossRef]
- Anacker, D.C.; Gautam, D.; Gillespie, K.A.; Chappell, W.H.; Moody, C.A. Productive replication of human papillomavirus 31 requires DNA repair factor nbs1. J. Virol. 2014, 88, 8528–8544. [Google Scholar] [CrossRef] [PubMed]
- Rockley, P.F.; Tyring, S.K. Interferon-alpha, interferon-beta and interferon-gamma therapy of anogenital human papillomavirus infections. Pharmacol. Ther. 1995, 65, 265–287. [Google Scholar] [CrossRef] [PubMed]
- Turek, L.P.; Byrne, J.C.; Lowy, D.R.; Dvoretzky, I.; Friedman, R.M.; Howley, P.M. Interferon induces morphologic reversion with elimination of extrachromosomal viral genomes in bovine papillomavirus-transformed mouse cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7914–7918. [Google Scholar] [CrossRef] [PubMed]
- Herdman, M.T.; Pett, M.R.; Roberts, I.; Alazawi, W.O.; Teschendorff, A.E.; Zhang, X.Y.; Stanley, M.A.; Coleman, N. Interferon-beta treatment of cervical keratinocytes naturally infected with human papillomavirus 16 episomes promotes rapid reduction in episome numbers and emergence of latent integrants. Carcinogenesis 2006, 27, 2341–2353. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.E.; Pena, L.; Sen, G.C.; Park, J.K.; Laimins, L.A. Long-term effect of interferon on keratinocytes that maintain human papillomavirus type 31. J. Virol. 2002, 76, 8864–8874. [Google Scholar] [CrossRef] [PubMed]
- Pett, M.R.; Herdman, M.T.; Palmer, R.D.; Yeo, G.S.; Shivji, M.K.; Stanley, M.A.; Coleman, N. Selection of cervical keratinocytes containing integrated HPV16 associates with episome loss and an endogenous antiviral response. Proc. Natl. Acad. Sci. USA 2006, 103, 3822–3827. [Google Scholar] [CrossRef] [PubMed]
- Stark, G.R.; Darnell, J.E. The jak-stat pathway at twenty. Immunity 2012, 36, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Y.; Labrecque, S.; Gauzzi, M.C.; Cuddihy, A.R.; Wong, A.H.T.; Pellegrini, S.; Matlashewski, G.J.; Koromilas, A.E. The human papilloma virus (HPV)-18 E6 oncoprotein physically associates with TYK2 and impairs JAK-stat activation by interferon-alpha. Oncogene 1999, 18, 5727–5737. [Google Scholar] [CrossRef] [PubMed]
- Ronco, L.V.; Karpova, A.Y.; Vidal, M.; Howley, P.M. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev. 1998, 12, 2061–2072. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Mehta, K.P.; Laimins, L.A. Suppression of STAT-1 expression by human papillomaviruses is necessary for differentiation-dependent genome amplification and plasmid maintenance. J. Virol. 2011, 85, 9486–9494. [Google Scholar] [CrossRef] [PubMed]
- Petersen-Mahrt, S. DNA deamination in immunity. Immunol. Rev. 2005, 203, 80–97. [Google Scholar] [CrossRef] [PubMed]
- Stenglein, M.D.; Burns, M.B.; Li, M.; Lengyel, J.; Harris, R.S. Apobec3 proteins mediate the clearance of foreign DNA from human cells. Nat. Struct. Mol. Biol. 2010, 17, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Mehta, H.V.; Jones, P.H.; Weiss, J.P.; Okeoma, C.M. IFN-alpha and lipopolysaccharide upregulate APOBEC3 mRNA through different signaling pathways. J. Immunol. 2012, 189, 4088–4103. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.L.; Greene, W.C. The APOBEC3 cytidine deaminases: An innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu. Rev. Immunol. 2008, 26, 317–353. [Google Scholar] [CrossRef] [PubMed]
- Vartanian, J.-P.; Guetard, D.; Henry, M.; Wain-Hobson, S. Evidence for editing of human papillomavirus DNA by APOBEC3 in benign and precancerous lesions. Science 2008, 320, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Warren, C.J.; Xu, T.; Guo, K.; Griffin, L.M.; Westrich, J.A.; Lee, D.; Lambert, P.F.; Santiago, M.L.; Pyeon, D. APOBEC3A functions as a restriction factor of human papillomavirus. J. Virol. 2014. [Google Scholar] [CrossRef]
- Wang, Z.; Wakae, K.; Kitamura, K.; Aoyama, S.; Liu, G.Y.; Koura, M.; Monjurul, A.M.; Kukimoto, I.; Muramatsu, M. APOBEC3 deaminases induce hypermutation in human papillomavirus 16 DNA upon beta interferon stimulation. J. Virol. 2014, 88, 1308–1317. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Campagna, M.; Qi, Y.; Zhao, X.; Guo, F.; Xu, C.; Li, S.; Li, W.; Block, T.M.; Chang, J.; et al. Alpha-interferon suppresses hepadnavirus transcription by altering epigenetic modification of cccdna minichromosomes. PLoS Pathog. 2013, 9, e1003613. [Google Scholar] [CrossRef] [PubMed]
- Reiser, J.; Hurst, J.; Voges, M.; Krauss, P.; Munch, P.; Iftner, T.; Stubenrauch, F. High-risk human papillomaviruses repress constitutive kappa interferon transcription via E6 to prevent pathogen recognition receptor and antiviral-gene expression. J. Virol. 2011, 85, 11372–11380. [Google Scholar] [CrossRef] [PubMed]
- Saikia, P.; Fensterl, V.; Sen, G.C. The inhibitory action of p56 on select functions of E1 mediates interferon’s effect on human papillomavirus DNA replication. J. Virol. 2010, 84, 13036–13039. [Google Scholar] [CrossRef] [PubMed]
- Terenzi, F.; Saikia, P.; Sen, G.C. Interferon-inducible protein, p56, inhibits HPV DNA replication by binding to the viral protein E1. Embo J. 2008, 27, 3311–3321. [Google Scholar] [CrossRef] [PubMed]
- Becker, Y.; Weinber, A. Distamycin a inhibition of epstein-barr virus replication in arginine-deprived burkitt lymphoblasts. Isr. J. Med. Sci. 1972, 8, 75–78. [Google Scholar] [PubMed]
- Verini, M.A.; Ghione, M. Activity of a distamycin a on vaccinia virus infection of cell cultures. Chemotherapy 1964, 9, 144–157. [Google Scholar] [CrossRef] [PubMed]
- Martinez, T.F.; Phillips, J.W.; Karanja, K.K.; Polaczek, P.; Wang, C.M.; Li, B.C.; Campbell, J.L.; Dervan, P.B. Replication stress by Py-Im polyamides induces a non-canonical ATR-dependent checkpoint response. Nucl. Acids Res. 2015, 42, 11546–11559. [Google Scholar] [CrossRef]
- Chenoweth, D.M.; Dervan, P.B. Allosteric modulation of DNA by small molecules. Proc. Natl. Acad. Sci. USA 2009, 106, 13175–13179. [Google Scholar] [CrossRef] [PubMed]
- Chenoweth, D.M.; Dervan, P.B. Structural basis for cyclic Py-Im polyamide allosteric inhibition of nuclear receptor binding. J. Am. Chem. Soc. 2010, 132, 14521–14529. [Google Scholar] [CrossRef] [PubMed]
- Hunt, R.A.; Munde, M.; Kumar, A.; Ismail, M.A.; Farahat, A.A.; Arafa, R.K.; Say, M.; Batista-Parra, A.; Tevis, D.; Boykin, D.W.; et al. Induced topological changes in DNA complexes: Influence of DNA sequences and small molecule structures. Nucl. Acids Res. 2011, 39, 4265–4274. [Google Scholar] [CrossRef] [PubMed]
- Drsata, T.; Zgarbova, M.; Špačková, N.; Jurecka, P.; Sponer, J.; Lankas, F. Mechanical model of DNA allostery. J. Phys. Chem. Lett. 2014, 5, 3831–3835. [Google Scholar] [CrossRef]
- Moretti, R.; Donato, L.J.; Brezinski, M.L.; Stafford, R.L.; Hoff, H.; Thorson, J.S.; Dervan, P.B.; Ansari, A.Z. Targeted chemical wedges reveal the role of allosteric DNA modulation in protein-DNA assembly. ACS Chem. Biol. 2008, 3, 220–229. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Vasilieva, E.; Harris, G.D., Jr.; Koeller, K.J.; Bashkin, J.K.; Dupureur, C.M. Binding studies of a large antiviral polyamide to a natural HPV sequence. Biochimie 2014, 102, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Koeller, K.J.; Harris, G.D.; Aston, K.; He, G.; Castaneda, C.H.; Thornton, M.A.; Edwards, T.G.; Wang, S.; Nanjunda, R.; Wilson, W.D.; et al. DNA binding polyamides and the importance of DNA recognition in their use as gene-specific and antiviral agents. Med. Chem. (Los Angeles) 2014, 4, 338–344. [Google Scholar]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kacmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Bai, L.; Gong, Y.; Xie, W.; Hang, H.; Jiang, T. Structure and functional implications of the human RAD9-HUS1-RAD1 cell cycle checkpoint complex. J. Biol. Chem. 2009, 284, 20457–20461. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.J.; Lees-Miller, S.P.; Tainer, J.A. MRE11-RAD50-NBS1 conformations and the control of sensing, signaling, and effector responses at DNA double-strand breaks. DNA Repair (Amst) 2010, 9, 1299–1306. [Google Scholar] [CrossRef]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human ctip promotes DNA end resection. Nature 2007, 450, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, E.E.; Morreale, R.J.; Werner, S.P.; Higginbotham, J.M.; Laimins, L.A.; Lambert, P.F.; Brown, D.R.; Gillison, M.L.; Nuovo, G.J.; Witte, D.P.; et al. The fanconi anemia pathway limits human papillomavirus replication. J. Virol. 2012, 86, 8131–8138. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, E.E.; Morris, T.A.; Higginbotham, J.M.; Spardy, N.; Cha, E.; Kelly, P.; Williams, D.A.; Wikenheiser-Brokamp, K.A.; Duensing, S.; Wells, S.I. Fanconi anemia deficiency stimulates HPV-associated hyperplastic growth in organotypic epithelial raft culture. Oncogene 2009, 28, 674–685. [Google Scholar] [CrossRef] [PubMed]
- Romick-Rosendale, L.E.; Lui, V.W.; Grandis, J.R.; Wells, S.I. The fanconi anemia pathway: Repairing the link between DNA damage and squamous cell carcinoma. Mutat. Res. 2013, 743–744, 78–88. [Google Scholar]
- Dupre, A.; Boyer-Chatenet, L.; Sattler, R.M.; Modi, A.P.; Lee, J.H.; Nicolette, M.L.; Kopelovich, L.; Jasin, M.; Baer, R.; Paull, T.T.; et al. A forward chemical genetic screen reveals an inhibitor of the MRE11-RAD50-NBS1 complex. Nat. Chem. Biol. 2008, 4, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Garner, K.M.; Pletnev, A.A.; Eastman, A. Corrected structure of mirin, a small-molecule inhibitor of the MRE11-RAD50-NBS1 complex. Nat. Chem. Biol. 2009, 5, 129–130. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, J.F.S.; Kanaar, R. Targeting homologous recombination-mediated DNA repair in cancer. Expert Opin. Ther. Targets 2014, 18, 427–458. [Google Scholar] [CrossRef] [PubMed]
- Kastan, M.B. DNA damage responses: Mechanisms and roles in human disease: 2007 g.H.A. Mol. Cancer Res. 2008, 6, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, S.; Urata, Y.; Fujiwara, T. Ataxia-telangiectasia mutated and the MRE11-RAD50-NBS1 complex: Promising targets for radiosensitization. Acta Med. Okayama 2012, 66, 83–92. [Google Scholar] [PubMed]
- Buis, J.; Wu, Y.P.; Deng, Y.B.; Leddon, J.; Westfield, G.; Eckersdorff, M.; Sekiguchi, J.M.; Chang, S.; Ferguson, D.O. MRE11 nuclease activity has essential roles in DNA repair and genomic stability distinct from atm activation. Cell 2008, 135, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.S.; Moncalian, G.; Williams, J.S.; Yamada, Y.; Limbo, O.; Shin, D.S.; Groocock, L.M.; Cahill, D.; Hitomi, C.; Guenther, G.; et al. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell 2008, 135, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Kadaja, M.; Isok-Paas, H.; Laos, T.; Ustav, E.; Ustav, M. Mechanism of genomic instability in cells infected with the high-risk human papillomaviruses. PLoS Pathog. 2009, 5, e1000397. [Google Scholar] [CrossRef] [PubMed]
- Daniel, J.A.; Pellegrini, M.; Lee, B.S.; Guo, Z.; Filsuf, D.; Belkina, N.V.; You, Z.; Paull, T.T.; Sleckman, B.P.; Feigenbaum, L.; et al. Loss of atm kinase activity leads to embryonic lethality in mice. J. Cell Biol. 2012, 198, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Wang, Y.; Jiang, W.; Liu, X.; Dubois, R.L.; Lin, C.S.; Ludwig, T.; Bakkenist, C.J.; Zha, S. Kinase-dead ATM protein causes genomic instability and early embryonic lethality in mice. J. Cell Biol. 2012, 198, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L.; Nitiss, K.C. Tdp2: A means to fixing the ends. PLoS Genet. 2013, 9, e1003370. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Huang, S.Y.; Gao, R.; Das, B.B.; Murai, J.; Marchand, C. Tyrosyl-DNA-phosphodiesterases (TDP1 and TDP2). DNA Repair (Amst) 2014, 19, 114–129. [Google Scholar] [CrossRef]
- Pype, S.; Declercq, W.; Ibrahimi, A.; Michiels, C.; van Rietschoten, J.G.; Dewulf, N.; de Boer, M.; Vandenabeele, P.; Huylebroeck, D.; Remacle, J.E. Ttrap, a novel protein that associates with cd40, tumor necrosis factor (TNF) receptor-75 and TNF receptor-associated factors (TRAFs), and that inhibits nuclear factor-kappa b activation. J. Biol. Chem. 2000, 275, 18586–18593. [Google Scholar] [CrossRef] [PubMed]
- Virgen-Slane, R.; Rozovics, J.M.; Fitzgerald, K.D.; Ngo, T.; Chou, W.; van der, H.; van Noort, G.J.; Filippov, D.V.; Gershon, P.D.; Semler, B.L. An RNA virus hijacks an incognito function of a DNA repair enzyme. Proc. Natl. Acad. Sci. USA 2012, 109, 14634–14639. [Google Scholar] [CrossRef] [PubMed]
- Koniger, C.; Wingert, I.; Marsmann, M.; Rosler, C.; Beck, J.; Nassal, M. Involvement of the host DNA-repair enzyme TDP2 in formation of the covalently closed circular DNA persistence reservoir of hepatitis B viruses. Proc. Natl. Acad. Sci. USA 2014, 111. [Google Scholar] [CrossRef]
- Tosi, A.; Haas, C.; Herzog, F.; Gilmozzi, A.; Berninghausen, O.; Ungewickell, C.; Gerhold, C.B.; Lakomek, K.; Aebersold, R.; Beckmann, R.; et al. Structure and subunit topology of the ino80 chromatin remodeler and its nucleosome complex. Cell 2013, 154, 1207–1219. [Google Scholar] [CrossRef] [PubMed]
- Nano, N.; Houry, W.A. Chaperone-like activity of the AAA+ proteins Rvb1 and Rvb2 in the assembly of various complexes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368. [Google Scholar] [CrossRef] [PubMed]
- Vannier, J.B.; Sarek, G.; Boulton, S.J. Rtel1: Functions of a disease-associated helicase. Trends Cell Biol. 2014, 24, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Uringa, E.J.; Youds, J.L.; Lisaingo, K.; Lansdorp, P.M.; Boulton, S.J. Rtel1: An essential helicase for telomere maintenance and the regulation of homologous recombination. Nucl. Acids Res. 2011, 39, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Barber, L.J.; Youds, J.L.; Ward, J.D.; McIlwraith, M.J.; OʼNeil, N.J.; Petalcorin, M.I.; Martin, J.S.; Collis, S.J.; Cantor, S.B.; Auclair, M.; et al. RTEL1 maintains genomic stability by suppressing homologous recombination. Cell 2008, 135, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Srivenugopal, K.S.; Ali-Osman, F. The DNA repair protein, O-6-methylguanine-DNA methyltransferase is a proteolytic target for the E6 human papillomavirus oncoprotein. Oncogene 2002, 21, 5940–5945. [Google Scholar] [CrossRef] [PubMed]
- Henken, F.E.; Wilting, S.M.; Overmeer, R.M.; van Rietschoten, J.G.; Nygren, A.O.; Errami, A.; Schouten, J.P.; Meijer, C.J.; Snijders, P.J.; Steenbergen, R.D. Sequential gene promoter methylation during hpv-induced cervical carcinogenesis. Br. J. Cancer 2007, 97, 1457–1464. [Google Scholar] [CrossRef] [PubMed]
- Craveiro, R.; Costa, S.; Pinto, D.; Salgado, L.; Carvalho, L.; Castro, C.; Bravo, I.; Lopes, C.; Silva, I.; Medeiros, R. Tp73 alterations in cervical carcinoma. Cancer Genet. Cytogenet. 2004, 150, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Cheng, Q.; Shen, J.; Zhou, C.; Chen, H. Mismatch repair gene MLH3 Pro844Leu and Thr942ile polymorphisms and the susceptibility to cervical carcinoma and HPV infection: A case-control study in a chinese population. PLoS One 2014, 9, e96224. [Google Scholar] [CrossRef] [PubMed]
- Famooto, A.; Almujtaba, M.; Dareng, E.; Akarolo-Anthony, S.; Ogbonna, C.; Offiong, R.; Olaniyan, O.; Wheeler, C.M.; Doumatey, A.; Rotimi, C.N.; et al. RPS19 and TYMS SNPs and prevalent high risk human papilloma virus infection in nigerian women. PLoS One 2013, 8, e66930. [Google Scholar] [CrossRef] [PubMed]
- De Araujo, M.R.; Rubira-Bullen, I.R.; Santos, C.F.; Dionisio, T.J.; Bonfim, C.M.; de Marco, L.; Gillio-Tos, A.; Merletti, F. High prevalence of oral human papillomavirus infection in fanconiʼs anemia patients. Oral Dis. 2011, 17, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Spangle, J.M.; Munger, K. The human papillomavirus type 16 E6 oncoprotein activates MTORC1 signaling and increases protein synthesis. J. Virol. 2010, 84, 9398–9407. [Google Scholar] [CrossRef] [PubMed]
- Surviladze, Z.; Sterk, R.T.; DeHaro, S.A.; Ozbun, M.A. Cellular entry of human papillomavirus type 16 involves activation of the phosphatidylinositol 3-kinase/Akt/Mtor pathway and inhibition of autophagy. J. Virol. 2013, 87, 2508–2517. [Google Scholar] [CrossRef] [PubMed]
- De Lange, T. Protection of mammalian telomeres. Oncogene 2002, 21, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.; Yan, N. Safeguard against DNA sensing: The role of TREX1 in HIV-1 infection and autoimmune diseases. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Mazur, D.J.; Perrino, F.W. Identification and expression of the trex1 and trex2 cdna sequences encoding mammalian 3′-->5′ exonucleases. J. Biol. Chem. 1999, 274, 19655–19660. [Google Scholar] [CrossRef] [PubMed]
- Stetson, D.B.; Ko, J.S.; Heidmann, T.; Medzhitov, R. TREX1 prevents cell-intrinsic initiation of autoimmunity. Cell 2008, 134, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.L.; Harvey, S.; Perrino, F.W.; Hollis, T. Defects in DNA degradation revealed in crystal structures of trex1 exonuclease mutations linked to autoimmune disease. DNA Repair (Amst) 2012, 11, 65–73. [Google Scholar] [CrossRef]
- Lieberman, J. Granzyme a activates another way to die. Immunol. Rev. 2010, 235, 93–104. [Google Scholar] [PubMed]
- Chowdhury, D.; Beresford, P.J.; Zhu, P.; Zhang, D.; Sung, J.S.; Demple, B.; Perrino, F.W.; Lieberman, J. The exonuclease TREX1 is in the set complex and acts in concert with NM23-H1 to degrade DNA during granzyme a-mediated cell death. Mol. Cell 2006, 23, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, E.; Freese, U.K.; Gissmann, L.; Mayer, W.; Roggenbuck, B.; Stremlau, A.; Hausen, H.Z. Structure and transcription of human papillomavirus sequences in cervical-carcinoma cells. Nature 1985, 314, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Bernard, B.A.; Bailly, C.; Lenoir, M.C.; Darmon, M.; Thierry, F.; Yaniv, M. The human papillomavirus type-18 (HPV18) E2 gene-product is a repressor of the HPV18 regulatory region in human keratinocytes. J. Virol. 1989, 63, 4317–4324. [Google Scholar] [PubMed]
- Thierry, F.; Howley, P.M. Functional-analysis of E2-mediated repression of the HPV18 p105 promoter. New Biol. 1991, 3, 90–100. [Google Scholar] [PubMed]
- Choo, K.B.; Pan, C.C.; Han, S.H. Integration of human papillomavirus type 16 into cellular DNA of cervical carcinoma: Preferential deletion of the E2 gene and invariable retention of the long control region and the E6/E7 open reading frames. Virology 1987, 161, 259–261. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.P.; Friedman, C.L.; Bryant, E.M.; McDougall, J.K. Viral integration and fragile sites in human papillomavirus-immortalized human keratinocyte cell lines. Genes Chromosom. Cancer 1992, 5, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Thorland, E.C.; Myers, S.L.; Gostout, B.S.; Smith, D.I. Common fragile sites are preferential targets for HPV16 integrations in cervical tumors. Oncogene 2003, 22, 1225–1237. [Google Scholar] [CrossRef] [PubMed]
- Durkin, S.G.; Glover, T.W. Chromosome fragile sites. Annu. Rev. Genet. 2007, 41, 169–192. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.K.; Shen, K.; McBride, A.A. Papillomavirus genomes associate with brd4 to replicate at fragile sites in the host genome. PLoS Pathog. 2014, 10, e1004117. [Google Scholar] [CrossRef] [PubMed]
- Matzner, I.; Savelyeva, L.; Schwab, M. Preferential integration of a transfected marker gene into spontaneously expressed fragile sites of a breast cancer cell line. Cancer Lett. 2003, 189, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Ferber, M.J.; Cheung, T.H.; Chung, T.K.; Wong, Y.F.; Smith, D.I. The role of viral integration in the development of cervical cancer. Cancer Genet. Cytogenet. 2005, 158, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Winder, D.M.; Pett, M.R.; Foster, N.; Shivji, M.K.; Herdman, M.T.; Stanley, M.A.; Venkitaraman, A.R.; Coleman, N. An increase in DNA double-strand breaks, induced by Ku70 depletion, is associated with human papillomavirus 16 episome loss and de novo viral integration events. J. Pathol. 2007, 213, 27–34. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fisher, C. Recent Insights into the Control of Human Papillomavirus (HPV) Genome Stability, Loss, and Degradation. J. Clin. Med. 2015, 4, 204-230. https://doi.org/10.3390/jcm4020204
Fisher C. Recent Insights into the Control of Human Papillomavirus (HPV) Genome Stability, Loss, and Degradation. Journal of Clinical Medicine. 2015; 4(2):204-230. https://doi.org/10.3390/jcm4020204
Chicago/Turabian StyleFisher, Chris. 2015. "Recent Insights into the Control of Human Papillomavirus (HPV) Genome Stability, Loss, and Degradation" Journal of Clinical Medicine 4, no. 2: 204-230. https://doi.org/10.3390/jcm4020204
APA StyleFisher, C. (2015). Recent Insights into the Control of Human Papillomavirus (HPV) Genome Stability, Loss, and Degradation. Journal of Clinical Medicine, 4(2), 204-230. https://doi.org/10.3390/jcm4020204