The Use of Patient-Specific Induced Pluripotent Stem Cells (iPSCs) to Identify Osteoclast Defects in Rare Genetic Bone Disorders

Abstract
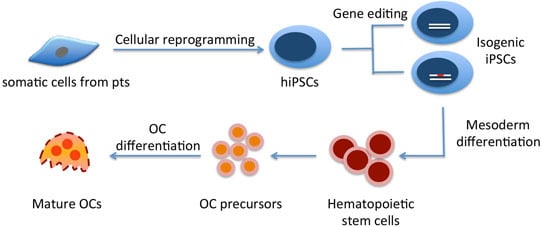
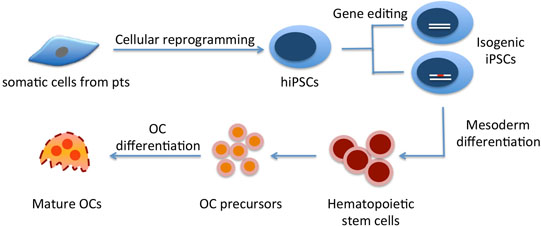
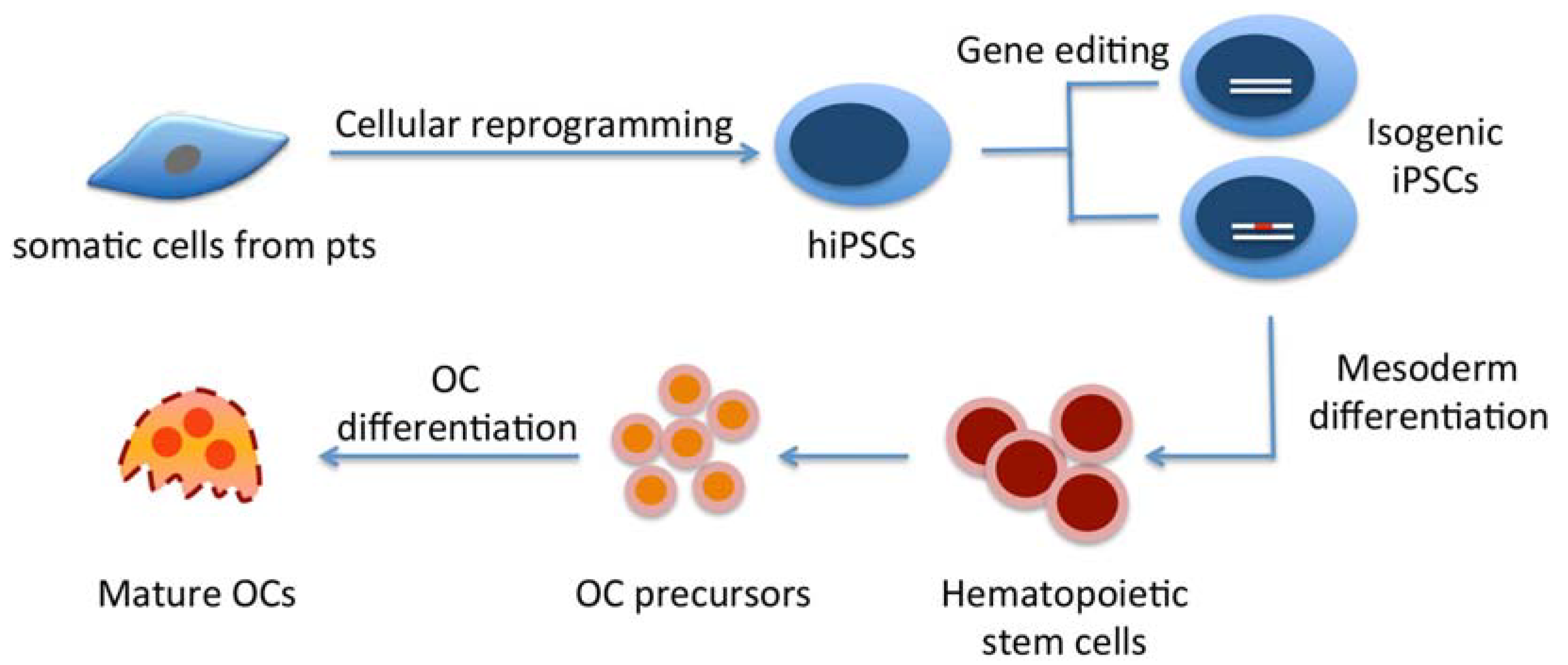
:
1. Introduction
2. Rare Genetic Bone Disorders with Osteoclast Defects
2.1. Diseases of Decreased Osteoclast Resorption
2.2. Diseases of Increased Osteoclast Resorption

{kind=link}
{kind=link}
| Diseases with Decreased Bone Resorption | ||||
|---|---|---|---|---|
| Disease | OMIM | Gene affected | Protein affected | Reference(s) |
| ARO | 259,700 | TCIRG1 | α3 Subunit of vacuolar proton pump H+ ATPase | [1,25] |
| ARO | 259,700 | CLCN7 | Chloride channel | [26] |
| ARO | 259,700 | OSTM1 | GL | [27] |
| IARO | 259,730 | CAII | Carbonic anhydrase II | [12] |
| ADOI | 166,600 | Lrp5 | Lrp5 | [28] |
| ADOII | 166,600 | CLCN7 | Chloride channel | [29] |
| Pycnodysostosis | 265,800 | CTSK | Cathepsin K | [14] |
| PDB | 6,002,080 | SQSTM1 | P62 | [30,31] |
| JPDB | 239,000 | TNFRSF11B | Osteoprotegerin (OPG) | [19] |
| FEO | 174,810 | TNFRSF11A | RANK | [23] |
| ESH | N/A | TNFRSF11A | RANK | [32] |
3. Generation of hiPSCs from Rare Genetic Bone Disorders
| Disease | Source of Somatic Cells | Method | Reprogramming Factors | Patient Numbers | Reference |
|---|---|---|---|---|---|
| OI | MSC derived from bone fragments | (1) lentivirus | (1) OCT4, SOX2, LIN28 or NANOG | 6 | [56] |
| (2) floxed, polycystronic foamy virus | (2) OCT4, SOX2, KLF4 and c-MYC | ||||
| CMD | 5–7 mL peripheral blood | Sendai virus | OCT3/4, SOX2, KLF4 and c-MYC | 8 | [57] |
| FOP | Dermal fibroblasts | (1) retrovirus | (1) OCT4, SOX2, KLF4 and c-MYC | 5 | [58] |
| (2) episomal vectors | (2) SOX2, KLF4, OCT4, L-MYC, LIN28, p53 | ||||
| MFS | Dermal fibroblasts | retrovirus | OCT4, SOX2, KLF4 and c-MYC | 2 | [59] |
4. Differentiating hiPSCs into Osteoclasts
4.1. Differentiating Mouse Embryonic Stem Cells (mESCs) into Osteoclasts
| Methods | Mouse ESC Lines | Factors Added in OC Medium | Results | Reference | Lessons Learned |
|---|---|---|---|---|---|
| mESCs on 24-well plates | D3, J1 | hM-CSF, hRANKL, A.A, VitD3, Dexa | TRAP+ cells (day 14) | [67] | A.A. increased total cell recovery and OC precursors through increasing Flk-1-positive cells when added during the initial 4 days. |
| Co-culture 1-step, 2-step, 3-step | D3 | hM-CSF (for OP9 coculture) VitD3, Dexa | TRAP+ cells (day 11–16) | [68] | ST2 supported osteoclastogenesis more efficiently than OP9. C-fms signaling is required for OC development from mESCs. |
| Co-culture 1-step, 2-step, 3-step | CCE, D3, J1, CJ7 | hM-CSF (for CFU assay) VitD3, Dexa | TRAP+ cells (day 11–16) | [69] | SCL is indispensable for osteoclastogenesis. GATA-2 is required for osteoclastogenesis at early but not terminal differentiation stage. |
| Co-culture 1-step | D3 | VitD3, Dexa, hRANKL, hM-CSF (for some exp.) | TRAP+ cells c-Kit, c-fms, β2-integrin, CD31 expression (day 3–17) | [70] | Temporal expression of markers: c-Kit → β2-integrin → c-fms, TRAP. Exogenous hM-CSF and hRANKL promote osteoclastogenesis. Continuous hM-CSF can reduce number of TRAP+ cells. |
| Co-culture 1-step, 2-step, 3-step | D3, CCE | VitD3, Dexa | TRAP+ cells | [71] | Blocking VEGFR-mediated signaling is inhibitory to OC development. |
| EB | mESCs | mM-CSF, mRANKL | TRAP+ (≥3 nuclei) (day 13) | [72] | Efficiency of OC generation: 3-step coculture > EB method > 1-step coculture. |
| EB, monolayer culture | J1, miPSCs (38c2, 20D17) | M-CSF, RANKL | TRAP+ (≥3 nuclei) (day 19) | [73] | A new in vitro culture method to differentiate mES/iPSCs into osteoclasts. |
4.2. Commitment of Human ESCs/hiPSCs into Hematopoietic Lineages/OC Precursors
4.3. Marker Genes for Mesodermal Formation and Hematopoietic Differentiation
| Methods | hES/iPSCs & Medium | Differentiation medium | Results | Reference | Protocol Time Line |
|---|---|---|---|---|---|
| Monolayer | KhES-1, KhES-3, 201B7, 253G4 mTeSR1, Stemline II | Stemline II + ITS | T + Mixl1+ cells (d4) KDR+ CD34+CD45− cells (d6) 36% CD235a+; 53% CD45+ (d30) | [82] |  |
| Monolayer (Collagen IV) | WA01 hiPSCs Matrigel/mTeSR1 | IMDM, BIT, MTG, NEAA, l-glu | 95% CD43+, 53% CD34+, 59% CD41a+, 60% CD235a+, 35% CD45+ (d14) | [83] | 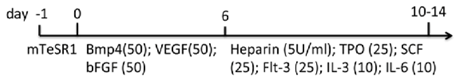 |
| EB monolayer (gelatin) | hiPSCs MEF/hESC medium | EB1 medium/monocyte differentiation medium | 90% CD14+ (d15 of attached, flatten EBs on gelatin plates) | [84] | 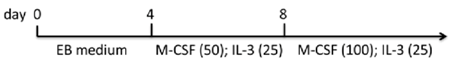 |
| EB | WA01, H9 Matrigel/condition medium | Knockout DMEM, FBS, NEAA, l-glu, ME | 9.3% CD45+ (d15) | [85] | 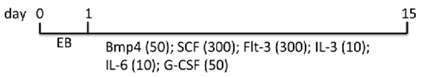 |
| EB | WA01, ES02 MEF/hESC medium | StemPro-34 + MTG + l-glu + A.A. | Mesoderm induction and hemangioblast development (d1-8), increased T (d3), CD34, SCL (d5), CD117+CD31+ (d8) | [76] | 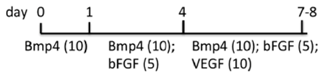 |
| EB | hFib2-iPS5 MEF/hESC medium | EB2 medium | 29% CD34+, 27% CD45+, 16% CD34+CD45+ (d17) | [86] | 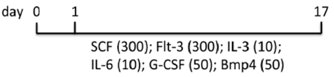 |
| EB | WA01, ES02, MSC-iPS1 matrigel/hESC medium | StemPro34 + MTG + l-glu + A.A. | 15%–59% CD45+ (d14); 38%–72% CD45+ (d22) | [87] | 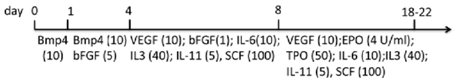 |
| EB | hiPSCs Matrigel/hESC medium | StemPro-34, l-glu, A.A., transferrin, MTG | Myeloid, erythroid, megakaryocytic cells released into the medium (d14) | [88] | 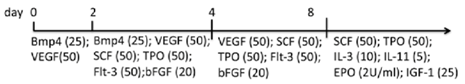 |
| Co-culture (S17/C166) | H1, H1.1, H9.2 MEF/hESC medium | DMEM, FBS, l-glu, ME, NEAA | 1%–2% CD34+CD38− (d17) | [89] |  |
| Co-culture (AM-20, UG26, IL08, AGM, FL) | H1, H9, hES-NCL1 MEF/hESC medium | Knockout DMEM, FCS, ME, l-glu, NEAA, antibiotics | 16% CD34+, 5%CD45+, 8% CD31+, 6% CD34+CD31+ (d18) | [90] |  |
| Co-culture (OP9) | WA01, WA09, iPS-1, iPSCs (SK46)-M-4-10 MEF/hESC medium | α-MEM, FBS, MTG | 9.8% CD43+, 14% CD45+ (d9) 94% CD43+, 78% CD45+ (d11) 98% CD43+, 97% CD45+ (d17) | [81] |  |
4.4. Factors and Cytokines to Promote Hematopoiesis
4.5. Variability among Hematopoietic Differentiation Protocols
4.6. Differentiating hiPSCs-Derived Osteoclast Progenitors into Osteoclasts
4.7. Strategies of using hiPSC-Osteoclasts to Study Rare Genetic Bone Diseases
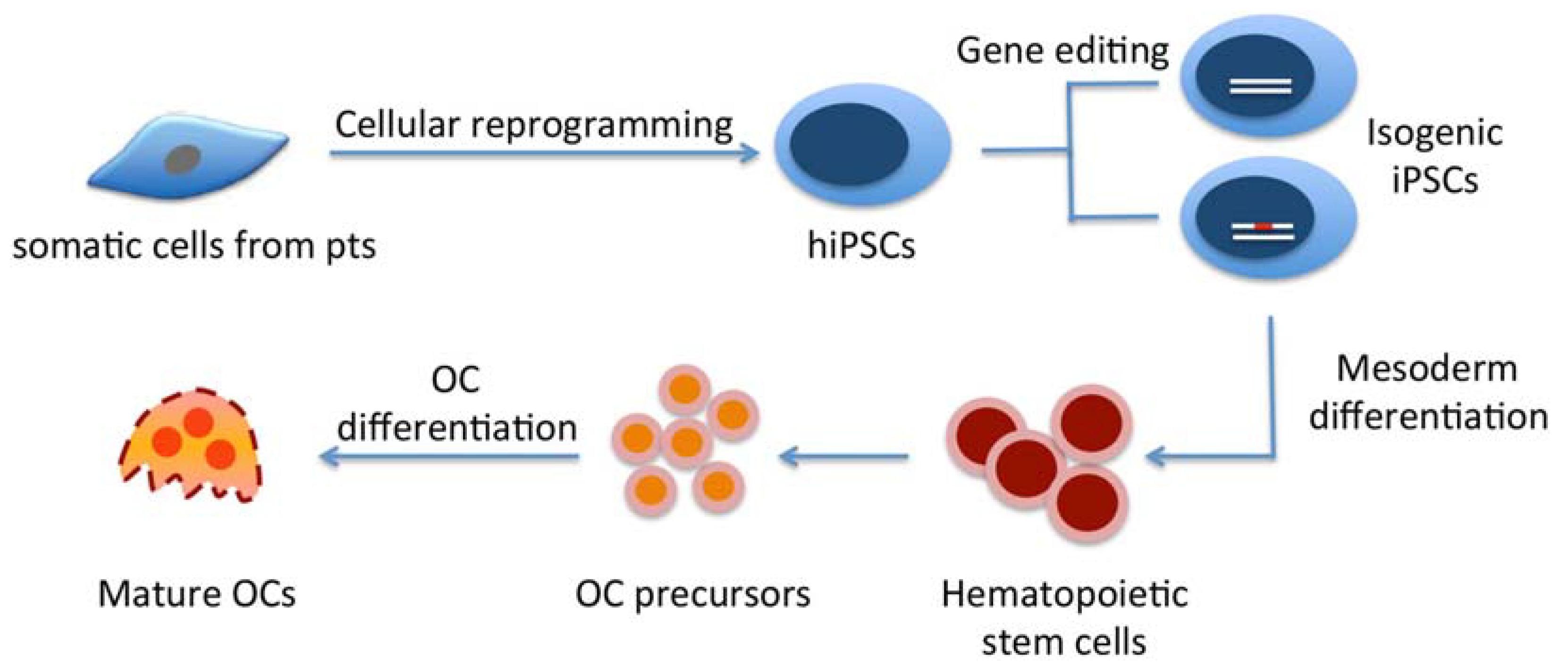
5. Conclusions
Acknowledgement
Conflicts of Interest
References
- Kornak, U.; Mundlos, S. Genetic disorders of the skeleton: A developmental approach. Am. J. Hum. Genet. 2003, 73, 447–474. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.P.; Wang, C.J.; Strecker, S.; Koczon-Jaremko, B.; Boskey, A.; Reichenberger, E.J. Introduction of a Phe377del mutation in ANK creates a mouse model for craniometaphyseal dysplasia. J. Bone Miner. Res. 2009, 24, 1206–1215. [Google Scholar] [CrossRef]
- Chen, I.P.; Wang, L.; Jiang, X.; Aguila, H.L.; Reichenberger, E.J. A Phe377del mutation in ANK leads to impaired osteoblastogenesis and osteoclastogenesis in a mouse model for craniometaphyseal dysplasia (CMD). Hum. Mol. Genet. 2011, 20, 948–961. [Google Scholar] [CrossRef] [PubMed]
- Laslo, P.; Spooner, C.J.; Warmflash, A.; Lancki, D.W.; Lee, H.J.; Sciammas, R.; Gantner, B.N.; Dinner, A.R.; Singh, H. Multilineage transcriptional priming and determination of alternate hematopoietic cell fates. Cell 2006, 126, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, N.; Noda, M. Mitf is expressed in osteoclast progenitors in vitro. Exp. Cell Res. 2000, 260, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, T.; Ohneda, O.; Arai, F.; Iwamoto, K.; Okada, S.; Takagi, K.; Anderson, D.M.; Suda, T. Bifurcation of osteoclasts and dendritic cells from common progenitors. Blood 2001, 98, 2544–2554. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.M.; Ryan, G.R.; Hapel, A.J.; Dominguez, M.G.; Russell, R.G.; Kapp, S.; Sylvestre, V.; Stanley, E.R. Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood 2002, 99, 111–120. [Google Scholar] [CrossRef]
- Kim, M.S.; Day, C.J.; Morrison, N.A. MCP-1 is induced by receptor activator of nuclear factor-κb ligand, promotes human osteoclast fusion, and rescues granulocyte macrophage colony-stimulating factor suppression of osteoclast formation. J. Biol. Chem. 2005, 280, 16163–16169. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Rho, J.; Jeong, D.; Sul, J.Y.; Kim, T.; Kim, N.; Kang, J.S.; Miyamoto, T.; Suda, T.; Lee, S.K.; et al. V-atpase V0 subunit d2-deficient mice exhibit impaired osteoclast fusion and increased bone formation. Nat. Med. 2006, 12, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Faccio, R.; Teitelbaum, S.L.; Fujikawa, K.; Chappel, J.; Zallone, A.; Tybulewicz, V.L.; Ross, F.P.; Swat, W. Vav3 regulates osteoclast function and bone mass. Nat. Med. 2005, 11, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Gerritsen, E.J.; Vossen, J.M.; van Loo, I.H.; Hermans, J.; Helfrich, M.H.; Griscelli, C.; Fischer, A. Autosomal recessive osteopetrosis: Variability of findings at diagnosis and during the natural course. Pediatrics 1994, 93, 247–253. [Google Scholar] [PubMed]
- Sly, W.S.; Hewett-Emmett, D.; Whyte, M.P.; Yu, Y.S.; Tashian, R.E. Carbonic anhydrase II deficiency identified as the primary defect in the autosomal recessive syndrome of osteopetrosis with renal tubular acidosis and cerebral calcification. Proc. Natl. Acad. Sci. USA 1983, 80, 2752–2756. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.E., Jr.; Bollerslev, J. Heterogeneity of autosomal dominant osteopetrosis. Radiology 1987, 164, 223–225. [Google Scholar] [CrossRef]
- Gelb, B.D.; Shi, G.P.; Chapman, H.A.; Desnick, R.J. Pycnodysostosis, a lysosomal disease caused by cathepsin K deficiency. Science 1996, 273, 1236–1238. [Google Scholar] [CrossRef] [PubMed]
- Selby, P.L.; Davie, M.W.; Ralston, S.H.; Stone, M.D. Guidelines on the management of paget’s disease of bone. Bone 2002, 31, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Ralston, S.H.; Langston, A.L.; Reid, I.R. Pathogenesis and management of paget’s disease of bone. Lancet 2008, 372, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Tiegs, R.D. Paget’s disease of bone: Indications for treatment and goals of therapy. Clin. Ther. 1997, 19, 1309–1329. [Google Scholar] [CrossRef] [PubMed]
- Golob, D.S.; McAlister, W.H.; Mills, B.G.; Fedde, K.N.; Reinus, W.R.; Teitelbaum, S.L.; Beeki, S.; Whyte, M.P. Juvenile paget disease: Life-long features of a mildly affected young woman. J. Bone Miner. Res. 1996, 11, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Whyte, M.P.; Obrecht, S.E.; Finnegan, P.M.; Jones, J.L.; Podgornik, M.N.; McAlister, W.H.; Mumm, S. Osteoprotegerin deficiency and juvenile paget’s disease. N. Engl. J. Med. 2002, 347, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Cundy, T.; Hegde, M.; Naot, D.; Chong, B.; King, A.; Wallace, R.; Mulley, J.; Love, D.R.; Seidel, J.; Fawkner, M.; et al. A mutation in the gene TNFRSF11B encoding osteoprotegerin causes an idiopathic hyperphosphatasia phenotype. Hum. Mol. Genet. 2002, 11, 2119–2127. [Google Scholar] [CrossRef] [PubMed]
- Dickson, G.R.; Shirodria, P.V.; Kanis, J.A.; Beneton, M.N.; Carr, K.E.; Mollan, R.A. Familial expansile osteolysis: A morphological, histomorphometric and serological study. Bone 1991, 12, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Takata, S.; Yasui, N.; Nakatsuka, K.; Ralston, S.H. Evolution of understanding of genetics of paget’s disease of bone and related diseases. J. Bone Miner. Res. 2004, 22, 519–523. [Google Scholar] [CrossRef]
- Hughes, A.E.; Ralston, S.H.; Marken, J.; Bell, C.; MacPherson, H.; Wallace, R.G.; van Hul, W.; Whyte, M.P.; Nakatsuka, K.; Hovy, L.; et al. Mutations in TNFRSF11A, affecting the signal peptide of rank, cause familial expansile osteolysis. Nat. Genet. 2000, 24, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Whyte, M.P.; Mills, B.G.; Reinus, W.R.; Podgornik, M.N.; Roodman, G.D.; Gannon, F.H.; Eddy, M.C.; McAlister, W.H. Expansile skeletal hyperphosphatasia: A new familial metabolic bone disease. J. Bone Miner. Res. 2000, 15, 2330–2344. [Google Scholar] [CrossRef] [PubMed]
- Frattini, A.; Orchard, P.J.; Sobacchi, C.; Giliani, S.; Abinun, M.; Mattsson, J.P.; Keeling, D.J.; Andersson, A.K.; Wallbrandt, P.; Zecca, L.; et al. Defects in TCIRG1 subunit of the vacuolar proton pump are responsible for a subset of human autosomal recessive osteopetrosis. Nat. Genet. 2000, 25, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Frattini, A.; Pangrazio, A.; Susani, L.; Sobacchi, C.; Mirolo, M.; Abinun, M.; Andolina, M.; Flanagan, A.; Horwitz, E.M.; Mihci, E.; et al. Chloride channel CICN7 mutations are responsible for severe recessive, dominant, and intermediate osteopetrosis. J. Bone Miner. Res. 2003, 18, 1740–1747. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, A.; Faupel, J.; Goebel, I.; Stiller, A.; Beyer, S.; Stockle, C.; Hasan, C.; Bode, U.; Kornak, U.; Kubisch, C. Identification of a novel mutation in the coding region of the grey-lethal gene OSTM1 in human malignant infantile osteopetrosis. Hum. Mutat. 2004, 23, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Pangrazio, A.; Boudin, E.; Piters, E.; Damante, G.; Lo Iacono, N.; D’Elia, A.V.; Vezzoni, P.; van Hul, W.; Villa, A.; Sobacchi, C. Identification of the first deletion in the LRP5 gene in a patient with autosomal dominant osteopetrosis type I. Bone 2011, 49, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Cleiren, E.; Benichou, O.; van Hul, E.; Gram, J.; Bollerslev, J.; Singer, F.R.; Beaverson, K.; Aledo, A.; Whyte, M.P.; Yoneyama, T.; et al. Albers-schonberg disease (autosomal dominant osteopetrosis, type II) results from mutations in the CICN7 chloride channel gene. Hum. Mol. Genet. 2001, 10, 2861–2867. [Google Scholar] [CrossRef] [PubMed]
- Hocking, L.J.; Lucas, G.J.; Daroszewska, A.; Mangion, J.; Olavesen, M.; Cundy, T.; Nicholson, G.C.; Ward, L.; Bennett, S.T.; Wuyts, W.; et al. Domain-specific mutations in sequestosome 1 (SQSTM1) cause familial and sporadic Paget’s disease. Hum. Mol. Genet. 2002, 11, 2735–2739. [Google Scholar] [CrossRef] [PubMed]
- Laurin, N.; Brown, J.P.; Morissette, J.; Raymond, V. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/P62) in paget disease of bone. Am. J. Hum. Genet. 2002, 70, 1582–1588. [Google Scholar] [CrossRef] [PubMed]
- Whyte, M.P.; Hughes, A.E. Expansile skeletal hyperphosphatasia is caused by a 15-base pair tandem duplication in TNFRSF11A encoding RANK and is allelic to familial expansile osteolysis. J. Bone Miner. Res. 2002, 17, 26–29. [Google Scholar] [CrossRef] [PubMed]
- Cherry, A.B.; Daley, G.Q. Reprogrammed cells for disease modeling and regenerative medicine. Ann. Rev. Med. 2013, 64, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Lee, Y.K.; Schaefer, E.A.; Peters, D.T.; Veres, A.; Kim, K.; Kuperwasser, N.; Motola, D.L.; Meissner, T.B.; Hendriks, W.T.; et al. A talen genome-editing system for generating human stem cell-based disease models. Cell Stem Cell 2013, 12, 238–251. [Google Scholar] [CrossRef] [PubMed]
- Emborg, M.E.; Liu, Y.; Xi, J.; Zhang, X.; Yin, Y.; Lu, J.; Joers, V.; Swanson, C.; Holden, J.E.; Zhang, S.C. Induced pluripotent stem cell-derived neural cells survive and mature in the nonhuman primate brain. Cell Rep. 2013, 3, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Park, I.H.; Arora, N.; Huo, H.; Maherali, N.; Ahfeldt, T.; Shimamura, A.; Lensch, M.W.; Cowan, C.; Hochedlinger, K.; Daley, G.Q. Disease-specific induced pluripotent stem cells. Cell 2008, 134, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Sommer, C.A.; Stadtfeld, M.; Murphy, G.J.; Hochedlinger, K.; Kotton, D.N.; Mostoslavsky, G. Induced pluripotent stem cell generation using a single lentiviral stem cell cassette. Stem Cells 2009, 27, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Stadtfeld, M.; Nagaya, M.; Utikal, J.; Weir, G.; Hochedlinger, K. Induced pluripotent stem cells generated without viral integration. Science 2008, 322, 945–949. [Google Scholar] [CrossRef] [PubMed]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A.; et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Woltjen, K.; Michael, I.P.; Mohseni, P.; Desai, R.; Mileikovsky, M.; Hamalainen, R.; Cowling, R.; Wang, W.; Liu, P.; Gertsenstein, M.; et al. Piggybac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature 2009, 458, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Hu, K.; Smuga-Otto, K.; Tian, S.; Stewart, R.; Slukvin, I.I.; Thomson, J.A. Human induced pluripotent stem cells free of vector and transgene sequences. Science 2009, 324, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wu, S.; Joo, J.Y.; Zhu, S.; Han, D.W.; Lin, T.; Trauger, S.; Bien, G.; Yao, S.; Zhu, Y.; et al. Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell 2009, 4, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T.; Raya, A.; Barrero, M.J.; Garreta, E.; Consiglio, A.; Gonzalez, F.; Vassena, R.; Bilic, J.; Pekarik, V.; Tiscornia, G.; et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat. Biotechnol. 2008, 26, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Giorgetti, A.; Montserrat, N.; Aasen, T.; Gonzalez, F.; Rodriguez-Piza, I.; Vassena, R.; Raya, A.; Boue, S.; Barrero, M.J.; Corbella, B.A.; et al. Generation of induced pluripotent stem cells from human cord blood using OCT4 and SOX2. Cell Stem Cell 2009, 5, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Qin, H.; Qu, C.; Tuan, R.S.; Shi, S.; Huang, G.T. IPS cells reprogrammed from human mesenchymal-like stem/progenitor cells of dental tissue origin. Stem Cells Dev. 2010, 19, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.Y.; Bouwman, B.A.; Ang, Y.S.; Kim, S.J.; Lee, D.F.; Lemischka, I.R.; Rendl, M. Single transcription factor reprogramming of hair follicle dermal papilla cells to induced pluripotent stem cells. Stem Cells 2011, 29, 964–971. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Benda, C.; Dunzinger, S.; Huang, Y.; Ho, J.C.; Yang, J.; Wang, Y.; Zhang, Y.; Zhuang, Q.; Li, Y.; et al. Generation of human induced pluripotent stem cells from urine samples. Nat. Protoc. 2012, 7, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wang, Y.; Goh, S.S.; Yang, J.; Lam, D.H.; Choudhury, Y.; Tay, F.C.; Du, S.; Tan, W.K.; Purwanti, Y.I.; et al. Inhibition of neuronal nitric oxide synthase activity promotes migration of human-induced pluripotent stem cell-derived neural stem cells toward cancer cells. J. Neurochem. 2013, 126, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Dimos, J.T.; Rodolfa, K.T.; Niakan, K.K.; Weisenthal, L.M.; Mitsumoto, H.; Chung, W.; Croft, G.F.; Saphier, G.; Leibel, R.; Goland, R.; et al. Induced pluripotent stem cells generated from patients with als can be differentiated into motor neurons. Science 2008, 321, 1218–1221. [Google Scholar] [CrossRef] [PubMed]
- Kudva, Y.C.; Ohmine, S.; Greder, L.V.; Dutton, J.R.; Armstrong, A.; de Lamo, J.G.; Khan, Y.K.; Thatava, T.; Hasegawa, M.; Fusaki, N.; et al. Transgene-free disease-specific induced pluripotent stem cells from patients with type 1 and type 2 diabetes. Stem Cells Trans. Med. 2012, 1, 451–461. [Google Scholar] [CrossRef]
- Maehr, R.; Chen, S.; Snitow, M.; Ludwig, T.; Yagasaki, L.; Goland, R.; Leibel, R.L.; Melton, D.A. Generation of pluripotent stem cells from patients with type 1 diabetes. Proc. Natl. Acad. Sci. USA 2009, 106, 15768–15773. [Google Scholar] [CrossRef] [PubMed]
- Teo, A.K.; Windmueller, R.; Johansson, B.B.; Dirice, E.; Njolstad, P.R.; Tjora, E.; Raeder, H.; Kulkarni, R.N. Derivation of human induced pluripotent stem cells from patients with maturity onset diabetes of the young. J. Biol. Chem. 2013, 288, 5353–5356. [Google Scholar] [CrossRef] [PubMed]
- Connelly, J.P.; Kwon, E.M.; Gao, Y.; Trivedi, N.S.; Elkahloun, A.G.; Horwitz, M.S.; Cheng, L.; Liu, P.P. Targeted correction of RUNX1 mutation in FPD patient-specific induced pluripotent stem cells rescues megakaryopoietic defects. Blood 2014, 124, 1926–1930. [Google Scholar] [CrossRef]
- Deyle, D.R.; Khan, I.F.; Ren, G.; Wang, P.R.; Kho, J.; Schwarze, U.; Russell, D.W. Normal collagen and bone production by gene-targeted human osteogenesis imperfecta iPSCs. Mol. Ther. 2012, 20, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.P.; Fukuda, K.; Fusaki, N.; Iida, A.; Hasegawa, M.; Lichtler, A.; Reichenberger, E.J. Induced pluripotent stem cell reprogramming by integration-free sendai virus vectors from peripheral blood of patients with craniometaphyseal dysplasia. Cell. Reprogram. 2013, 15, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Hayashi, Y.; Schlieve, C.R.; Ikeya, M.; Kim, H.; Nguyen, T.D.; Sami, S.; Baba, S.; Barruet, E.; Nasu, A.; et al. Induced pluripotent stem cells from patients with human fibrodysplasia ossificans progressiva show increased mineralization and cartilage formation. Orphanet J. Rare Dis. 2013, 8. [Google Scholar] [CrossRef]
- Quarto, N.; Leonard, B.; Li, S.; Marchand, M.; Anderson, E.; Behr, B.; Francke, U.; Reijo-Pera, R.; Chiao, E.; Longaker, M.T. Skeletogenic phenotype of human marfan embryonic stem cells faithfully phenocopied by patient-specific induced-pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Cole, W.G. Advances in osteogenesis imperfecta. Clin. Orthop. Relat. Res. 2002, 401, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Reichenberger, E.; Tiziani, V.; Watanabe, S.; Park, L.; Ueki, Y.; Santanna, C.; Baur, S.T.; Shiang, R.; Grange, D.K.; Beighton, P.; et al. Autosomal dominant craniometaphyseal dysplasia is caused by mutations in the transmembrane protein ANK. Am. J. Hum. Genet. 2001, 68, 1321–1326. [Google Scholar] [CrossRef] [PubMed]
- Nurnberg, P.; Thiele, H.; Chandler, D.; Hohne, W.; Cunningham, M.L.; Ritter, H.; Leschik, G.; Uhlmann, K.; Mischung, C.; Harrop, K.; et al. Heterozygous mutations in ANKH, the human ortholog of the mouse progressive ankylosis gene, result in craniometaphyseal dysplasia. Nat. Genet. 2001, 28, 37–41. [Google Scholar] [PubMed]
- Hu, Y.; Chen, I.P.; de Almeida, S.; Tiziani, V.; Do Amaral, C.M.; Gowrishankar, K.; Passos-Bueno, M.R.; Reichenberger, E.J. A novel autosomal recessive GJA1 missense mutation linked to craniometaphyseal dysplasia. PLoS One 2013, 8, e73576. [Google Scholar] [CrossRef] [PubMed]
- Shore, E.M.; Xu, M.; Feldman, G.J.; Fenstermacher, D.A.; Cho, T.J.; Choi, I.H.; Connor, J.M.; Delai, P.; Glaser, D.L.; LeMerrer, M.; et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat. Genet. 2006, 38, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; D’Alessio, M.; Ramirez, F.; Lynch, J.R.; Sykes, B.; Pangilinan, T.; Bonadio, J. Genomic organization of the sequence coding for fibrillin, the defective gene product in marfan syndrome. Hum. mol. Genet. 1993, 2, 961–968. [Google Scholar] [CrossRef] [PubMed]
- Quarto, N.; Li, S.; Renda, A.; Longaker, M.T. Exogenous activation of BMP-2 signaling overcomes TGFβ-mediated inhibition of osteogenesis in marfan embryonic stem cells and marfan patient-specific induced pluripotent stem cells. Stem Cells 2012, 30, 2709–2719. [Google Scholar] [CrossRef] [PubMed]
- Tsuneto, M.; Yamazaki, H.; Yoshino, M.; Yamada, T.; Hayashi, S. Ascorbic acid promotes osteoclastogenesis from embryonic stem cells. Biochem. Biophys. Res. Commun. 2005, 335, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Yamane, T.; Kunisada, T.; Yamazaki, H.; Era, T.; Nakano, T.; Hayashi, S.I. Development of osteoclasts from embryonic stem cells through a pathway that is c-fms but not c-kit dependent. Blood 1997, 90, 3516–3523. [Google Scholar] [PubMed]
- Yamane, T.; Kunisada, T.; Yamazaki, H.; Nakano, T.; Orkin, S.H.; Hayashi, S.I. Sequential requirements for SCL/tal-1, GATA-2, macrophage colony-stimulating factor, and osteoclast differentiation factor/osteoprotegerin ligand in osteoclast development. Exp. Hematol. 2000, 28, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Hemmi, H.; Okuyama, H.; Yamane, T.; Nishikawa, S.; Nakano, T.; Yamazaki, H.; Kunisada, T.; Hayashi, S. Temporal and spatial localization of osteoclasts in colonies from embryonic stem cells. Biochem. Biophys. Res. Commun. 2001, 280, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Okuyama, H.; Tsuneto, M.; Yamane, T.; Yamazaki, H.; Hayashi, S. Discrete types of osteoclast precursors can be generated from embryonic stem cells. Stem Cells 2003, 21, 670–680. [Google Scholar] [CrossRef] [PubMed]
- Goodman, M.L.; Chen, S.; Yang, F.C.; Chan, R.J. Novel method of murine embryonic stem cell-derived osteoclast development. Stem Cells Dev. 2009, 18, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, K.; Iwamoto, Y.; Ishii, M. Development of an in vitro culture method for stepwise differentiation of mouse embryonic stem cells and induced pluripotent stem cells into mature osteoclasts. J. Bone Miner. Metab. 2014, 32, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, B.G. Expression pattern of the brachyury gene in whole-mount TWis/TWis mutant embryos. Development 1991, 113, 913–917. [Google Scholar] [PubMed]
- Davis, R.P.; Ng, E.S.; Costa, M.; Mossman, A.K.; Sourris, K.; Elefanty, A.G.; Stanley, E.G. Targeting a GFP reporter gene to the MIXL1 locus of human embryonic stem cells identifies human primitive streak-like cells and enables isolation of primitive hematopoietic precursors. Blood 2008, 111, 1876–1884. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, M.; D’Souza, S.L.; Lynch-Kattman, M.; Schwantz, S.; Keller, G. Development of the hemangioblast defines the onset of hematopoiesis in human ES cell differentiation cultures. Blood 2007, 109, 2679–2687. [Google Scholar] [PubMed]
- Porcher, C.; Swat, W.; Rockwell, K.; Fujiwara, Y.; Alt, F.W.; Orkin, S.H. The T cell leukemia oncoprotein SCL/tal-1 is essential for development of all hematopoietic lineages. Cell 1996, 86, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Lacaud, G.; Gore, L.; Kennedy, M.; Kouskoff, V.; Kingsley, P.; Hogan, C.; Carlsson, L.; Speck, N.; Palis, J.; Keller, G. Runx1 is essential for hematopoietic commitment at the hemangioblast stage of development in vitro. Blood 2002, 100, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Orkin, S.H. Gata-binding transcription factors in hematopoietic cells. Blood 1992, 80, 575–581. [Google Scholar] [PubMed]
- Woodford-Thomas, T.; Thomas, M.L. The leukocyte common antigen, CD45 and other protein tyrosine phosphatases in hematopoietic cells. Semin. Cell Biol. 1993, 4, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.D.; Vodyanik, M.A.; Slukvin, I.I. Generation of mature human myelomonocytic cells through expansion and differentiation of pluripotent stem cell-derived lin−CD34+CD43+CD45+ progenitors. J. Clin. Invest. 2009, 119, 2818–2829. [Google Scholar] [CrossRef] [PubMed]
- Niwa, A.; Heike, T.; Umeda, K.; Oshima, K.; Kato, I.; Sakai, H.; Suemori, H.; Nakahata, T.; Saito, M.K. A novel serum-free monolayer culture for orderly hematopoietic differentiation of human pluripotent cells via mesodermal progenitors. PLoS One 2011, 6, e22261. [Google Scholar] [CrossRef] [PubMed]
- Salvagiotto, G.; Burton, S.; Daigh, C.A.; Rajesh, D.; Slukvin, I.I.; Seay, N.J. A defined, feeder-free, serum-free system to generate in vitro hematopoietic progenitors and differentiated blood cells from hESCs and hiPSCs. PLoS One 2011, 6, e17829. [Google Scholar] [CrossRef] [PubMed]
- Panicker, L.M.; Miller, D.; Park, T.S.; Patel, B.; Azevedo, J.L.; Awad, O.; Masood, M.A.; Veenstra, T.D.; Goldin, E.; Stubblefield, B.K.; et al. Induced pluripotent stem cell model recapitulates pathologic hallmarks of gaucher disease. Proc. Natl. Acad. Sci. USA 2012, 109, 18054–18059. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, K.; Wang, L.; Li, L.; Menendez, P.; Murdoch, B.; Rouleau, A.; Bhatia, M. Cytokines and BMP-4 promote hematopoietic differentiation of human embryonic stem cells. Blood 2003, 102, 906–915. [Google Scholar] [CrossRef] [PubMed]
- Lengerke, C.; Grauer, M.; Niebuhr, N.I.; Riedt, T.; Kanz, L.; Park, I.H.; Daley, G.Q. Hematopoietic development from human induced pluripotent stem cells. Ann. NY Acad. Sci. 2009, 1176, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Grigoriadis, A.E.; Kennedy, M.; Bozec, A.; Brunton, F.; Stenbeck, G.; Park, I.H.; Wagner, E.F.; Keller, G.M. Directed differentiation of hematopoietic precursors and functional osteoclasts from human es and ips cells. Blood 2010, 115, 2769–2776. [Google Scholar] [CrossRef] [PubMed]
- Gandre-Babbe, S.; Paluru, P.; Aribeana, C.; Chou, S.T.; Bresolin, S.; Lu, L.; Sullivan, S.K.; Tasian, S.K.; Weng, J.; Favre, H.; et al. Patient-derived induced pluripotent stem cells recapitulate hematopoietic abnormalities of juvenile myelomonocytic leukemia. Blood 2013, 121, 4925–4929. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, D.S.; Hanson, E.T.; Lewis, R.L.; Auerbach, R.; Thomson, J.A. Hematopoietic colony-forming cells derived from human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2001, 98, 10716–10721. [Google Scholar] [CrossRef] [PubMed]
- Ledran, M.H.; Krassowska, A.; Armstrong, L.; Dimmick, I.; Renstrom, J.; Lang, R.; Yung, S.; Santibanez-Coref, M.; Dzierzak, E.; Stojkovic, M.; et al. Efficient hematopoietic differentiation of human embryonic stem cells on stromal cells derived from hematopoietic niches. Cell Stem Cell 2008, 3, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Pick, M.; Azzola, L.; Mossman, A.; Stanley, E.G.; Elefanty, A.G. Differentiation of human embryonic stem cells in serum-free medium reveals distinct roles for bone morphogenetic protein 4, vascular endothelial growth factor, stem cell factor, and fibroblast growth factor 2 in hematopoiesis. Stem Cells 2007, 25, 2206–2214. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, M.; Bonnet, D.; Kapp, U.; Wang, J.C.; Murdoch, B.; Dick, J.E. Quantitative analysis reveals expansion of human hematopoietic repopulating cells after short-term ex vivo culture. J. Exp. Med. 1997, 186, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Zandstra, P.W.; Conneally, E.; Piret, J.M.; Eaves, C.J. Ontogeny-associated changes in the cytokine responses of primitive human haemopoietic cells. Br. J. Haematol. 1998, 101, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, R.; Tong, J.; Brandt, J.; Traycoff, C.; Bruno, E.; McGuire, B.W.; Gordon, M.S.; McNiece, I.; Srour, E.F. The in vitro and in vivo effects of stem cell factor on human hematopoiesis. Stem Cells 1993, 11 (Suppl. 2), 76–82. [Google Scholar] [CrossRef]
- Kardel, M.D.; Eaves, C.J. Modeling human hematopoietic cell development from pluripotent stem cells. Exp. Hematol. 2012, 40, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Bar-Nur, O.; Russ, H.A.; Efrat, S.; Benvenisty, N. Epigenetic memory and preferential lineage-specific differentiation in induced pluripotent stem cells derived from human pancreatic islet beta cells. Cell Stem Cell 2011, 9, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Papapetrou, E.P.; Tomishima, M.J.; Chambers, S.M.; Mica, Y.; Reed, E.; Menon, J.; Tabar, V.; Mo, Q.; Studer, L.; Sadelain, M. Stoichiometric and temporal requirements of Oct4, Sox2, Klf4, and c-Myc expression for efficient human iPSC induction and differentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 12759–12764. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.; Kiskinis, E.; Verstappen, G.; Gu, H.; Boulting, G.; Smith, Z.D.; Ziller, M.; Croft, G.F.; Amoroso, M.W.; Oakley, D.H.; et al. Reference maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell 2011, 144, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Urnov, F.D.; Rebar, E.J.; Holmes, M.C.; Zhang, H.S.; Gregory, P.D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 2010, 11, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Wood, A.J.; Lo, T.W.; Zeitler, B.; Pickle, C.S.; Ralston, E.J.; Lee, A.H.; Amora, R.; Miller, J.C.; Leung, E.; Meng, X.; et al. Targeted genome editing across species using ZFNs and TALENs. Science 2011, 333. [Google Scholar] [CrossRef] [PubMed]
- Soldner, F.; Laganiere, J.; Cheng, A.W.; Hockemeyer, D.; Gao, Q.; Alagappan, R.; Khurana, V.; Golbe, L.I.; Myers, R.H.; Lindquist, S.; et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset parkinson point mutations. Cell 2011, 146, 318–331. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, I.-P. The Use of Patient-Specific Induced Pluripotent Stem Cells (iPSCs) to Identify Osteoclast Defects in Rare Genetic Bone Disorders. J. Clin. Med. 2014, 3, 1490-1510. https://doi.org/10.3390/jcm3041490
Chen I-P. The Use of Patient-Specific Induced Pluripotent Stem Cells (iPSCs) to Identify Osteoclast Defects in Rare Genetic Bone Disorders. Journal of Clinical Medicine. 2014; 3(4):1490-1510. https://doi.org/10.3390/jcm3041490
Chicago/Turabian StyleChen, I-Ping. 2014. "The Use of Patient-Specific Induced Pluripotent Stem Cells (iPSCs) to Identify Osteoclast Defects in Rare Genetic Bone Disorders" Journal of Clinical Medicine 3, no. 4: 1490-1510. https://doi.org/10.3390/jcm3041490
APA StyleChen, I.-P. (2014). The Use of Patient-Specific Induced Pluripotent Stem Cells (iPSCs) to Identify Osteoclast Defects in Rare Genetic Bone Disorders. Journal of Clinical Medicine, 3(4), 1490-1510. https://doi.org/10.3390/jcm3041490