iPSC-Based Models to Unravel Key Pathogenetic Processes Underlying Motor Neuron Disease Development

 and
and 
Abstract
:
1. Introduction
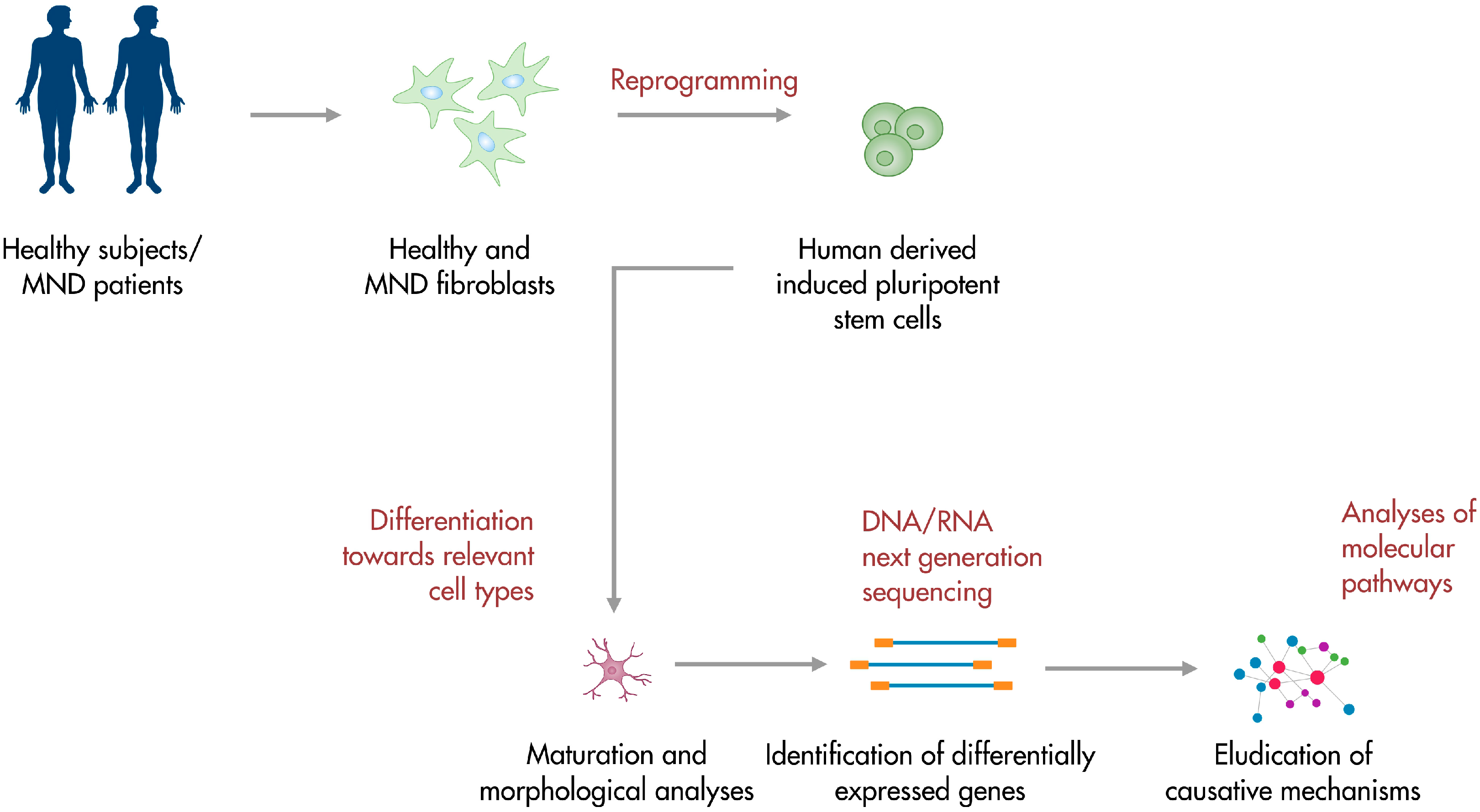
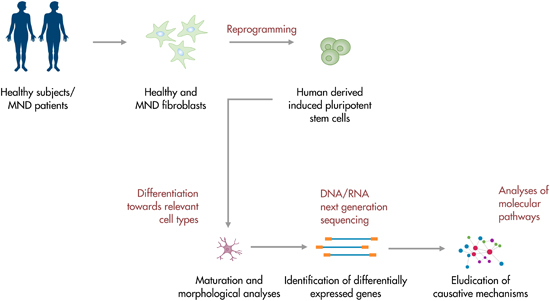
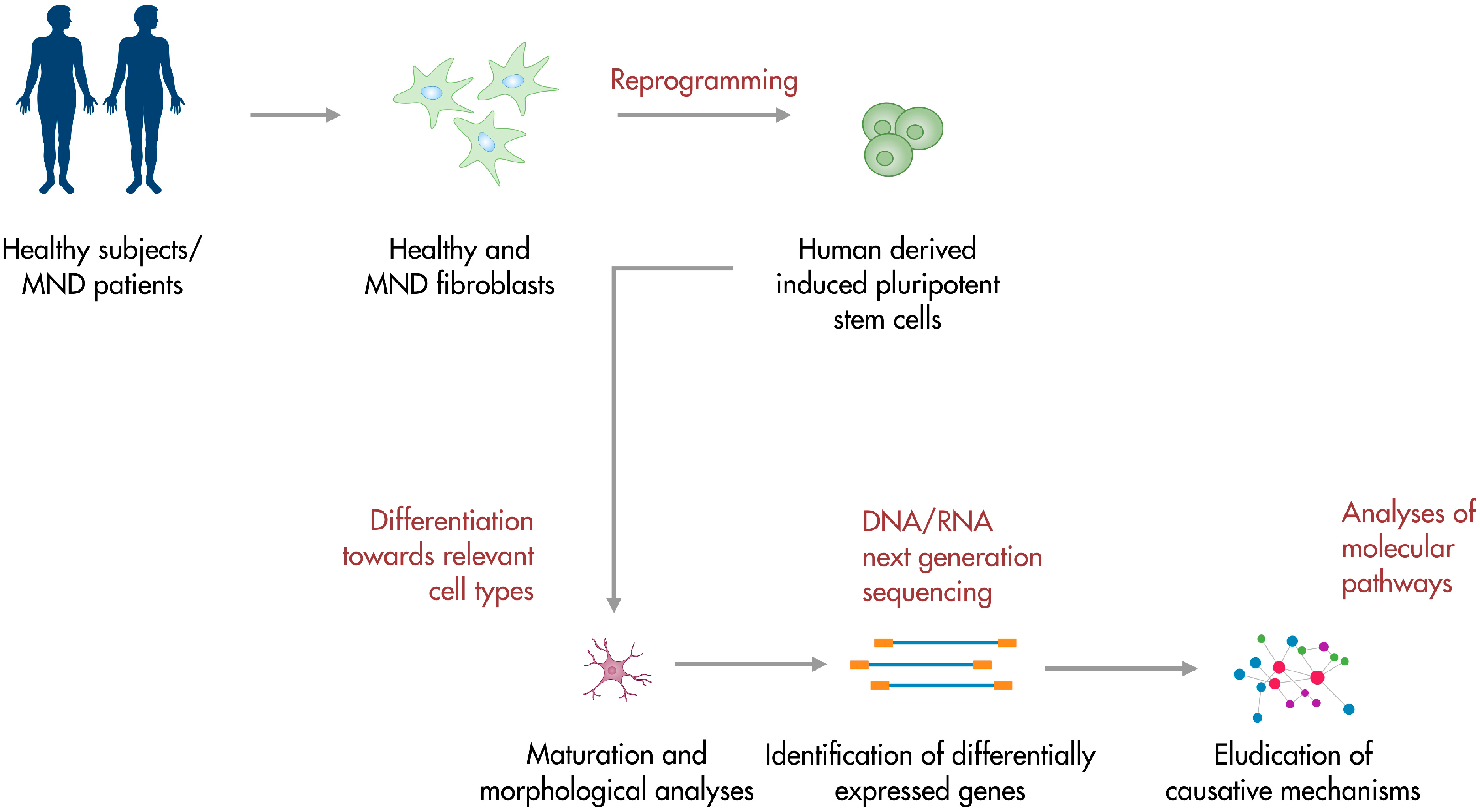
2. Modeling and Studying Amyotrophic Lateral Sclerosis Using IPSCs

{kind=link}
{kind=link}
| Reference | Cells | Reprogramming Method | Differentiation Protocol | Mechanism |
|---|---|---|---|---|
| Di Giorgio et al. 2008 [37] | Human ESC-derived MNs | - | Human ESC media without FGF2 or plasmanate + RA (Sigma) (1 μM) and an agonist of the SHH signaling pathway (1 μM) in N2 media: 1:1 DMEM:F12 + Glutamate (Gibco), penicillin (10,000 units) and streptomycin (Gibco) (1 mg/mL), N2 Supplement (Gibco) (1%), AA (Sigma-Aldrich) (0.2 mM), d-(+)-Glucose (Sigma-Aldrich) (0.16%), BDNF (R&D) (10 ng/mL), for 14 days. | MNs co-cultured with SOD1G93A astrocytes undergo cell death. |
| Prostaglandin D2 is responsible for the decrease in MN survival. | ||||
| Kiskinis et al. 2014 [26] | Fibroblasts from SOD1A4V ALS patients → iPSC-derived MNs | Retroviral transduction (KLF4, SOX2, OCT4, and c-MYC) | DMEM/F12, KSR (15%) on days 1–4; | SOD1 iPSCs and MNs suffer a reduction in survival when co-cultured with SOD1 glia. SOD1 MNs exhibit shorter processes and reduced soma. Gene correction of SOD1 mutation rescues both morphology and survival. |
| DMEM/F12 with l-glutamine, NEAAs, HE (2 μg/mL), N2 supplement (Gibco) on days 5–24; | SOD1 MNs show a down-regulation of genes implied in mitochondria homeostasis. | |||
| SB431542 (Sigma) (10 μM) + DM (Segment) (1 μM) on days 1–6; | Oxidative and ER stress and the up-regulation of UPR may contribute to neuronal toxicity. | |||
| BDNF (R&D) (10 ng/mL), AA (Sigma) (0.4 mg/mL), RA (Sigma) (1 μM) and SAG 1.3 (Calbiochem) (1 μM) on days 5–24. | C9ORF72 and SOD1 MNs share common disrupted pathways leading to enhanced oxidative stress response and decreased mitochondria activity. | |||
| Sareen et al. 2013 [39] | Fibroblasts from C9ORF72 ALS patients → iPSC-derived MNs | Episomal plasmid nucleofection (OCT4, SOX2, KLF4, L-MYC, LIN28, and p53 shRNA) | IMDM supplemented with B27-vitamin A (2%) and N2 (1%) on days 1–6; | RNA foci in C9ORF72 MNs co-localize with hnRNAP1, suggesting an indirect connection between C9ORF72 and TDP43 ALS forms. |
| addition of all-trans RA (0.1 μM) on days 6–25; | The toxicity linked to C9ORF72 hexanucleotide expansion may be due to a “gain of function” mechanism. | |||
| Neurobasal, B27 (2%) and N2 (1%) + RA (0.1 μM) and PMN (1 μM) on days 17–25; | ||||
| DMEM/F12, B27 (2%), RA (0.1 μM), PMN (1 μM), db-cAMP (1 μM), AA (200 ng/mL), BDNF (10 ng/mL), and GDNF (10 ng/mL) for a further 2–7 weeks; | The downregulation of the mutated allele with antisense oligonucleotides corrects the cell transcriptional profile. | |||
| Bilican et al. 2012 [42] | Fibroblasts from TDP43 M337V ALS patients → iPSC-derived MNs | Retroviral transduction (KLF4, SOX2, OCT4, and c-MYC) | Chemically defined medium supplemented with SB431542 (Tocris) (10 μM), DM (Calbiochem) (2.5 μM), and NAC (Sigma) (0.5 μM) for 5–7 days; | TDP43 MNs present a reduced survival in culture and high levels of TDP43 due to aberrant post-translational mechanisms. |
| chemically defined medium with RA (Sigma) (0.1 μM) for 7–12 days; | ||||
| Neurobasal medium (Invitrogen), RA (0.1 μM), PMN (Calbiochem) (1 μM), N2 supplement (Invitrogen) (1%), NEAAs (Invitrogen) (1%), penicillin/streptomycin (Invitrogen) (1%), GlutaMAX (Invitrogen) (1%), and basic FGF (5 ng/mL) for 7–10 days; | Neuronal response to neurotrophic factors involved in PI3K pathways influences TDP43 MN survival. | |||
| Neurobasal medium (Invitrogen), N2 supplement (Invitrogen) (0.5%), NEAAs (Invitrogen) (1%), penicillin/streptomycin (Invitrogen) (1%), GlutaMAX (Invitrogen) (0.5%), BDNF (PeproTech) (10 ng/mL), GDNF (PeproTech) (10 ng/mL), and F (Tocris) (10 μM) for 3–6 weeks. | ||||
| Alami et al. 2014 [43] | Fibroblasts from TDP43 A315T and TPD43 G298S ALS patients → iPSC-derived MNs | Retroviral transduction (OCT4, SOX2, and KLF4) | KSR medium (KO-DMEM (Life Technologies) supplemented with KSR (Life Technologies) (15%), 1 × Gibco GlutaMAX (Life Technologies) and NEAAs (100 μM) on days 0–10; | The microtubule-dependent transport of NEFL mRNA granules along the axon is impaired in TDP43 MNs. |
| N2 medium (Neurobasal (Life Technologies)) supplemented with 1 × N2 (Life Technologies), 1X Gibco GlutaMAX (Life Technologies) and NEAAs (100 μM)) on days 4–14; | ||||
| SB431542 (Sigma) (10 μM) and LDN-193189 (Segment) (100 nM) on days 0–5; | TDP43 domain affected by the mutation is involved in the assembly of RNA granules. | |||
| RA (Sigma) (1 μM), SAG (EMD Millipore) (1 μM), DAPT (EMD Millipore) (5 μM) and SU-5402 (Biovision) (4 μM) on days 2–14; | ||||
| murine glia-conditioned N2 medium supplemented with 1 × B-27 (Life Technologies), and BDNF (10 ng/mL), GDNF (10 ng/mL) and CNTF (R&D) (10 ng/mL). | ||||
| Chen et al. 2014 [44] | Fibroblasts from SOD1A4V and SOD1D90A ALS patients → iPSC-derived MNs | Non-integrating Sendai virus transduction (OCT3/4, SOX2, KLF4, and c-MYC) | DMEM/F12, N2 supplement, NEAAs, SB431542 (2 μM), LDN193189 (300 nM), and CHIR99021 (3 μM, all from Stemgent, Cambridge, MA, USA) on days 1–7; | Binding of SOD1 to 3′ UTR of NF-L mRNA may be responsible for neurofilament tendency to aggregate leading to neurite degeneration. |
| addition of RA (0.1 μM) and PMN (0.5 μM) on days 8–14; | Unlike mice SOD1 MNs, human SOD1 MNs have lower or normal levels of SOD1 compared to control MNs. | |||
| DMEM/F12, N2 supplement, and NEAAs on days 14–21. | ||||
| Ebert et al. 2009 [48] | Fibroblasts from a SMN1 SMA type I patient → iPSC-derived MNs | Lentiviral transduction (OCT4, SOX2, NANOG, and LIN28) | NIM (1:1 DMEM/F12 and N2 supplement (Gibco) (1%)) supplemented with RA (0.1 μM) for 1 week; | SMA iPSCs show reduced levels of SMN full-length transcripts, due to SMN1 loss, and a few truncated transcripts lacking exon 7. |
| addition of SHH (R&D) (100 ng/mL) for 1 week; | After a robust production, SMA iPSC-derived MNs undergo a reduction in number and size compared to WT iPSC-derived MNs. | |||
| RA and SHH medium supplemented with cAMP (1 mM), AA (200 ng/mL), BDNF and GDNF (both 10 ng/mL, PeproTech Inc., Rocky Hill, USA) for 2–6 weeks. | MN ontogenesis in SMA is disrupted by post-development damage. | |||
| Sareen et al. 2012 [49] | Fibroblasts from a SMN1 SMA type I patient → iPSC-derived MNs | Episomal plasmid nucleofection (OCT4, SOX2, NANOG, and LIN28) | Stemlin Neural Expansion Media (Sigma) supplemented with EGF (100 g/mL), FGF-2 (100 ng/mL), and HE (5 μg/mL) for 3 weeks; | SMA MNs show increased levels of cleaved caspase-3 and caspase-8 and membrane-bound Fas ligand, suggesting that apoptosis is implied in MN dysfunction and loss in SMA. |
| NIM (1:1 DMEM/F12 and N2 (1%)) in the presence of all-trans RA (0.1 μM) for 1 week; | The administration of Anti Fas-Ab rescues MN survival in in vitro models of SMA. | |||
| addition of PMN (1 μM) or SHH (10 ng/mL) for 1–3 weeks. | ||||
| Corti et al. 2012 [20] | Fibroblasts from SMN1 SMA type I patients → iPSC-derived MNs | Episomal plasmid nucleofection (OCT4, SOX2, NANOG, LIN28, c-MYC, and KLF4) | DMEM/F12 (Gibco, Invitrogen), supplemented with MEM NEAAs solution, N2, and HE (Sigma-Aldrich) (2 mg/mL) for 10 days; | SMA MNs show a reduction in size, axonal elongation, neuromuscular junction production and overall decreased survival. |
| addition of RA (Sigma-Aldrich) (0.1 μM) for 7 days; | SMA MNs exhibit a different splicing profile in a subset of genes encoding proteins involved in RNA metabolism, MN differentiation, axonal guidance and signal transduction. | |||
| same medium with RA (0.1 μM) and SHH (R&D) (100–200 ng/mL) for 7 days; | Gene correction of SMN2 with antisense oligodeoxynucleotides rescues the cellular damage and the altered splicing profile secondary to SMN1 deficiency in vitro. | |||
| addition of BDNF, GDNF, and IGF-1 (PeproTech) (10 ng/mL) on day 24. | ||||
| McGivern et al. 2013 [50] | Fibroblasts from SMN1 SMA patients → iPSC-derived glia | Lentiviral transduction (OCT4, SOX2, NANOG, and LIN28); Episomal plasmid nucleofection (OCT4, SOX2, NANOG, LIN28) | Human neural progenitor growth medium (Stemline, Sigma-Aldrich) supplemented with basic FGF-2 (Chemicon) (100 ng/mL), EGF (Chemicon) (100 ng/mL), and HE (Sigma-Aldrich) (5 μg/mL); | SMA astrocytes show increased basal calcium levels with a minimal response to ATP and an activated state that precedes MN loss. |
| DMEM: Nutrient Mixture F12 (Invitrogen) supplemented with B27 (Invitrogen) (2%) with or without CNTF for 2–8 weeks. | The ERK apoptosis pathways of SMA MNs may be initiated by the defective calcium homeostasis and the deficiency of trophic factors. |
3. Modeling and Studying Spinal Muscular Atrophy Using IPSCs
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of interest
References
- McDermott, C.J.; Shaw, P.J. Diagnosis and management of motor neurone disease. BMJ 2008, 336, 658–662. [Google Scholar]
- Gordon, P.H. Amyotrophic lateral sclerosis: An update for 2013 clinical features, pathophysiology, management and therapeutic trials. Aging Dis. 2013, 4, 295–310. [Google Scholar]
- Patten, S.A.; Armstrong, G.A.; Lissouba, A.; Kabashi, E.; Parker, J.A.; Drapeau, P. Fishing for causes and cures of motor neuron disorders. Dis. Model. Mech. 2014, 7, 799–809. [Google Scholar]
- Rafiq, M.K.; Proctor, A.R.; McDermott, C.J.; Shaw, P.J. Respiratory management of motor neurone disease: A review of current practice and new developments. Pract. Neurol. 2012, 12, 166–176. [Google Scholar]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar]
- Finsterer, J.; Burgunder, J.M. Recent progress in the genetics of motor neuron disease. Eur. J. Med. Genet. 2014, 57, 103–112. [Google Scholar]
- Logroscino, G.; Traynor, B.J.; Hardiman, O.; Chio, A.; Mitchell, D.; Swingler, R.J.; Millul, A.; Benn, E.; Beghi, E. Incidence of amyotrophic lateral sclerosis in europe. J. Neurol. Neurosurg. Psychiatry 2010, 81, 385–390. [Google Scholar]
- Beghi, E.; Chio, A.; Couratier, P.; Esteban, J.; Hardiman, O.; Logroscino, G.; Millul, A.; Mitchell, D.; Preux, P.M.; Pupillo, E.; et al. The epidemiology and treatment of als: Focus on the heterogeneity of the disease and critical appraisal of therapeutic trials. Amyotroph. Lateral Scler. 2011, 12, 1–10. [Google Scholar]
- Al-Chalabi, A.; Hardiman, O. The epidemiology of ALS: A conspiracy of genes, environment and time. Nat. Rev. Neurol. 2013, 9, 617–628. [Google Scholar]
- Majounie, E.; Renton, A.E.; Mok, K.; Dopper, E.G.; Waite, A.; Rollinson, S.; Chio, A.; Restagno, G.; Nicolaou, N.; Simon-Sanchez, J.; et al. Frequency of the C9ORF72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: A cross-sectional study. Lancet Neurol. 2012, 11, 323–330. [Google Scholar]
- Lorson, C.L.; Rindt, H.; Shababi, M. Spinal muscular atrophy: Mechanisms and therapeutic strategies. Hum. Mol. Genet. 2010, 19. [Google Scholar] [CrossRef]
- Prior, T.W.; Snyder, P.J.; Rink, B.D.; Pearl, D.K.; Pyatt, R.E.; Mihal, D.C.; Conlan, T.; Schmalz, B.; Montgomery, L.; Ziegler, K.; et al. Newborn and carrier screening for spinal muscular atrophy. Am. J. Med. Genet. A 2010, 152A, 1608–1616. [Google Scholar]
- Russman, B.S. Spinal muscular atrophy: Clinical classification and disease heterogeneity. J. Child Neurol. 2007, 22, 946–951. [Google Scholar]
- Lefebvre, S.; Burlet, P.; Liu, Q.; Bertrandy, S.; Clermont, O.; Munnich, A.; Dreyfuss, G.; Melki, J. Correlation between severity and smn protein level in spinal muscular atrophy. Nat. Genet. 1997, 16, 265–269. [Google Scholar]
- Monani, U.R.; Lorson, C.L.; Parsons, D.W.; Prior, T.W.; Androphy, E.J.; Burghes, A.H.; McPherson, J.D. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 1999, 8, 1177–1183. [Google Scholar]
- Vitte, J.; Fassier, C.; Tiziano, F.D.; Dalard, C.; Soave, S.; Roblot, N.; Brahe, C.; Saugier-Veber, P.; Bonnefont, J.P.; Melki, J.; et al. Refined characterization of the expression and stability of the smn gene products. Am. J. Pathol. 2007, 171, 1269–1280. [Google Scholar]
- Wang, C.H.; Finkel, R.S.; Bertini, E.S.; Schroth, M.; Simonds, A.; Wong, B.; Aloysius, A.; Morrison, L.; Main, M.; Crawford, T.O.; et al. Consensus statement for standard of care in spinal muscular atrophy. J. Child Neurol. 2007, 22, 1027–1049. [Google Scholar]
- Wee, C.D.; Kong, L.; Sumner, C.J. The genetics of spinal muscular atrophies. Curr. Opin. Neurol. 2010, 23, 450–458. [Google Scholar]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar]
- Corti, S.; Nizzardo, M.; Simone, C.; Falcone, M.; Nardini, M.; Ronchi, D.; Donadoni, C.; Salani, S.; Riboldi, G.; Magri, F.; et al. Genetic correction of human induced pluripotent stem cells from patients with spinal muscular atrophy. Sci. Transl. Med. 2012, 4. [Google Scholar] [CrossRef]
- Chipman, P.H.; Toma, J.S.; Rafuse, V.F. Generation of motor neurons from pluripotent stem cells. Prog. Brain Res. 2012, 201, 313–331. [Google Scholar]
- Sandoe, J.; Eggan, K. Opportunities and challenges of pluripotent stem cell neurodegenerative disease models. Nat. Neurosci. 2013, 16, 780–789. [Google Scholar]
- Kondo, T.; Asai, M.; Tsukita, K.; Kutoku, Y.; Ohsawa, Y.; Sunada, Y.; Imamura, K.; Egawa, N.; Yahata, N.; Okita, K.; et al. Modeling alzheimer’s disease with ipscs reveals stress phenotypes associated with intracellular abeta and differential drug responsiveness. Cell Stem Cell 2013, 12, 487–496. [Google Scholar]
- Camnasio, S.; Delli Carri, A.; Lombardo, A.; Grad, I.; Mariotti, C.; Castucci, A.; Rozell, B.; Lo Riso, P.; Castiglioni, V.; Zuccato, C.; et al. The first reported generation of several induced pluripotent stem cell lines from homozygous and heterozygous huntington’s disease patients demonstrates mutation related enhanced lysosomal activity. Neurobiol. Dis. 2012, 46, 41–51. [Google Scholar]
- Chung, C.Y.; Khurana, V.; Auluck, P.K.; Tardiff, D.F.; Mazzulli, J.R.; Soldner, F.; Baru, V.; Lou, Y.; Freyzon, Y.; Cho, S.; et al. Identification and rescue of alpha-synuclein toxicity in parkinson patient-derived neurons. Science 2013, 342, 983–987. [Google Scholar]
- Kiskinis, E.; Sandoe, J.; Williams, L.A.; Boulting, G.L.; Moccia, R.; Wainger, B.J.; Han, S.; Peng, T.; Thams, S.; Mikkilineni, S.; et al. Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell 2014, 14, 781–795. [Google Scholar]
- Miller, R.G.; Brooks, B.R.; Swain-Eng, R.J.; Basner, R.C.; Carter, G.T.; Casey, P.; Cohen, A.B.; Dubinsky, R.; Forshew, D.; Jackson, C.E.; et al. Quality improvement in neurology: Amyotrophic lateral sclerosis quality measures: Report of the quality measurement and reporting subcommittee of the american academy of neurology. Neurology 2013, 81, 2136–2140. [Google Scholar]
- Tovar, Y.R.L.B.; Ramirez-Jarquin, U.N.; Lazo-Gomez, R.; Tapia, R. Trophic factors as modulators of motor neuron physiology and survival: Implications for ALS therapy. Front. Cell. Neurosci. 2014, 8. [Google Scholar] [CrossRef]
- Brockington, A.; Ning, K.; Heath, P.R.; Wood, E.; Kirby, J.; Fusi, N.; Lawrence, N.; Wharton, S.B.; Ince, P.G.; Shaw, P.J.; et al. Unravelling the enigma of selective vulnerability in neurodegeneration: Motor neurons resistant to degeneration in als show distinct gene expression characteristics and decreased susceptibility to excitotoxicity. Acta Neuropathol. 2013, 125, 95–109. [Google Scholar]
- Sreedharan, J.; Brown, R.H., Jr. Amyotrophic lateral sclerosis: Problems and prospects. Ann. Neurol. 2013, 74, 309–316. [Google Scholar]
- Vucic, S.; Rothstein, J.D.; Kiernan, M.C. Advances in treating amyotrophic lateral sclerosis: Insights from pathophysiological studies. Trends Neurosci. 2014, 37, 433–442. [Google Scholar]
- Rosen, D.R.; Sapp, P.; O’Regan, J.; McKenna-Yasek, D.; Schlumpf, K.S.; Haines, J.L.; Gusella, J.F.; Horvitz, H.R.; Brown, R.H., Jr. Genetic linkage analysis of familial amyotrophic lateral sclerosis using human chromosome 21 microsatellite DNA markers. Am. J. Med. Genet. 1994, 51, 61–69. [Google Scholar]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar]
- Su, X.W.; Broach, J.R.; Connor, J.R.; Gerhard, G.S.; Simmons, Z. Genetic heterogeneity of amyotrophic lateral sclerosis: Implications for clinical practice and research. Muscle Nerve 2014, 49, 786–803. [Google Scholar]
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438. [Google Scholar]
- Renton, A.E.; Chio, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar]
- Di Giorgio, F.P.; Boulting, G.L.; Bobrowicz, S.; Eggan, K.C. Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell 2008, 3, 637–648. [Google Scholar]
- Wainger, B.J.; Kiskinis, E.; Mellin, C.; Wiskow, O.; Han, S.S.; Sandoe, J.; Perez, N.P.; Williams, L.A.; Lee, S.; Boulting, G.; et al. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 2014, 7, 1–11. [Google Scholar]
- Sareen, D.; O’Rourke, J.G.; Meera, P.; Muhammad, A.K.; Grant, S.; Simpkinson, M.; Bell, S.; Carmona, S.; Ornelas, L.; Sahabian, A.; et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Sanchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar]
- Buratti, E.; Brindisi, A.; Giombi, M.; Tisminetzky, S.; Ayala, Y.M.; Baralle, F.E. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail. J. Biol. Chem. 2005, 280, 37572–37584. [Google Scholar]
- Bilican, B.; Serio, A.; Barmada, S.J.; Nishimura, A.L.; Sullivan, G.J.; Carrasco, M.; Phatnani, H.P.; Puddifoot, C.A.; Story, D.; Fletcher, J.; et al. Mutant induced pluripotent stem cell lines recapitulate aspects of TDP-43 proteinopathies and reveal cell-specific vulnerability. Proc. Natl. Acad. Sci. USA 2012, 109, 5803–5808. [Google Scholar]
- Alami, N.H.; Smith, R.B.; Carrasco, M.A.; Williams, L.A.; Winborn, C.S.; Han, S.S.; Kiskinis, E.; Winborn, B.; Freibaum, B.D.; Kanagaraj, A.; et al. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 2014, 81, 536–543. [Google Scholar]
- Chen, H.; Qian, K.; Du, Z.; Cao, J.; Petersen, A.; Liu, H.; Blackbourn, L.W., IV; Huang, C.L.; Errigo, A.; Yin, Y.; et al. Modeling als with ipscs reveals that mutant SOD1 misregulates neurofilament balance in motor neurons. Cell Stem Cell 2014, 14, 796–809. [Google Scholar]
- Poppe, L.; Rue, L.; Robberecht, W.; van Den Bosch, L. Translating biological findings into new treatment strategies for amyotrophic lateral sclerosis (ALS). Exp. Neurol. 2014. [Google Scholar] [CrossRef]
- Egawa, N.; Kitaoka, S.; Tsukita, K.; Naitoh, M.; Takahashi, K.; Yamamoto, T.; Adachi, F.; Kondo, T.; Okita, K.; Asaka, I.; et al. Drug screening for ALS using patient-specific induced pluripotent stem cells. Sci. Transl. Med. 2012, 4. [Google Scholar] [CrossRef]
- Kondo, T.; Funayama, M.; Tsukita, K.; Hotta, A.; Yasuda, A.; Nori, S.; Kaneko, S.; Nakamura, M.; Takahashi, R.; Okano, H.; et al. Focal transplantation of human iPSC-derived glial-rich neural progenitors improves lifespan of als mice. Stem Cell Rep. 2014, 3, 242–249. [Google Scholar]
- Ebert, A.D.; Yu, J.; Rose, F.F., Jr.; Mattis, V.B.; Lorson, C.L.; Thomson, J.A.; Svendsen, C.N. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 2009, 457, 277–280. [Google Scholar]
- Sareen, D.; Ebert, A.D.; Heins, B.M.; McGivern, J.V.; Ornelas, L.; Svendsen, C.N. Inhibition of apoptosis blocks human motor neuron cell death in a stem cell model of spinal muscular atrophy. PLoS One 2012, 7, e39113. [Google Scholar]
- McGivern, J.V.; Patitucci, T.N.; Nord, J.A.; Barabas, M.E.; Stucky, C.L.; Ebert, A.D. Spinal muscular atrophy astrocytes exhibit abnormal calcium regulation and reduced growth factor production. Glia 2013, 61, 1418–1428. [Google Scholar]
- Lafont, E.; Milhas, D.; Teissie, J.; Therville, N.; Andrieu-Abadie, N.; Levade, T.; Benoist, H.; Segui, B. Caspase-10-dependent cell death in Fas/CD95 signalling is not abrogated by caspase inhibitor zVAD-fmk. PLoS One 2010, 5, e13638. [Google Scholar]
- Zhang, Z.; Lotti, F.; Dittmar, K.; Younis, I.; Wan, L.; Kasim, M.; Dreyfuss, G. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell 2008, 133, 585–600. [Google Scholar]
- Baumer, D.; Lee, S.; Nicholson, G.; Davies, J.L.; Parkinson, N.J.; Murray, L.M.; Gillingwater, T.H.; Ansorge, O.; Davies, K.E.; Talbot, K.; et al. Alternative splicing events are a late feature of pathology in a mouse model of spinal muscular atrophy. PLoS Genet. 2009, 5, e1000773. [Google Scholar]
- Brites, D.; Vaz, A.R. Microglia centered pathogenesis in als: Insights in cell interconnectivity. Front. Cell. Neurosci. 2014, 8, 117. [Google Scholar] [CrossRef]
- Benkler, C.; Ben-Zur, T.; Barhum, Y.; Offen, D. Altered astrocytic response to activation in SOD1(G93A) mice and its implications on amyotrophic lateral sclerosis pathogenesis. Glia 2013, 61, 312–326. [Google Scholar]
- Hirsch, E.C.; Hunot, S.; Hartmann, A. Neuroinflammatory processes in parkinson’s disease. Park. Relat. Disord. 2005, 11 (Suppl. 1), 9–15. [Google Scholar]
- Wang, D.D.; Bordey, A. The astrocyte odyssey. Prog. Neurobiol. 2008, 86, 342–367. [Google Scholar]
- Garbes, L.; Heesen, L.; Holker, I.; Bauer, T.; Schreml, J.; Zimmermann, K.; Thoenes, M.; Walter, M.; Dimos, J.; Peitz, M.; et al. VPA response in sma is suppressed by the fatty acid translocase CD36. Hum. Mol. Genet. 2013, 22, 398–407. [Google Scholar]
- Van Den Bosch, L. Genetic rodent models of amyotrophic lateral sclerosis. J. Biomed. Biotechnol. 2011, 2011. [Google Scholar] [CrossRef]
- Schmid, A.; DiDonato, C.J. Animal models of spinal muscular atrophy. J. Child Neurol. 2007, 22, 1004–1012. [Google Scholar]
- Re, D.B.; Le Verche, V.; Yu, C.; Amoroso, M.W.; Politi, K.A.; Phani, S.; Ikiz, B.; Hoffmann, L.; Koolen, M.; Nagata, T.; et al. Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron 2014, 81, 1001–1008. [Google Scholar]
- Vaskova, E.A.; Stekleneva, A.E.; Medvedev, S.P.; Zakian, S.M. “Epigenetic memory” phenomenon in induced pluripotent stem cells. Acta Naturae 2013, 5, 15–21. [Google Scholar]
- Hermann, A.; Storch, A. Induced neural stem cells (iNSCs) in neurodegenerative diseases. J. Neural Transm. 2013, 120 (Suppl. 1), 19–25. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faravelli, I.; Frattini, E.; Ramirez, A.; Stuppia, G.; Nizzardo, M.; Corti, S. iPSC-Based Models to Unravel Key Pathogenetic Processes Underlying Motor Neuron Disease Development. J. Clin. Med. 2014, 3, 1124-1145. https://doi.org/10.3390/jcm3041124
Faravelli I, Frattini E, Ramirez A, Stuppia G, Nizzardo M, Corti S. iPSC-Based Models to Unravel Key Pathogenetic Processes Underlying Motor Neuron Disease Development. Journal of Clinical Medicine. 2014; 3(4):1124-1145. https://doi.org/10.3390/jcm3041124
Chicago/Turabian StyleFaravelli, Irene, Emanuele Frattini, Agnese Ramirez, Giulia Stuppia, Monica Nizzardo, and Stefania Corti. 2014. "iPSC-Based Models to Unravel Key Pathogenetic Processes Underlying Motor Neuron Disease Development" Journal of Clinical Medicine 3, no. 4: 1124-1145. https://doi.org/10.3390/jcm3041124
APA StyleFaravelli, I., Frattini, E., Ramirez, A., Stuppia, G., Nizzardo, M., & Corti, S. (2014). iPSC-Based Models to Unravel Key Pathogenetic Processes Underlying Motor Neuron Disease Development. Journal of Clinical Medicine, 3(4), 1124-1145. https://doi.org/10.3390/jcm3041124