Non-Invasive Prenatal Testing Using Cell Free DNA in Maternal Plasma: Recent Developments and Future Prospects

Abstract
:
1. Introduction
2. Monogenic Disorders
2.1. Current Approaches
2.1.1. Paternally Inherited Autosomal Dominant and De Novo Mutation
2.1.2. Autosomal Recessive
2.1.3. Sex-linked or Sex Limited Conditions
2.2. Future Prospects, Generalized Approaches

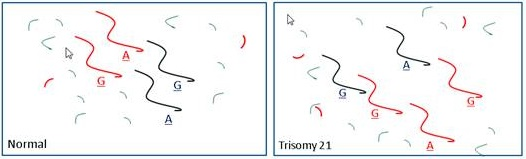
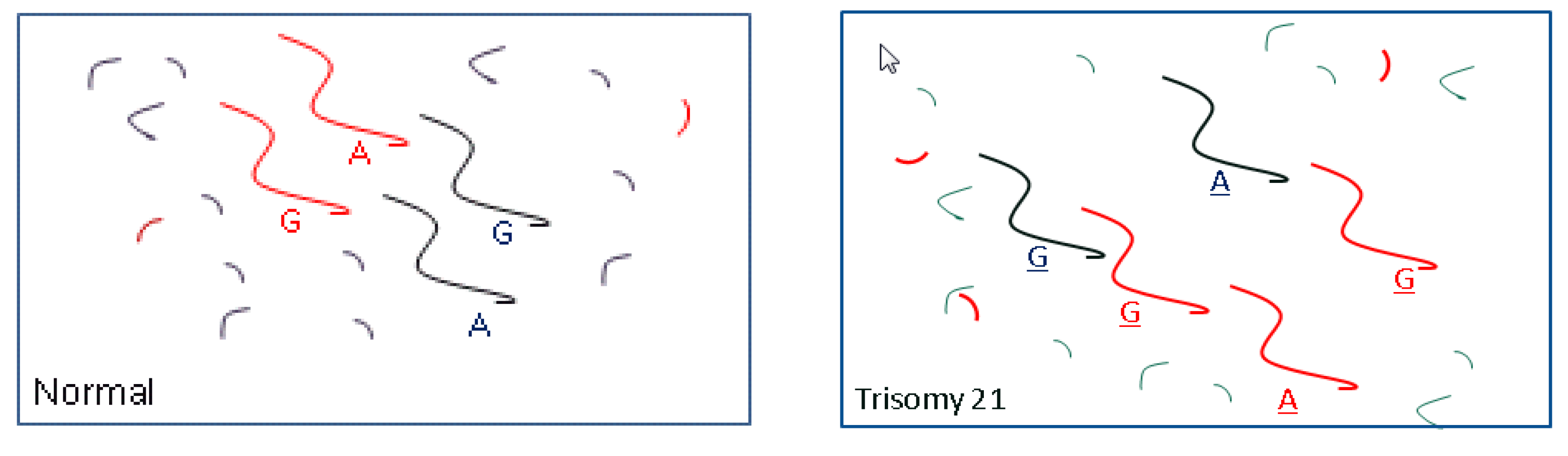
3. Aneuploidy
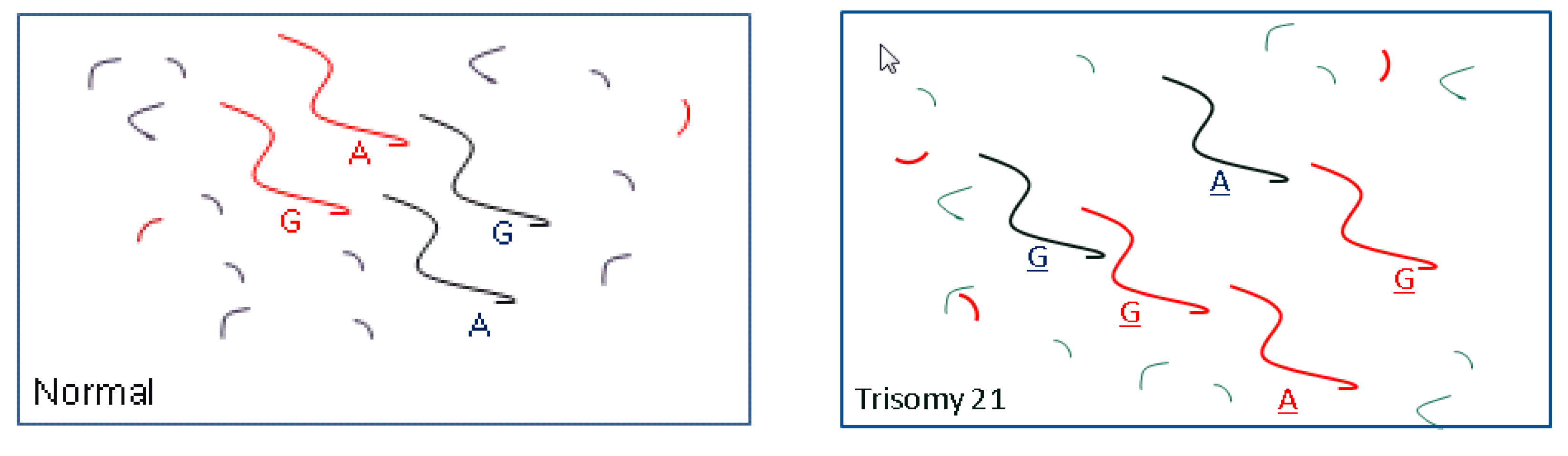
3.1. Methods
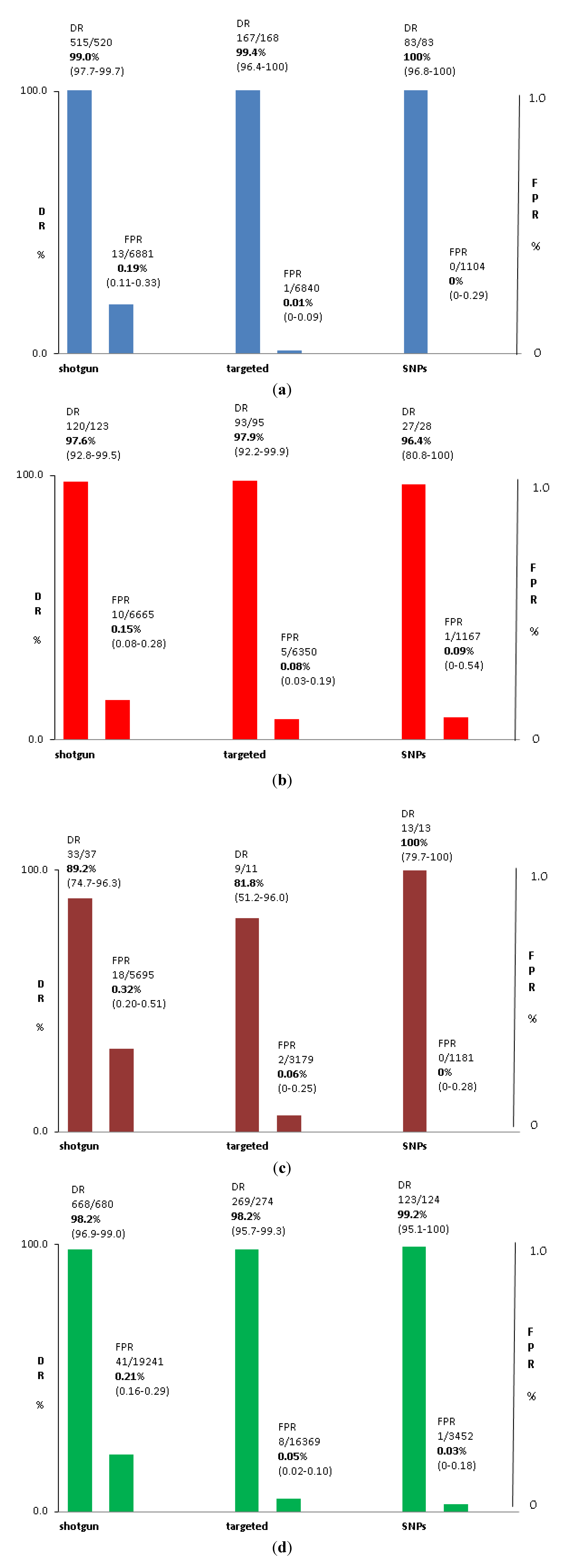
3.2. Test Performance: Autosomal Aneuploidy
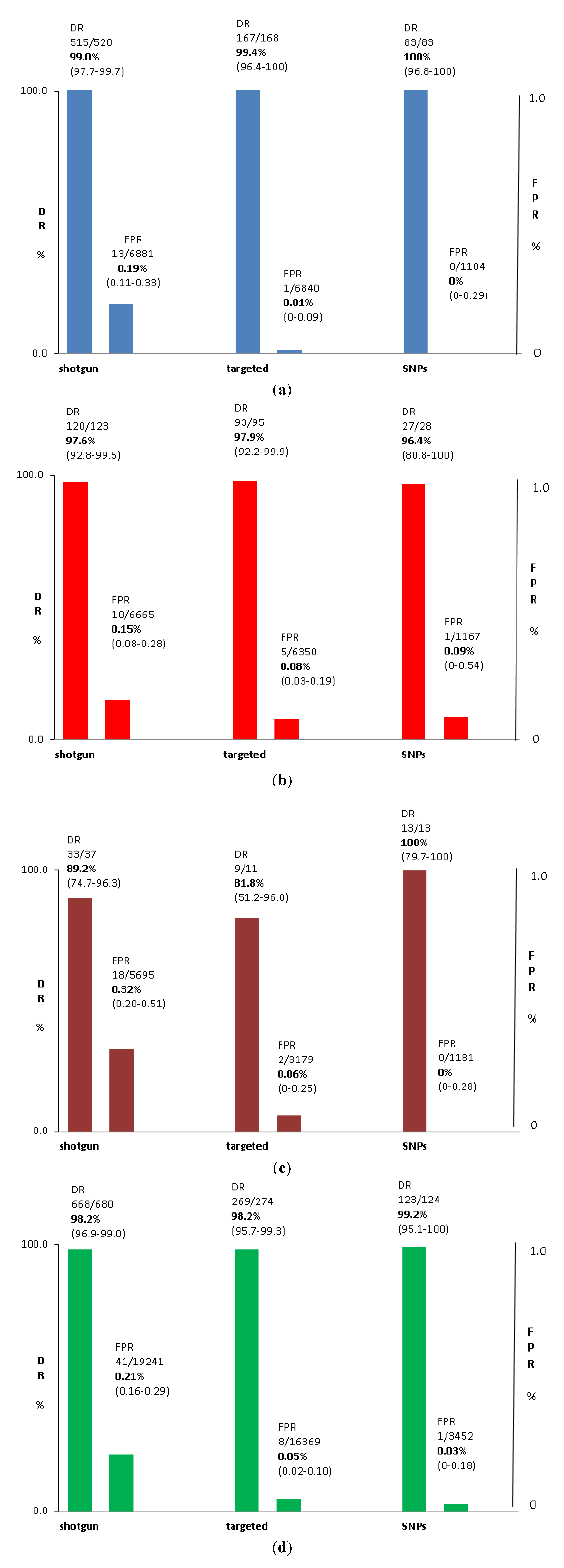

| Trial | DS DR | DS FPR | c21 NR | t18 DR | t18 FPR | c18 NR | t13 DR | t13 FPR | c13 NR |
|---|---|---|---|---|---|---|---|---|---|
| Chiu et al. [49] | 86/86 (100%) | 3/146 (2.05%) | 11/764 (1.4%) | ||||||
| Ehrich et al. [50] | 39/39 (100%) | 1/410 (0.24%) | 18/467 (3.9%) | ||||||
| Palomaki et al. [51,52] | 209/212 (98.6%) | 3/1471 (0.20%) | 13/1686 (0.8%) | 59/59 (100%) | 5/1688 (0.30%) | 17/1988 (0.9%) | 11/12 (91.7%) | 16/1688 (0.95%) | 17/1988 (0.9%) |
| Bianchi et al. [53] | 89/90 (98.9%) | 0/410 (0.00%) | 16/532 (3.0%) a | 35/38 (92.1%) | 0/463 (0.00%) | 16/532 (3.0%) b | 11/14 (78.6%) | 0/488 (0.00%) | 16/532 (3.0%) c |
| Liang et al. [40] | 40/40 (100%) | 0/372 (0.00%) | 12/435 (2.8%) | 14/14 (100%) | 0/372 (0.00%) | 12/435 (2.8%) | 4/4 (100%) | 1/408 (0.25%) | 12/435 (2.8%) |
| Song et al. [45] | 8/8 (100%) | 0/1733 (0.00%) | 73/1916 (3.8%) | 2/2 (100%) | 1/1739 (0.01%) | 73/1916 (3.8%) | 1/1 (100%) | 0/1740 (0.00%) | 73/1916 (3.8%) |
| Stumm et al. [54] | 39/40 (97.5%) | 0/430 (0.00%) | 32/504 (6.3%) | 8/8 (100%) | 1/472 (0.21%) | 32/504 (6.3%) | 5/5 (100%) | 0/472 (0.00%) | 32/504 (6.3%) |
| Bianchi et al. [46] | 5/5 (100%) | 6/1909 (0.31%) | 17/2042 (0.8%) | 2/2 (100%) | 3/1905 (0.16%) | 17/2042 (0.8%) | 1/1 (100%) | 1/899 (0.11%) | |
| Total | 99.0% | 0.19% | 2.30% | 97.6% | 0.15% | 2.3% | 89.2% | 0.32% | 2.8% |
| (95% CI) | (97.7%–99.7%) | (0.11%–0.33%) | (2.0%–2.7%) | (92.8%–99.5%) | (0.08%–0.28%) | (1.9%–2.6%) | (74.7%–96.3%) | (0.02%–0.51%) | (2.4%–3.3%) |
| Trial | DS DR | DS FPR | c21 NR | t18 DR | t18 FPR | c18 NR | t13 DR | t13 FPR | c13 NR |
|---|---|---|---|---|---|---|---|---|---|
| Ashoor et al. [55,56] | 50/50 (100%) | 0/297 (0.00%) | 3/400 (0.8%) | 49/50 (98.0%) | 0/297 (0.00%) | 3/400 (0.8%) | 8/10 (80%) | 2/1939 (0.05%) | 53/2002 (2.6%) |
| Verweij et al. [57] | 17/18 (94.4%) | 0/486 (0.00%) | 16/520 (3.1%) | ||||||
| Norton et al. [58] | 81/81 (100%) | 1/2888 (0.03%) | 148/3228 (4.6%) | 37/38 (97.4%) | 2/2888 (0.06%) | 148/3228 (4.6%) | |||
| Nicolaides et al. [47] | 8/8 (100%) | 0/1939 (0.00%) | 100/2049 (4.9%) | 2/2 (100%) | 2/1929 (0.01%) | 100/2049 (4.9%) | |||
| Fairbrother et al. [48] | - | 0/284 (0.00%) | 4/288 (1.4%) | - | 0/284 (0.00%) | 4/288 (1.4%) | - | 0/284 (0.00%) | 4/288 (1.4%) |
| Gil et al. [59] | 11/11 (100%) | 0/946 (0.00%) | 48/1005 (4.8%) | 5/5 (100%) | 1/952 (0.11%) | 48/1005 (4.8%) | 1/1 (100%) | 0/956 (0.00%) | 48/1005 (4.8%) |
| Total | 99.4% | 0.01% | 4.3% | 97.9% | 0.08% | 4.3% | 81.8% | 0.06% | 3.2% |
| (95% CI) | (96.4%–100%) | (0.00%–0.09%) | (3.8%–4.7%) | (92.2%–99.9%) | (0.03%–0.19%) | (3.9%–4.9%) | (51.2%–96.0%) | (0.00%–0.25%) | (2.6%–3.9%) |
| Trial | DS DR | DS FPR | c21 NR | t18 DR | t18 FPR | c18 NR | t13 DR | t13 FPR | c13 NR |
|---|---|---|---|---|---|---|---|---|---|
| Nicolaides et al. [60] | 25/25 (100%) | 0/204 (0.00%) | 13/242 (5.4%) | 3/3 (100%) | 0/226 (0.00%) | 13/242 (5.4%) | 1/1 (100%) | 0/228 (0.00%) | 13/242 (5.4%) |
| Pergament et al. [61] | 58/58 (100%) | 0/905 (0.00%) | 88/1051 (8.4%) | 24/25 (96%) | 1/939 (0.11%) | 87/1051 (8.3%) | 12/12 (100%) | 0/953 (0.00%) | 86/1051 (8.2%) |
| Total | 100.0% | 0.00% | 7.7% | 96.4% | 0.09% | 7.6% | 100.0% | 0.00% | 7.7% |
| (95% CI) | (96.8%–100%) | (0.00%–0.29%) | (6.3%–9.2%) | (80.8%–100%) | (0.00%–0.54%) | (6.3%–9.2%) | (79.7%–100%) | (0.00%–0.28%) | (6.3%–9.2%) |
3.3. Test Performance: Sex Chromosome Aneuploidy
3.4. Multiple Pregnancies
3.5. Triploidy
3.6. Test Failures
3.7. Reasons for Discordant Results
- (a)
- Low fetal fraction and/or insufficient depth of sequencing. For NIPT based on counting DNA fragments (i.e., s-MPS and t-MPS but not SNP based methods), test performance will be strongly dependent on a combination of the fetal fraction and the total numberof DNA fragments counted (i.e., the depth of sequencing) [83]. The effect of fetal fraction was also illustrated by Canick et al. [84] who showed that the z-values (the test statistic for distinguishing between affected and unaffected cases) for Down syndrome affected pregnancies were relatively close to normal when the fetal fraction was low. Consistent with this, Allen et al. [85] described a trisomy 21 NIPT false-negative result that appeared to be attributable to low fetal fraction.
- (b)
- Fetal and placental mosaicism. It is well known from cytogenetic studies that in mosaic cases, the proportion of cells showing each cell type can vary substantially from tissue to tissue and the proportions of cells seen in amniotic fluid cells and chorionic villi specimens can be a poor reflection of that present in individual tissues from the fetus [86]. Sometimes, an abnormal cell line is detected in some tissues but not others. NIPT relies on the analysis of DNA from trophoblasts with results generally reported as either positive or negative. The presence of two cell lines should therefore occasionally cause the NIPT result to appear to be discordant with the cytogenetic result [82]. In the most extreme situation, a cell line may be substantially confined to the placenta (confined placental mosaicism, CPM) with no evidence in other tissues. For some seemingly non-mosaic fetal aneuploidies (for example 45,X), the presence of a second cell line in the placenta may actually be the normal situation in surviving cases [87]. Numerous cases of discrepant NIPT results have now been attributed to CPM. For example, Pan et al. [88] described a case in which NIPT was positive for trisomy 21, karyotyping and QF-PCR indicated only two copies of chromosome 21 (but with uniparental disomy), and analysis of placenta showed evidence of a trisomy 21 cell population. Conversely, Wang et al. [89] described two cases in which NIPT was negative but where invasive test samples showed apparent non-mosaic trisomy 21 and subsequent analysis of placental biopsies showed mosaicism. Discordancy due to undetected mosaicism can be expected to arise regardless of which NIPT methodology is used.
- (c)
- Maternal chromosome abnormality. Another cause of discrepancy can be attributed to the presence of an abnormal karyotype or cell line in the mother, mistakenly attributed to the fetus. For example, the mother may have a non-mosaic 47,XXX karyotype [64,90], may have a low level constitutional mosaicism involving an autosome [45], a small imbalance [91], or have a malignancy that is karyotypically abnormal [92]. Maternal somatic cell mosaicism is expected to be quite common for loss or gain of an X-chromosome [63]. Wang et al. [64] reported evidence for maternal X-chromosome abnormality in 16 of 181 (8.6%) of cases with a NIPT result positive for a SCA. These causes for discordancy confound counting methods for NIPT but the approach based on SNP analyses should potentially be able to distinguish between at least some situations where a maternal mosaicism is present versus a fetal abnormality.
- (d)
- Multiple pregnancy and vanishing twin. As noted above, there is less confidence in testing for dizygotic and higher multiple pregnancies. If an aneuploid twin fetus is non-viable and unrecognized (vanishing twin) this could cause a discrepancy between NIPT and the invasive test result or outcome. Since placental tissue can persist long after a fetus is no longer evident and many fetal deaths can be attributed to aneuploidy, it seems likely that this will be increasingly recognized as a cause of false-positive results [44].
- (e)
- Laboratory error. Hopefully, errors in test and reporting procedures are rare and when they do occur corrective actions are implemented to prevent recurrence [44].
4. Future Developments in NIPT for Cytogenetic Abnormality
4.1. Women at Low Prior Risk
4.2. Other Aneuploidy and Chromosome Imbalances
4.3. Mosaicism
4.4. Microdeletions and Microduplications
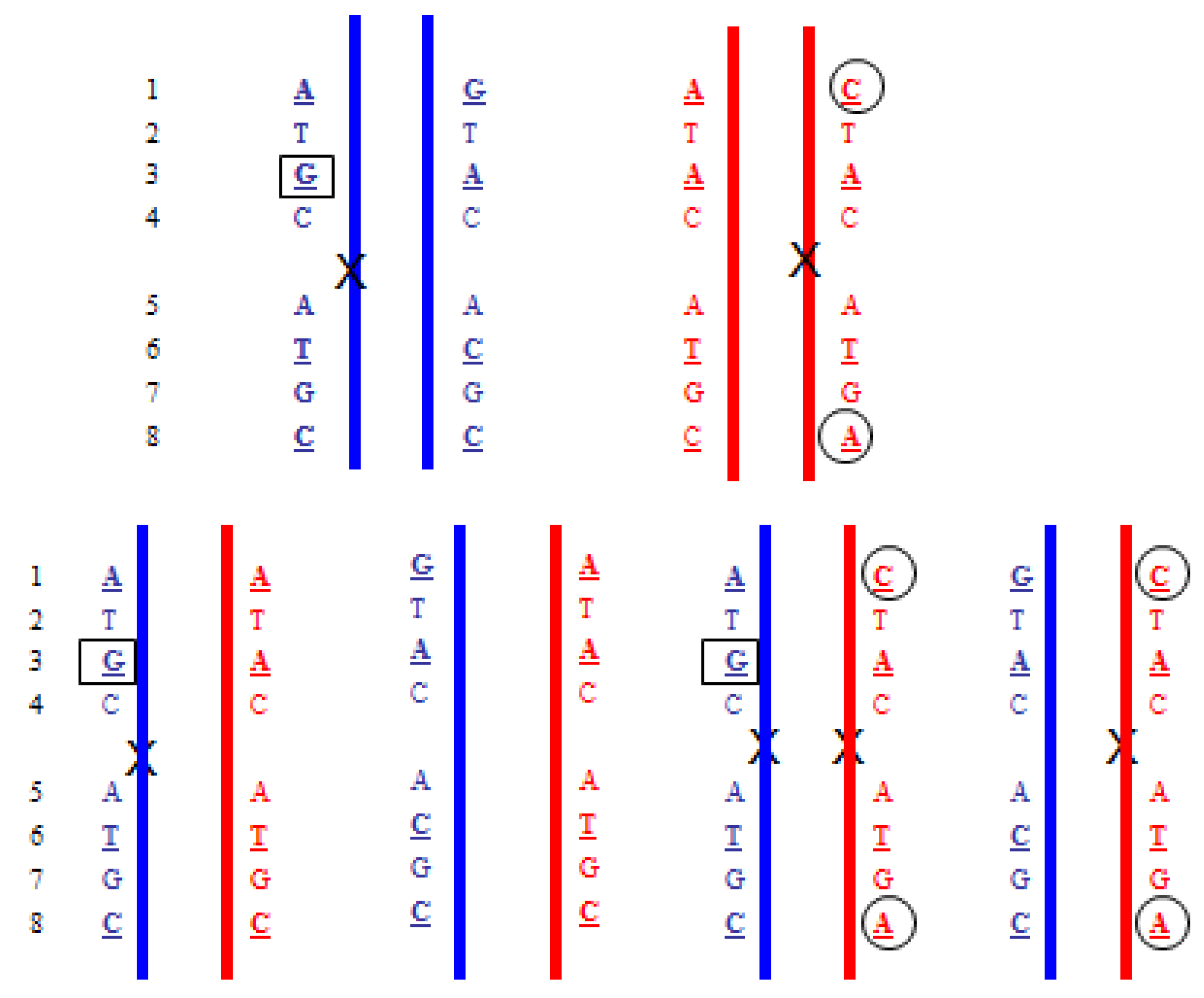
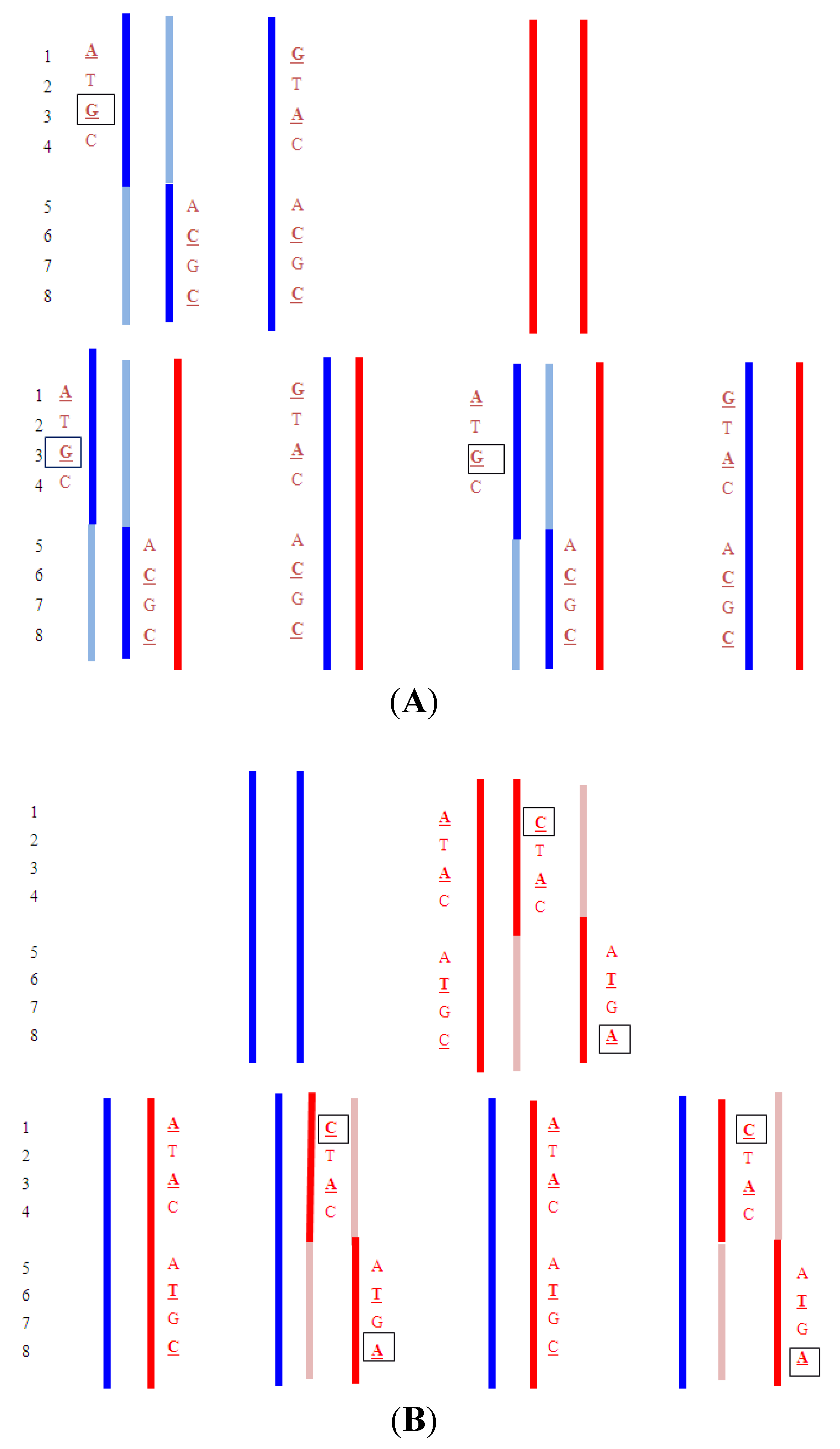
4.5. Treanslocations
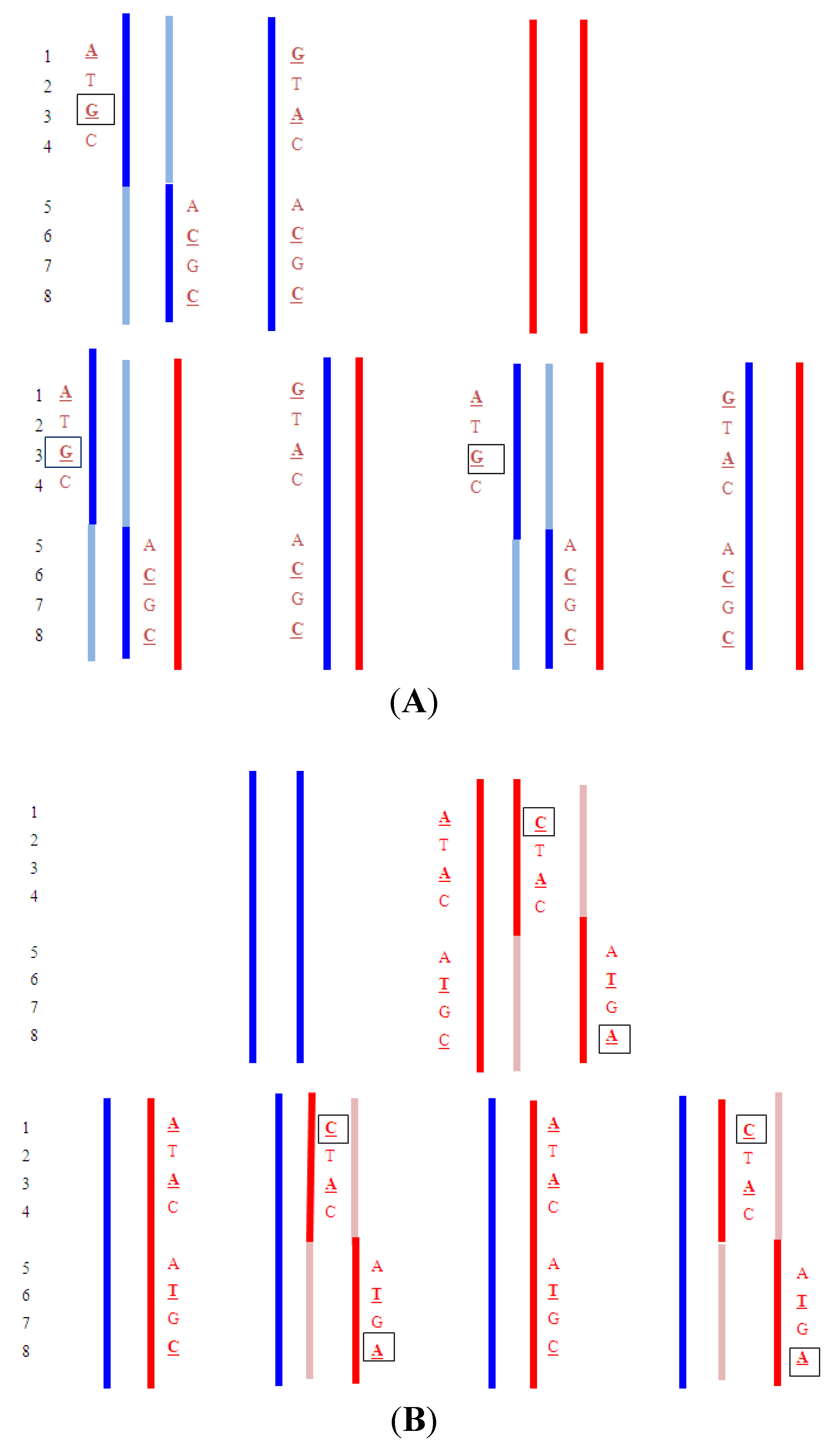
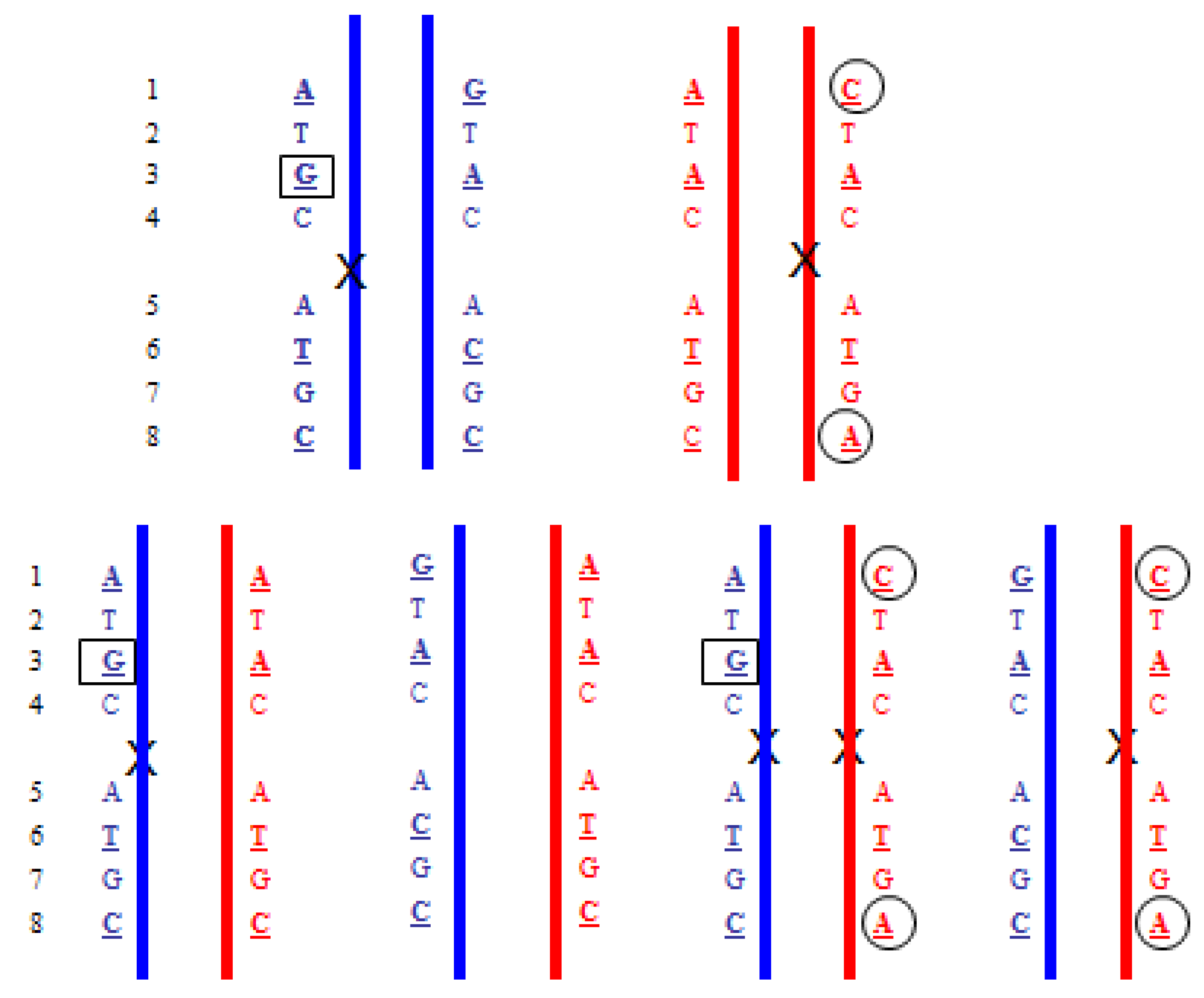
- Upper: Paternal reciprocal translocation between two chromosomes (light blue and dark blue). The normal homolog (solid dark blue chromosome) and the maternal copies of the same chromosomes (red) are shown. Haplotypes for the blue chromosome adjacent to the breakpoint are shown. Underlined bases are polymorphic, and boxed are informative. Lower: The four possible segregation products. The detection of the DNA fragments with
![]() indicates that the translocation chromosome was inherited. In practice, multiple linked SNPs would be used to be certain which chromosome was segregating. Similar analyses can be carried out for the light blue chromosome (haplotypes not shown).
indicates that the translocation chromosome was inherited. In practice, multiple linked SNPs would be used to be certain which chromosome was segregating. Similar analyses can be carried out for the light blue chromosome (haplotypes not shown). - Upper: Maternal reciprocal translocation between two chromosomes (red and pink). The normal homolog for the red chromosome together with the paternal copies of the same chromosome (blue are shown). Haplotypes for the red chromosome adjacent to the breakpoint are shown. Underlined bases are polymorphic, and boxed are informative. Lower: The four possible segregation products. The detection of an excess of the DNA fragments with
![]() and
and ![]() indicate that the translocation chromosomes were inherited. The maternally inherited haplotypes need to be detected in a background of maternal DNA and therefore the testing relies on detecting a relative excess (or deficiency) of particular fragments with the particular polymorphism.
indicate that the translocation chromosomes were inherited. The maternally inherited haplotypes need to be detected in a background of maternal DNA and therefore the testing relies on detecting a relative excess (or deficiency) of particular fragments with the particular polymorphism.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lo, Y.M.; Corbetta, N.; Chamberlain, P.F.; Rai, V.; Sargent, I.L.; Redman, C.W.; Wainscoat, J.S. Presence of fetal DNA in maternal plasma and serum. Lancet 1997, 350, 485–487. [Google Scholar] [CrossRef]
- Lo, Y.M.; Tein, M.S.; Lau, T.K.; Haines, C.J.; Leung, T.N.; Poon, P.M.; Wainscoat, J.S.; Johnson, P.J.; Chang, A.M.; Hjelm, N.M. Quantitative analysis of fetal DNA in maternal plasma and serum: Implications for noninvasive prenatal diagnosis. Am. J. Hum. Genet. 1998, 62, 768–775. [Google Scholar] [CrossRef]
- Lo, Y.M.; Hjelm, N.M.; Fidler, C.; Sargent, I.L.; Murphy, M.F.; Chamberlain, P.F.; Poon, P.M.; Redman, C.W.; Wainscoat, J.S. Prenatal diagnosis of fetal RhD status by molecular analysis of maternal plasma. N. Engl. J. Med. 1998, 339, 1734–1738. [Google Scholar] [CrossRef]
- Faas, B.H.W.; Beuling, E.A.; Christiaens, G.C.; von dem Borne, A.E.; van der Schoot, C.E. Detection of fetal RHD-specific sequences in maternal plasma. Lancet 1998, 352, 1196. [Google Scholar]
- Alberry, M.; Maddocks, D.; Jones, M.; Abdel Hadi, M.; Abdel-Fattah, S.; Avent, N.; Soothill, P.W. Free fetal DNA in maternal plasma in anembryonic pregnancies: Confirmation that the origin is the trophoblast. Prenat. Diagn. 2007, 27, 415–418. [Google Scholar] [CrossRef]
- Lo, Y.M.; Zhang, J.; Leung, T.N.; Lau, T.K.; Chang, A.M.; Hjelm, N.M. Rapid clearance of fetal DNA from maternal plasma. Am. J. Hum. Genet. 1999, 64, 218–224. [Google Scholar] [CrossRef]
- Smid, M.; Galbiati, S.; Vassallo, A.; Gambini, D.; Ferrari, A.; Viora, E.; Pagliano, M.; Restagno, G.; Ferrari, M.; Cremonesi, L. No evidence of fetal DNA persistence in maternal plasma after pregnancy. Hum. Genet. 2003, 112, 617–618. [Google Scholar]
- Hui, L.; Vaughan, J.I.; Nelson, M. Effect of labor on postpartum clearance of cell-free fetal DNA from the maternal circulation. Prenat. Diagn. 2008, 28, 304–308. [Google Scholar] [CrossRef]
- Benn, P.; Cuckle, H.; Pergament, E. Non-invasive prenatal testing for aneuploidy: Current status and future prospects. Ultrasound Obstet. Gynecol. 2013, 42, 15–33. [Google Scholar] [CrossRef]
- Gonzalez-Gonzalez, M.C.; Trujillo, M.J.; Rodriguez de Alba, M.; Garcia-Hoyos, M.; Lorda-Sanchez, I.; Diaz-Recasens, J.; Ayuso, C.; Ramos, C. Huntington disease-unaffected fetus diagnosed from maternal plasma using QF-PCR. Prenat. Diagn. 2003, 23, 232–234. [Google Scholar] [CrossRef]
- Rodríguez de Alba, M.; Bustamante-Aragonés, A.; Perlado, S.; Trujillo-Tiebas, M.J.; Díaz-Recasens, J.; Plaza-Arranz, J.; Ramos, C. Noninvasive prenatal diagnosis of monogenic disorders. Expert Opin. Biol. Ther. 2012, 12, S171–S179. [Google Scholar] [CrossRef]
- Amicucci, P.; Gennarelli, M.; Novelli, G.; Dallapiccola, B. Prenatal diagnosis of myotonic dystrophy using fetal DNA obtained from maternal plasma. Clin. Chem. 2000, 46, 301–302. [Google Scholar]
- Meaney, C.; Norbury, G. Noninvasive prenatal diagnosis of early onset primary dystonia I in maternal plasma. Prenat. Diagn. 2009, 29, 1218–1221. [Google Scholar] [CrossRef]
- González-González, M.C.; Garcia-Hoyos, M.; Trujillo-Tiebas, M.J.; Bustamante Aragonés, A.; Rodriguez de Alba, M.; Diego Alvarez, D.; Diaz-Recasens, J.; Ayuso, C.; Ramos, C. Improvement in strategies for the non-invasive prenatal diagnosis of Huntington disease. J. Assist. Reprod. Genet. 2008, 25, 477–481. [Google Scholar] [CrossRef]
- Avant, N.D. RHD genotyping from maternal plasma: Guidelines and technical challenges. Methods Mol. Biol. 2008, 444, 185–201. [Google Scholar] [CrossRef]
- Pauli, R.M. Achondroplasia. In GeneReviewsTM; Pagon, R.A., Adam, M.P., Bird, T.D., Dolan, C.R., Fong, C.T., Smith, R.J.H., Stephens, K., Eds.; University of Washington Seattle: Seattle, WA, USA.
- Saito, H.; Sekizawa, A.; Morimoto, T.; Suzuki, M.; Yanaihara, T. Prenatal DNA diagnosis of a single-gene disorder from maternal plasma. Lancet 2000, 356, 1170. [Google Scholar] [CrossRef]
- Chitty, L.S.; Griffin, D.R.; Meaney, C.; Barrett, A.; Khalil, A.; Pajkrt, E.; Cole, T.J. New aids for the non-invasive prenatal diagnosis of achondroplasia: Dysmorphic features, charts of fetal size and molecular confirmation using cell-free fetal DNA in maternal plasma. Ultrasound Obstet. Gynecol. 2011, 37, 283–289. [Google Scholar] [CrossRef]
- Chitty, L.S.; Khalil, A.; Barrett, A.N.; Pajkrt, E.; Griffin, D.R.; Cole, T.J. Safe, accurate, prenatal diagnosis of thanatophoric dysplasia using ultrasound and free fetal DNA. Prenat. Diagn. 2013, 33, 416–423. [Google Scholar] [CrossRef]
- Bustamante-Aragonés, A.; Rodríguez de Alba, M.; Perlado, S.; Trujillo-Tiebas, M.J.; Arranz, J.P.; Díaz-Recasens, J.; Troyano-Luque, J.; Ramos, C. Non-invasive prenatal diagnosis of single-gene disorders from maternal blood. Gene 2012, 504, 144–149. [Google Scholar] [CrossRef]
- Phylipsen, M.; Yamsri, S.; Treffers, E.E.; Jansen, D.T.; Kanhai, W.A.; Boon, E.M.; Giordano, P.C.; Fucharoen, S.; Bakker, E.; Harteveld, C.L. Non-invasive prenatal diagnosis of beta-thalassemia and sickle-cell disease using pyrophosphorolysis-activated polymerization and melting curve analysis. Prenat. Diagn. 2012, 32, 578–587. [Google Scholar] [CrossRef]
- Lun, F.M.; Nancy, B.Y.; Tsui, N.B.; Chan, K.C.; Leung, T.K.; Lau, T.K.; Charoenkwan, P.; Chow, K.C.; Lo, W.Y.; Wanapirak, C.; et al. Noninvasive prenatal diagnosis of monogenic diseases by digital size selection and relative mutation dosage on DNA in maternal plasma. Proc. Nat. Acad. Sci. USA 2008, 105, 19920–19925. [Google Scholar] [CrossRef]
- Lench, N.; Barrett, A.; Fielding, S.; McKay, F.; Hill, M.; Jenkins, L.; White, H.; Chitty, L.S. The clinical implementation of non-invasive prenatal diagnosis for single-gene disorders: Challenges and progress made. Prenat. Diagn. 2013, 33, 555–562. [Google Scholar] [CrossRef]
- Barrett, A.N.; McDonnell, T.C.; Chan, K.C.; Chitty, L.S. Digital PCR analysis of maternal plasma for noninvasive detection of sickle cell anemia. Clin. Chem. 2012, 58, 1026–1032. [Google Scholar] [CrossRef]
- Liao, G.J.; Lun, F.M.; Zheng, Y.W.; Chan, K.C.; Leung, T.Y.; Lau, T.K.; Chiu, R.W.; Lo, Y.M. Targeted massively parallel sequencing of maternal plasma DNA permits efficient and unbiased detection of fetal alleles. Clin. Chem. 2011, 57, 92–101. [Google Scholar] [CrossRef]
- Gu, W.; Koh, W.; Blumenfeld, Y.J.; El-Sayed, Y.Y.; Hudgins, L.; Hintz, S.R.; Quake, S.R. Noninvasive prenatal diagnosis in a fetus at risk for methylmalonic academia. Genet. Med. 2014. [Google Scholar] [CrossRef]
- Devaney, S.A.; Palomaki, G.E.; Scott, J.A.; Bianchi, D.W. Noninvasive fetal sex determination using cell-free fetal DNA. J. Am. Med. Assoc. 2011, 306, 627–636. [Google Scholar]
- Colmant, C.; Morin-Surroca, M.; Fuchs, F.; Fernandez, H.; Senat, M.V. Non-invasive prenatal testing for fetal sex determination: Is ultrasound still relevant? Eur. J. Obstet. Gynecol. Reprod. Biol. 2013, 171, 197–204. [Google Scholar]
- Madan, K.; Breuning, M.H. Impact of prenatal technologies on the sex ratio in India: An overview. Genet. Med. 2013. [Google Scholar] [CrossRef]
- Benn, P.A. Prenatal technologies and the sex ratio. Genet. Med. 2013. [Google Scholar] [CrossRef]
- Chapman, A.R.; Benn, P.A. Noninvasive prenatal testing for early sex identification: A few benefits and many concerns. Perspect. Biol. Med. 2013, 56, 530–547. [Google Scholar] [CrossRef]
- Lo, Y.M.D.; Chan, K.C.; Sun, H.; Chen, E.Z.; Jiang, P.; Lun, F.M.; Zheng, W.; Leung, T.Y.; Lau, T.K.; Cantor, C.R.; et al. Maternal plasma sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci. Trans. Med. 2010, 2. [Google Scholar] [CrossRef]
- Kitzman, J.O.; Snyder, M.W.; Ventura, M.; Lewis, A.; Qiu, R.; Simmons, L.E.; Gammill, H.S.; Rubens, C.E.; Santillan, D.A.; Murray, J.C.; et al. Noninvasive whole-genome sequencing of a human fetus. Sci. Transl. Med. 2012, 4. [Google Scholar] [CrossRef]
- Fan, H.C.; Gu, W.; Wang, J.; Blumenfeld, Y.J.; El-Sayed, Y.Y.; Quake, S.R. Non-invasive prenatal measurement of the fetal genome. Nature 2012, 487, 320–324. [Google Scholar] [CrossRef]
- Fan, H.C.; Blumenfeld, Y.J.; Chitkara, U.; Hudgins, L.; Quake, S.R. Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc. Natl. Acad. Sci. USA 2008, 105, 16266–16271. [Google Scholar] [CrossRef]
- Chiu, R.W.; Chan, K.C.; Gao, Y.; Lau, V.Y.; Zheng, W.; Leung, T.Y.; Foo, C.H.; Xie, B.; Tsui, N.B.; Lun, F.M.; et al. Noninvasive prenatal diagnosis of fetal chromosomal aneuploidy by massively parallel genomic sequencing of DNA in maternal plasma. Proc. Natl. Acad. Sci. USA 2008, 105, 20458–20463. [Google Scholar] [CrossRef]
- Sehnert, A.J.; Rhees, B.; Comstock, D.; de Feo, E.; Heilek, G.; Burke, J.; Rava, R.P. Optimal detection of fetal chromosomal abnormalities by massively parallel DNA sequencing: Of cell-free fetal DNA from maternal blood. Clin. Chem. 2011, 57, 1042–1049. [Google Scholar] [CrossRef]
- Dan, S.; Wang, W.; Ren, J.; Li, Y.; Hu, H.; Xu, Z.; Lau, T.K.; Xie, J.; Zhao, W.; Huang, H.; et al. Clinical application of massively parallel sequencing-based prenatal noninvasive fetal trisomy test for trisomies 21 and 18 in 11,105 pregnancies with mixed risk factors. Prenat. Diagn. 2012, 32, 1225–1232. [Google Scholar] [CrossRef]
- Fan, H.C.; Quake, S.R. Sensitivity of noninvasive prenatal detection of fetal aneuploidy from maternal plasma using shotgun sequencing is limited only by counting statistics. PLoS One 2010, 5, e10439. [Google Scholar] [CrossRef]
- Liang, D.; Lv, W.; Wang, H.; Xu, L.; Liu, J.; Li, H.; Hu, L.; Peng, Y.; Wu, L. Non-invasive prenatal testing of fetal whole chromosome aneuploidy by massively parallel sequencing. Prenat. Diagn. 2013, 33, 409–415. [Google Scholar] [CrossRef]
- Sparks, A.B.; Wang, E.T.; Struble, C.A.; Barrett, W.; Stokowski, R.; McBride, C.; Zahn, J.; Lee, K.; Shen, N.; Doshi, J.; et al. Selective analysis of cell-free DNA in maternal blood for evaluation of fetal trisomy. Prenat. Diagn. 2012, 32, 3–9. [Google Scholar] [CrossRef]
- Zimmermann, B.; Hill, M.; Gemelos, G.; Demko, Z.; Banjevic, M.; Baner, J.; Ryan, A.; Sigurjonsson, S.; Chopra, N.; Dodd, M.; et al. Noninvasive prenatal aneuploidy testing of chromosomes 13, 18, 21, X, and Y, using targeted sequencing of polymorphic loci. Prenat. Diagn. 2012, 32, 1233–1241. [Google Scholar] [CrossRef]
- Samango-Sprouse, C.; Banjevic, M.; Ryan, A.; Sigurjonsson, S.; Zimmermann, B.; Hill, M.; Hall, M.P.; Westemeyer, M.; Saucier, J.; Demko, Z.; et al. SNP-based non-invasive prenatal testing detects sex chromosome aneuploidies with high accuracy. Prenat. Diagn. 2013, 33, 643–649. [Google Scholar] [CrossRef]
- Futch, T.; Spinosa, J.; Bhatt, S.; de Feo, E.; Rava, R.P.; Sehnert, A.J. Initial clinical laboratory experience in noninvasive prenatal testing for fetal aneuploidy from maternal plasma DNA samples. Prenat. Diagn. 2013, 33, 569–574. [Google Scholar] [CrossRef]
- Song, Y.; Liu, C.; Qi, H.; Zhang, Y.; Bian, X.; Liu, J. Noninvasive prenatal testing of fetal aneuploidies by massively parallel sequencing in a prospective Chinese population. Prenat. Diagn. 2013, 33, 700–706. [Google Scholar] [CrossRef]
- Bianchi, D.W.; Parker, L.; Wentworth, J.; Madenkumar, R.; Saffer, C.; Das, A.F.; Craig, J.A.; Chudova, D.I.; Devers, P.L.; Jones, K.W.; et al. DNA sequencing versus standard prenatal aneuploidy screening. N. Engl. J. Med. 2014, 370, 799–808. [Google Scholar] [CrossRef]
- Nicolaides, K.H.; Syngelaki, A.; Ashoor, G.; Birdir, C.; Touzet, G. Noninvasive prenatal testing for fetal trisomies in a routinely screened first-trimester population. Am. J. Obstet. Gynecol. 2012, 207, e1–e6. [Google Scholar]
- Fairbrother, G.; Johnson, S.; Musci, T.J.; Song, K. Clinical experience of noninvasive prenatal testing with cell-free DNA for fetal trisomies 21, 18, and 13, in a general screening population. Prenat. Diagn. 2013, 33, 580–583. [Google Scholar] [CrossRef]
- Chiu, R.W.; Akolekar, R.; Zheng, Y.W.; Leung, T.Y.; Sun, H.; Chan, K.C.; Lun, F.M.; Go, A.T.; Lau, E.T.; To, W.W.; et al. Non-invasive prenatal assessment of trisomy 21 by multiplexed maternal plasma DNA sequencing: Large scale validation study. Br. Med. J. 2011, 342. [Google Scholar] [CrossRef]
- Ehrich, M.; Deciu, C.; Zweifellhofer, T.; Tynan, J.A.; Cagasan, L.; Tim, R.; Lu, V.; McCullough, R.; McCarthy, E.; Nygren, A.O.H.; et al. Noninvasive detection of fetal trisomy 21 by sequencing of DNA in maternal blood: A study in a clinical setting. Am. J. Obstet. Gynecol. 2011, 204, e1–e11. [Google Scholar] [CrossRef]
- Palomaki, G.E.; Kloza, E.M.; Lambert-Messerlian, G.M.; Haddow, J.E.; Neveux, L.M.; Ehrich, M.; van den Boom, D.; Bombard, A.T.; Deciu, C.; Grody, W.W.; et al. DNA sequencing of maternal plasma to detect Down syndrome: An international clinical validation study. Genet. Med. 2011, 13, 913–920. [Google Scholar] [CrossRef]
- Palomaki, G.E.; Deciu, C.; Kloza, E.M.; Lambert-Messerlian, G.M.; Haddow, J.E.; Neveux, L.M.; Ehrich, M.; van den Boom, D.; Bombard, A.T.; Grody, W.W.; et al. DNA sequencing of maternal plasma reliably identifies trisomy 18 and trisomy 13 as well as Down syndrome: An international collaborative study. Genet. Med. 2012, 14, 296–305. [Google Scholar] [CrossRef]
- Bianchi, D.W.; Platt, L.D.; Goldberg, J.D.; Abuhamad, A.Z.; Sehnert, A.J.; Rava, R.P. Genome-wide fetal aneuploidy detection by maternal plasma DNA sequencing. Obstet. Gynecol. 2012, 119, 890–901. [Google Scholar] [CrossRef]
- Stumm, M.; Entezami, M.; Haug, K.; Blank, C.; Wüstemann, M.; Schulze, B.; Raabe-Meyer, G.; Hempel, M.; Schelling, M.; Ostermayer, E.; et al. Diagnostic accuracy of random massively parallel sequencing for non-invasive prenatal detection of common autosomal aneuploidies: A collaborative study in Europe. Prenat. Diagn. 2013, 34, 185–191. [Google Scholar]
- Ashoor, G.; Syngelaki, A.; Wagner, M.; Birdir, C.; Nicolaides, K.H. Chromosome-selective sequencing of maternal plasma cell-free DNA for first-trimester detection of trisomy 21 and trisomy 18. Am. J. Obset. Gynecol. 2012, 206, e1–e5. [Google Scholar] [CrossRef]
- Ashoor, G.; Syngelaki, A.; Wang, E.; Struble, C.; Oliphant, A.; Song, K.; Nicolaides, K.H. Trisomy 13 detection in the first trimester of pregnancy using a chromosome-selective cell-free DNA analysis method. Ultrasound Obstet. Gynecol. 2013, 41, 21–25. [Google Scholar] [CrossRef]
- Verweij, E.J.; Jacobsson, B.; van Scheltema, P.A.; de Boer, M.A.; Hoffer, M.J.; Hollemon, D.; Westgren, M.; Song, K.; Oepkes, D. European non-invasive trisomy evaluation (EU-NITE) study: A multicenter prospective cohort study for non-invasive fetal trisomy 21 testing. Prenat. Diagn. 2013, 33, 996–1001. [Google Scholar]
- Norton, M.E.; Brar, H.; Weiss, J.; Karimi, A.; Laurent, L.C.; Caughey, A.B.; Rodriguez, M.H.; Williams, J., III; Mitchell, M.E.; Adair, C.D.; et al. Non-invasive chromosomal evaluation (NICE) study: Results of a multicenter prospective cohort study for detection of fetal trisomy 21 and trisomy 18. Am. J. Obstet. Gynecol. 2012, 207, e1–e8. [Google Scholar]
- Gil, M.M.; Quezada, M.S.; Bregant, B.; Ferraro, M.; Nicolaides, K.H. Implementation of maternal blood cell-free DNA testing in early screening for aneuploidies. Ultrasound Obstet. Gynecol. 2013, 42, 34–40. [Google Scholar] [CrossRef]
- Nicolaides, K.H.; Syngelaki, A.; Gil, M.; Atanasova, V.; Markova, D. Validation of targeted sequencing of single-nucleotide polymorphisms for non-invasive prenatal detection of aneuploidy of chromosomes 13, 18, 21, X and Y. Prenat. Diagn. 2013, 33, 575–579. [Google Scholar] [CrossRef]
- Pergament, E.; Cuckle, H.; Zimmermann, B.; Banjevic, M.; Sigurjonsson, S.; Ryan, A.; Dodd, M.; Lacroute, P.; Hall, M.P.; McAdoo, S.; et al. Single-nucleotide polymorphism-based non-invasive prenatal aneuploidy testing in a high- and low-risk cohort. Obstet. Gynecol. submitted for publication. 2014. [Google Scholar]
- Mazloom, A.R.; Džakula, Ž.; Oeth, P.; Wang, H.; Jensen, T.; Tynan, J.; McCullough, R.; Saldivar, J.S.; Ehrich, M.; van den Boom, D.; et al. Noninvasive prenatal detection of sex chromosomal aneuploidies by sequencing circulating cell-free DNA from maternal plasma. Prenat. Diagn. 2013, 33, 591–597. [Google Scholar] [CrossRef]
- Nicolaides, K.H.; Musci, T.J.; Struble, C.A.; Syngelaki, A.; Gil, M.M. Assessment of fetal sex chromosome aneuploidy using directed cell-free DNA analysis. Fetal Diagn. Ther. 2013, 35, 1–6. [Google Scholar]
- Wang, Y.; Chen, Y.; Tian, F.; Zhang, J.; Song, Z.; Wu, Y.; Han, X.; Hu, W.; Ma, D.; Cram, D.; et al. Maternal mosaicism is a significant contributor to discordant sex chromosomal aneuploidies associated with noninvasive prenatal testing. Clin. Chem. 2014, 60, 251–259. [Google Scholar] [CrossRef]
- Srinivasan, A.; Bianchi, D.; Liao, W.; Sehnert, A.; Rava, R. Maternal plasma DNA sequencing: Effects of multiple gestation on aneuploidy detection and the relative cell-free fetal DNA (cffDNA) per fetus. Am. J. Obstet. Gynecol. 2013, 208. [Google Scholar] [CrossRef]
- Struble, C.A.; Syngelaki, A.; Oliphant, A.; Song, K.; Nicolaides, K.H. Fetal fraction estimate in twin pregnancies using directed cell-free DNA analysis. Fetal Diagn. Ther. 2013, 35. [Google Scholar] [CrossRef]
- Canick, J.A.; Kloza, E.M.; Lambert-Messerlian, G.M.; Haddow, J.E.; Ehrich, M.; van den Boom, D.; Bombard, A.T.; Deciu, C.; Palomaki, G.E. DNA sequencing of maternal plasma to identify Down syndrome and other trisomies in multiple gestations. Prenat. Diagn. 2012, 32, 1–5. [Google Scholar]
- Lau, T.K.; Jiang, F.; Chan, M.K.; Zhang, H.; Salome Lo, P.S.; Wang, W. Non-invasive prenatal screening of fetal Down syndrome by maternal plasma DNA sequencing in twin pregnancies. J. Matern. Fetal Neonatal Med. 2013, 26, 434–437. [Google Scholar] [CrossRef]
- Gil, M.M.; Quezada, M.S.; Bregant, B.; Syngelaki, A.; Nicolaides, K.H. Cell-free DNA analysis for trisomy risk assessment in first-trimester twin pregnancies. Fetal Diagn. Ther. 2013, 35. [Google Scholar] [CrossRef]
- Huang, X.; Zheng, J.; Chen, M.; Zhao, Y.; Zhang, C.; Liu, L.; Xie, W.; Shi, S.; Wei, Y.; Lei, D.; et al. Non-invasive prenatal testing of trisomies 21 and 18 by massively parallel sequencing of maternal plasma DNA in twin pregnancies. Prenat. Diagn. 2013. [Google Scholar] [CrossRef]
- Nicolaides, K.H.; Syngelaki, A.; Gil, M.M.; Quezada, M.S.; Zinevich, Y. Prenatal detection of fetal triploidy from cell-free DNA testing in maternal blood. Fetal Diagn. Ther. 2013, 35. [Google Scholar] [CrossRef]
- Spencer, K.; Liao, A.W.; Skentou, H.; Cicero, S.; Nicolaides, K.H. Screening for triploidy by fetal nuchal translucency and maternal serum free beta-hCG and PAPP-A at 10–14 weeks of gestation. Prenat. Diagn. 2000, 20, 495–499. [Google Scholar] [CrossRef]
- Benn, P.A.; Gainey, A.; Ingardia, C.J.; Rodis, J.F.; Egan, J.F. Second trimester maternal serum analytes in triploid pregnancies: Correlation with phenotype and sex chromosome complement. Prenat. Diagn. 2001, 21, 680–686. [Google Scholar]
- Hancock, B.W.; Nazir, K.; Everard, J.E. Persistent gestational trophoblastic neoplasia after partial hydatidiform mole incidence and outcome. J. Reprod. Med. 2006, 51, 764–766. [Google Scholar]
- Vora, N.L.; Johnson, K.L.; Lambert-Messerlian, G.; Tighiouart, H.; Peter, I.; Urato, A.C.; Bianchi, D.W. Relationships between cell-free DNA and serum analytes in the first and second trimesters of pregnancy. Obstet. Gynecol. 2010, 116, 673–678. [Google Scholar] [CrossRef]
- Ashoor, G.; Poon, L.; Syngelaki, A.; Mosimann, B.; Nicolaides, K.H. Fetal fraction in maternal plasma cell-free DNA at 11–13 weeks’ gestation: Effect of maternal and fetal factors. Fetal Diagn. Ther. 2012, 31, 237–243. [Google Scholar] [CrossRef]
- Wang, E.; Batey, A.; Struble, C.; Musci, T.; Song, K.; Oliphant, A. Gestational age and maternal weight effects on fetal cell-free DNA in maternal plasma. Prenat. Diagn. 2013, 33, 662–666. [Google Scholar] [CrossRef]
- Vora, N.L.; Johnson, K.L.; Basu, S.; Catalano, P.M.; Hauguel-De Mouzon, S.; Bianchi, D.W. A multifactorial relationship exists between total circulating cell-free DNA levels and maternal BMI. Prenat. Diagn. 2012, 32, 912–914. [Google Scholar]
- Wegrzyn, P.; Faro, C.; Falcon, O.; Peralta, C.F.; Nicolaides, K.H. Placental volume measured by three-dimensional ultrasound at 11 to 13 + 6 weeks of gestation: Relation to chromosomal defects. Ultrasound Obstet. Gynecol. 2005, 26, 28–32. [Google Scholar] [CrossRef]
- Rava, R.P.; Srinivasan, A.; Sehnert, A.J.; Bianchi, D.W. Circulating fetal cell-free DNA fractions differ in autosomal aneuploidies and monosomy X. Clin. Chem. 2014, 60, 243–250. [Google Scholar] [CrossRef]
- Reiss, R.E.; Cherry, A.M. Still a screening test: More attention needed to noninvasive prenatal test false-positive rates. Am. J. Obstet. Gynecol. 2013, 209, 160–161. [Google Scholar] [CrossRef]
- Mennuti, M.T.; Cherry, A.M.; Morrissette, J.J.; Dugoff, L. Is it time to sound an alarm about false-positive cell-free DNA testing for fetal aneuploidy? Am. J. Obstet. Gynecol. 2013, 209, 415–419. [Google Scholar] [CrossRef]
- Benn, P.; Cuckle, H. Modeled performance of non-invasive prenatal testing for chromosome imbalances using counting. Of cell-free DNA fragments in maternal plasma. Prenat. Diagn. 2014. [Google Scholar] [CrossRef]
- Canick, J.A.; Palomaki, G.E.; Kloza, E.M.; Lambert-Messerlian, G.M.; Haddow, J.E. The impact of maternal plasma DNA fetal fraction on next generation sequencing tests for common fetal aneuploidies. Prenat. Diagn. 2013, 33, 667–674. [Google Scholar] [CrossRef]
- Allen, R.; Kezmarsky, K.; Lescale, L. False Negative NIPT and Potential Implications for Genetic Counseling. ACMG Annual Clinical Genetics Meeting 2013, Abstract 47. Available online: http://ww2.aievolution.com/acm1301/index.cfm?do=abs.viewAbs&abs=1427 (accessed on 22 March 2013).
- Benn, P.A. Prenatal diagnosis of chromosome abnormalities through amniocentesis. In Genetic Disorders and the Fetus, 6th ed.; Milunsky, A., Milunsky, J.M., Eds.; Wiley-Blackwell: Chichester, UK, 2010; pp. 194–272. [Google Scholar]
- Hook, E.B.; Warburton, D. Turner syndrome revisited: Review of new data supports the hypothesis that all viable 45,X cases are cryptic mosaics with a rescue cell line, implying an origin by mitotic loss. Hum. Genet. 2014, 133, 417–424. [Google Scholar] [CrossRef]
- Pan, M.; Li, F.T.; Li, Y.; Jiang, F.M.; Li, D.Z.; Lau, T.K.; Liao, C. Discordant results between fetal karyotyping and non-invasive prenatal testing by maternal plasma sequencing in a case of uniparental disomy 21 due to trisomic rescue. Prenat. Diagn. 2013, 33, 598–601. [Google Scholar] [CrossRef]
- Wang, Y.; Zhu, J.; Chen, Y.; Lu, S.; Chen, B.; Zhao, X.; Wu, Y.; Han, X.; Ma, D.; Liu, Z.; et al. Two cases of placental T21 mosaicism: Challenging the detection limits of non-invasive prenatal testing. Prenat. Diagn. 2013, 33, 1207–1210. [Google Scholar] [CrossRef]
- Yao, H.; Zhang, L.; Zhang, H.; Jiang, F.; Hu, H.; Chen, F.; Jiang, H.; Mu, F.; Zhao, L.; Liang, Z.; et al. Noninvasive prenatal genetic testing for fetal aneupliody detects maternal trisomy X. Prenat. Diagn. 2012, 32, 1114–1116. [Google Scholar] [CrossRef]
- Lau, T.K.; Jiang, F.M.; Stevenson, R.J.; Lo, T.K.; Chan, L.W.; Chan, M.K.; Lo, P.S.; Wang, W.; Zhang, H.Y.; Chen, F.; et al. Secondary findings from non-invasive prenatal testing for common fetal aneuploidies by whole genome sequencing as a clinical service. Prenat. Diagn. 2013, 33, 602–608. [Google Scholar] [CrossRef]
- Osborne, C.M.; Hardisty, E.; Devers, P.; Kaiser-Rogers, K.; Hayden, M.A.; Goodnight, W.; Vora, N.L. Discordant noninvasive prenatal testing results in a patient subsequently diagnosed with metastatic disease. Prenat. Diagn. 2013, 33, 609–611. [Google Scholar] [CrossRef]
- Brar, H.; Wang, E.; Struble, C.; Musci, T.; Norton, M. The fetal fraction of cell-free DNA in maternal plasma is not affected by a priori risk of fetal trisomy. J. Matern. Fetal Neonatal Med. 2013, 26, 143–145. [Google Scholar] [CrossRef]
- Benn, P.; Cuckle, H.; Pergament, E. Non-invasive prenatal diagnosis for Down syndrome: The paradigm will shift, but slowly. Ultrasound Obstet. Gynecol. 2012, 39, 127–130. [Google Scholar] [CrossRef]
- Norton, M.E.; Rose, N.C.; Benn, P. Noninvasive Prenatal Testing for Fetal Aneuploidy: Clinical Assessment and a Plea for Restraint. Obstet. Gynecol. 2013, 121, 847–850. [Google Scholar] [CrossRef]
- Morain, S.; Greene, M.F.; Mello, M.M. A new era in noninvasive prenatal testing. N. Engl. J. Med. 2013, 369, 499–501. [Google Scholar] [CrossRef]
- The American College of Obstetricians and Gynecologists Committee on Genetics and the Society for Maternal-Fetal Medicine Publications Committee. Noninvasive prenatal testing for fetal aneuploidy. Obstet. Gynecol. 2012, 120, 1532–1534. [Google Scholar] [CrossRef]
- Langlois, S.; Brock, J.-A. Current Status in Non-Invasive Prenatal Detection of Down Syndrome, Trisomy 18, and Trisomy 13 Using Cell-Free DNA in Maternal Plasm. J. Obstet. Gynaecol. Can. 2013, 35, 177–181. [Google Scholar]
- Wilson, K.L.; Czerwinski, J.L.; Hoskovec, J.M.; Noblin, S.J.; Sullivan, C.M.; Harbison, A.; Campion, M.W.; Devary, K.; Devers, P.; Singletary, C.N. NSGC practice guideline: Prenatal screening and diagnostic testing options for chromosome aneuploidy. J. Genet. Couns. 2013, 22, 4–15. [Google Scholar] [CrossRef]
- Benn, P.; Borell, A.; Chiu, R.; Cuckle, H.; Dugoff, L.; Faas, B.; Gross, S.; Johnson, J.; Maymon, R.; Norton, M.; et al. Position statement from the aneuploidy screening committee on behalf of the board of the international society for prenatal diagnosis. Prenat. Diagn. 2013, 33, 622–629. [Google Scholar] [CrossRef]
- Garfield, S.S.; Armstrong, S.O. Clinical and cost consequences of incorporating a novel non-invasive prenatal test into the diagnostic pathway for fetal trisomies. J. Manag. Care Med. 2012, 15, 34–41. [Google Scholar]
- Song, K.; Musci, T.; Caughey, A.B. Clinical utility and cost of non-invasive prenatal testing with cf-DNA analysis in high risk women based on a U.S. population. J. Matern. Fetal Neonatal Med. 2013, 26, 1180–1185. [Google Scholar] [CrossRef]
- Cuckle, H.; Benn, P.; Pergament, E. Clinical utility and cost of non-invasive prenatal testing. J. Matern. Fetal Neonatal Med. 2014, 27, 320–321. [Google Scholar] [CrossRef]
- Cuckle, H.; Benn, P.; Pergament, E. Maternal cf-DNA screening for Down’s syndrome—A cost sensitivity analysis. Prenat. Diagn. 2013, 33, 636–642. [Google Scholar] [CrossRef]
- Nicolaides, K.H.; Wright, D.; Poon, L.C.; Syngelaki, A.; Gil, M.M. First-trimester contingent screening for trisomy 21 by biomarkers and maternal blood cell-free DNA testing. Ultrasound Obstet. Gynecol. 2013, 42, 41–50. [Google Scholar] [CrossRef]
- Nicolaides, K.H.; Syngelaki, A.; Poon, L.C.; Gil, M.M.; Wright, D. First-trimester contingent screening for trisomies 21, 18 and 13 by biomarkers and maternal blood cell-free DNA testing. Fetal Diagn. Ther. 2013. [Google Scholar] [CrossRef]
- Johnson, J.; Pastuck, M.; Metcalfe, A.; Connors, G.; Krause, R.; Wilson, D.; Cuckle, H. First-trimester Down syndrome screening using additional serum markers with and without nuchal translucency and cell-free DNA. Prenat. Diagn. 2013, 33, 1044–1049. [Google Scholar] [CrossRef]
- Guex, N.; Iseli, C.; Syngelaki, A.; Deluen, C.; Pescia, G.; Nicolaides, K.H.; Xenarios, I.; Conrad, B. A robust second-generation genome-wide test for fetal aneuploidy based on shotgun sequencing cell-free DNA in maternal blood. Prenat. Diagn. 2013, 33, 707–710. [Google Scholar] [CrossRef]
- Chen, S.; Lau, T.K.; Zhang, C.; Xu, C.; Xu, Z.; Hu, P.; Xu, J.; Huang, H.; Pan, L.; Jiang, F.; et al. A method for noninvasive detection of fetal large deletions/duplications by low coverage massively parallel sequencing. Prenat. Diagn. 2013, 33, 584–590. [Google Scholar] [CrossRef]
- Dhallan, R.; Au, W.C.; Mattagajasingh, S.; Emche, S.; Bayliss, P.; Damewood, M.; Cronin, M.; Chon, V.; Mohr, M. Methods to increase the percentage of free fetal DNA recovered from the maternal circulation. JAMA 2004, 291, 1114–1119. [Google Scholar] [CrossRef]
- Go, A.T.; van Vugt, J.M.; Oudejans, C.B. Non-invasive aneuploidy detection using free fetal DNA and RNA in maternal plasma: Recent progress and future possibilities. Hum. Reprod. Update 2011, 17, 372–382. [Google Scholar] [CrossRef]
- Papageorgiou, E.A.; Fiegler, H.; Rakyan, V.; Beck, S.; Hulten, M.; Lamnissou, K.; Carter, N.P.; Patsalis, P.C. Sites of differential DNA methylation between placenta and peripheral blood: Molecular markers for noninvasive prenatal diagnosis of aneuploidies. Am. J. Pathol. 2009, 174, 1609–1618. [Google Scholar] [CrossRef]
- Forabosco, A.; Percesepe, A.; Santucci, S. Incidence of non-age-dependent chromosomal abnormalities: A population-based study on 88965 amniocenteses. Eur. J. Hum. Genet. 2009, 17, 897–903. [Google Scholar] [CrossRef]
- Ledbetter, D.H.; Zachary, J.M.; Simpson, J.L.; Golbus, M.S.; Pergament, E.; Jackson, L.; Mahoney, M.; Desnick, R.J.; Schulman, J.; Copeland, K.L.; et al. Cytogenetic results from the U.S. Collaborative Study on CVS. Prenat. Diagn. 1992, 12, 317–345. [Google Scholar] [CrossRef]
- Peters, D.; Chu, T.; Yatsenko, S.A.; Hendrix, N.; Hogge, W.A.; Surti, U.; Bunce, K.; Dunkel, M.; Shaw, P.; Rajkovic, A. Noninvasive prenatal diagnosis of a fetal microdeletion syndrome. N. Engl. J. Med. 2011, 365, 1847–1848. [Google Scholar] [CrossRef]
- Jensen, T.J.; Dzakula, Z.; Deciu, C.; van den Boom, D.; Ehrich, M. Detection of microdeletion 22q11.2 in a fetus by next-generation sequencing of maternal plasma. Clin. Chem. 2012, 58, 1148–1151. [Google Scholar] [CrossRef]
- Srinivasan, A.; Bianchi, D.W.; Huang, H.; Sehnert, A.J.; Rava, R.P. Noninvasive detection of fetal subchromosome abnormalities via deep sequencing of maternal plasma. Am. J. Hum. Genet. 2013, 92, 167–176. [Google Scholar] [CrossRef]
- Yu, S.C.; Jiang, P.; Choy, K.W.; Chan, K.C.; Won, H.S.; Leung, W.C.; Lau, E.T.; Tang, M.H.; Leung, T.Y.; Lo, Y.M.; et al. Noninvasive prenatal molecular karyotyping from maternal plasma. PLoS One 2013, 8, e60968. [Google Scholar] [CrossRef]
- Sequenom Inc. Available online: http://laboratories.sequenom.com/maternit21plus/maternit (accessed on 17 January 2014).
- Levy, B.; Wapner, R.; Melissa Savage, M.; Maisenbacher, M.; Sigurjonsson, S.; Hill, M.; Zimmermann, B.; Rabinowitz, M. Non-invasive Cell-free DNA-based Prenatal Detection of Microdeletions Using Single Nucleotide Polymorphism Targeted Sequencing. American Coledge of Medical Genetics 2014 Annual Meeting. Abstract 297. Available online: http://ww2.aievolution.com/acm1401/index.cfm?do=abs.viewAbs&abs=2518 (accessed on 17 May 2014).
- Yan, T.Z.; Mo, Q.H.; Cai, R.; Chen, X.; Zhang, C.M.; Liu, Y.-H.; Chen, Y.-J.; Zhou, W.J.; Xiong, F.; Xu, X.M. Reliable detection of paternal SNPs within deletion breakpoints for non-invasive prenatal exclusion of homozygous α-thalassemia in maternal plasma. PLoS One 2011, 6, e24779. [Google Scholar] [CrossRef]
- Lam, K.W.; Jiang, P.; Liao, G.J.; Chan, K.C.; Leung, T.Y.; Chiu, R.W.; Lo, Y.M. Noninvasive prenatal diagnosis of monogenic diseases by targeted massively parallel sequencing of maternal plasma: Application to β-thalassemia. Clin. Chem. 2012, 58, 1467–1475. [Google Scholar] [CrossRef]
- Ge, H.; Huang, X.; Li, X.; Chen, S.; Zheng, J.; Jiang, H.; Zhang, C.; Pan, X.; Guo, J.; Chen, F.; et al. Noninvasive prenatal detection for pathogenic CNVs: The application in α-thalassemia. PLoS One 2013, 8, e67464. [Google Scholar] [CrossRef]
- Benn, P.; Chapman, A.R.; Erickson, K.; Defrancesco, M.S.; Wilkins-Haug, L.; Egan, J.F.; Schulkin, J. Obstetricians’ and gynecologists’ practice and opinions of expanded carrier testing and non-invasive prenatal testing. Prenat. Diagn. 2013. [Google Scholar] [CrossRef]
- Bianchi, D.W. From prenatal genomic diagnosis to fetal personalized medicine: Progress and challenges. Nat. Med. 2012, 18, 1041–1051. [Google Scholar] [CrossRef]
- Tabor, A.; Alfirevic, Z. Update on procedure-related risks for prenatal diagnosis techniques. Fetal Diagn. Ther. 2010, 27, 1–7. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Benn, P. Non-Invasive Prenatal Testing Using Cell Free DNA in Maternal Plasma: Recent Developments and Future Prospects. J. Clin. Med. 2014, 3, 537-565. https://doi.org/10.3390/jcm3020537
Benn P. Non-Invasive Prenatal Testing Using Cell Free DNA in Maternal Plasma: Recent Developments and Future Prospects. Journal of Clinical Medicine. 2014; 3(2):537-565. https://doi.org/10.3390/jcm3020537
Chicago/Turabian StyleBenn, Peter. 2014. "Non-Invasive Prenatal Testing Using Cell Free DNA in Maternal Plasma: Recent Developments and Future Prospects" Journal of Clinical Medicine 3, no. 2: 537-565. https://doi.org/10.3390/jcm3020537
APA StyleBenn, P. (2014). Non-Invasive Prenatal Testing Using Cell Free DNA in Maternal Plasma: Recent Developments and Future Prospects. Journal of Clinical Medicine, 3(2), 537-565. https://doi.org/10.3390/jcm3020537