
Atypical Hemolytic Uremic Syndrome: A Review of Complement Dysregulation, Genetic Susceptibility and Multiorgan Involvement

 , , ,
, , ,  , and
, and
Abstract
1. Introduction
2. Genetic Factors and Disease Mechanisms
3. aHUS Manifestations
3.1. Cardiovascular and Pulmonary Involvement
3.2. Dermatologic and Systemic Signs
3.3. Neurological and Ocular Manifestations
3.4. Gastrointestinal Involvement
4. aHUS Diagnosis Criteria
- o Confirming the TMA triad;
- o Testing ADAMTS13 activity;
- o Evaluating for Shiga toxin-producing organisms (via stool PCR or serology);
- o Assessing secondary causes (e.g., pregnancy, autoimmune disease, malignancy);
5. Management Strategy
6. Discussions
7. Conclusions
8. Future Directions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Manrique-Caballero, C.L.; Peerapornratana, S.; Formeck, C.; Del Rio-Pertuz, G.; Gomez Danies, H.; Kellum, J.A. Typical and Atypical Hemolytic Uremic Syndrome in the Critically Ill. Crit. Care Clin. 2020, 36, 333–356. [Google Scholar] [CrossRef]
- George, J.N.; Nester, C.M. Syndromes of Thrombotic Microangiopathy. N. Engl. J. Med. 2014, 371, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Avila Bernabeu, A.I.; Cavero Escribano, T.; Cao Vilarino, M. Atypical Hemolytic Uremic Syndrome: New Challenges in the Complement Blockage Era. Nephron 2020, 144, 537–549. [Google Scholar] [PubMed]
- Sheerin, N.S.; Glover, E. Haemolytic uremic syndrome: Diagnosis and management. F1000Research 2019, 8, 1690. [Google Scholar]
- Zhang, K.; Lu, Y.; Harley, K.; Tran, M.H. Atypical Hemolytic Uremic Syndrome: A Brief Review. Hematol Rep. 2017, 9, 7053. [Google Scholar] [CrossRef]
- Bayer, G.; Von Tokarski, F.; Thoreau, B.; Bauvois, A.; Barbet, C.; Cloarec, S.; Mérieau, E.; Lachot, S.; Garot, D.; Bernard, L.; et al. Etiology and Outcomes of Thrombotic Microangiopathies. Clin. J. Am. Soc. Nephrol. 2019, 14, 557–566. [Google Scholar] [CrossRef]
- Tseng, M.H.; Lin, S.H.; Tsai, J.D.; Wu, M.S.; Tsai, I.J.; Chen, Y.C.; Chang, M.-C.; Chou, W.-C.; Chiou, Y.-H.; Huang, C.-C. Atypical hemolytic uremic syndrome: Consensus of diagnosis and treatment in Taiwan. J. Formos. Med. Assoc. 2023, 122, 366–375. [Google Scholar] [PubMed]
- Loirat, C.; Saland, J.; Bitzan, M. Management of hemolytic uremic syndrome. Presse Médicale 2012, 41, e115–e135. [Google Scholar]
- Tsai, H.M. Atypical Hemolytic Uremic Syndrome: Beyond Hemolysis and Uremia. Am. J. Med. 2019, 132, 161–167. [Google Scholar] [CrossRef]
- Loirat, C.; Frémeaux-Bacchi, V. Atypical hemolytic uremic syndrome. Orphanet J. Rare Dis. 2011, 6, 60. [Google Scholar]
- Story, C.M.; Gerber, G.F.; Chaturvedi, S. Medical consult: aHUS, TTP? How to distinguish and what to do. Hematology 2023, 2023, 745–753. [Google Scholar] [PubMed]
- Fakhouri, F.; Schwotzer, N.; Frémeaux-Bacchi, V. How I diagnose and treat atypical hemolytic uremic syndrome. Blood 2023, 141, 984–995. [Google Scholar] [PubMed]
- Brocklebank, V.; Wood, K.M.; Kavanagh, D. Thrombotic Microangiopathy and the Kidney. Clin. J. Am. Soc. Nephrol. 2018, 13, 300–317. [Google Scholar] [PubMed]
- Noris, M.; Remuzzi, G. Atypical Hemolytic–Uremic Syndrome. N. Engl. J. Med. 2009, 361, 1676–1687. [Google Scholar]
- Global aHUS Registry [Internet]. Available online: https://ahusregistry.com/ (accessed on 24 March 2025).
- Zimmerhackl, L.; Besbas, N.; Jungraithmayr, T.; Kar, N.; Karch, H.; Karpman, D.; Landau, D.; Loirat, C.; Proesmans, W.; Prüfer, F.; et al. Epidemiology, Clinical Presentation, and Pathophysiology of Atypical and Recurrent Hemolytic Uremic Syndrome. Semin. Thromb. Hemost. 2006, 32, 113–120. [Google Scholar]
- Constantinescu, A.R.; Bitzan, M.; Weiss, L.S.; Christen, E.; Kaplan, B.S.; Cnaan, A.; Trachtman, H. Non-enteropathic hemolytic uremic syndrome: Causes and short-term course. Am. J. Kidney Dis. 2004, 43, 976–982. [Google Scholar]
- Fremeaux-Bacchi, V.; Fakhouri, F.; Garnier, A.; Bienaimé, F.; Dragon-Durey, M.A.; Ngo, S.; Moulin, B.; Servais, A.; Provot, F.; Rostaing, L.; et al. Genetics and Outcome of Atypical Hemolytic Uremic Syndrome: A Nationwide French Series Comparing Children and Adults. Clin. J. Am. Soc. Nephrol. 2013, 8, 554–562. [Google Scholar]
- Licht, C.; Ardissino, G.; Ariceta, G.; Cohen, D.; Cole, J.A.; Gasteyger, C.; Greenbaum, L.A.; Johnson, S.; Ogawa, M.; Schaefer, F.; et al. The global aHUS registry: Methodology and initial patient characteristics. BMC Nephrol. 2015, 16, 207. [Google Scholar]
- Stevens, P.E.; Ahmed, S.B.; Carrero, J.J.; Foster, B.; Francis, A.; Hall, R.K.; Herrington, W.G.; Hill, G.; Inker, L.A.; Kazancıoğlu, R.; et al. KDIGO 2024 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int. 2024, 105, S117–S314. [Google Scholar]
- Formeck, C.; Swiatecka-Urban, A. Extra-renal manifestations of atypical hemolytic uremic syndrome. Pediatr. Nephrol. 2019, 34, 1337–1348. [Google Scholar]
- Chaturvedi, S.; Moliterno, A.R.; Merrill, S.A.; Braunstein, E.M.; Yuan, X.; Sperati, C.J.; Khneizer, G.; Brodsky, R.A. Chronic Kidney Disease, Hypertension and Cardiovascular Sequelae during Long Term Follow up of Adults with Atypical Hemolytic Uremic Syndrome. Blood 2018, 132 (Suppl. S1), 3754. [Google Scholar]
- Fakhouri, F.; Zuber, J.; Frémeaux-Bacchi, V.; Loirat, C. Haemolytic uraemic syndrome. Lancet 2017, 390, 681–696. [Google Scholar] [PubMed]
- Loirat, C.; Fakhouri, F.; Ariceta, G.; Besbas, N.; Bitzan, M.; Bjerre, A.; Coppo, R.; Emma, F.; Johnson, S.; Karpman, D.; et al. An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr. Nephrol. 2016, 31, 15–39. [Google Scholar] [PubMed]
- Noris, M.; Bresin, E.; Mele, C.; Remuzzi, G. Genetic Atypical Hemolytic-Uremic Syndrome. In GeneReviews® [Internet]; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. Available online: http://www.ncbi.nlm.nih.gov/books/NBK1367/ (accessed on 20 February 2025).
- Kavanagh, D.; Goodship, T.H.; Richards, A. Atypical Hemolytic Uremic Syndrome. Semin. Nephrol. 2013, 33, 508–530. [Google Scholar]
- Noris, M.; Caprioli, J.; Bresin, E.; Mossali, C.; Pianetti, G.; Gamba, S.; Daina, E.; Fenili, C.; Castelletti, F.; Sorosina, A.; et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin. J. Am. Soc. Nephrol. CJASN 2010, 5, 1844–1859. [Google Scholar]
- Sepúlveda Palamara, R.A.; Modelli De Andrade, L.G.; Fortunato, R.M.; Gómez, B.; Nieto-Ríos, J.F. Clinical presentation and management of atypical hemolytic uremic syndrome in Latin America: A narrative review of the literature. Expert. Rev. Hematol. 2024, 17, 361–374. [Google Scholar]
- Ávila, A.; Gavela, E.; Sancho, A. Thrombotic Microangiopathy After Kidney Transplantation: An Underdiagnosed and Potentially Reversible Entity. Front. Med. 2021, 8, 642864. [Google Scholar]
- Fakhouri, F.; Frémeaux-Bacchi, V. Thrombotic microangiopathy in aHUS and beyond: Clinical clues from complement genetics. Nat. Rev. Nephrol. 2021, 17, 543–553. [Google Scholar] [CrossRef]
- Leon, J.; LeStang, M.; Sberro-Soussan, R.; Servais, A.; Anglicheau, D.; Frémeaux-Bacchi, V.; Zuber, J. Complement-driven hemolytic uremic syndrome. Am. J. Hematol. 2023, 98 (Suppl. S4), S44–S56. [Google Scholar] [CrossRef]
- Raina, R.; Krishnappa, V.; Blaha, T.; Kann, T.; Hein, W.; Burke, L.; Bagga, A. Atypical Hemolytic-Uremic Syndrome: An Update on Pathophysiology, Diagnosis, and Treatment. Ther. Apher. Dial. 2019, 23, 4–21. [Google Scholar] [CrossRef]
- Smith-Jackson, K.; Walsh, P.; Zelek, W.M.; Hoyler, T.; Martinic, M.M.; Thompson, G.; Gibson, B.G.; Connelly, C.; Pappworth, I.Y.; Murphy, M.J.; et al. The membrane attack complex drives thrombotic microangiopathy in complement mediated atypical hemolytic uremic syndrome. Kidney Int. 2025, 107, 700–713. [Google Scholar]
- Yoshida, Y.; Kato, H.; Ikeda, Y.; Nangaku, M. Pathogenesis of Atypical Hemolytic Uremic Syndrome. J. Atheroscler. Thromb. 2019, 26, 99–110. [Google Scholar] [PubMed]
- Wada, T.; Nangaku, M. Novel roles of complement in renal diseases and their therapeutic consequences. Kidney Int. 2013, 84, 441–450. [Google Scholar] [PubMed]
- Lemaire, M.; Frémeaux-Bacchi, V.; Schaefer, F.; Choi, M.; Tang, W.H.; Le Quintrec, M.; Fakhouri, F.; Taque, S.; Nobili, F.; Martinez, F.; et al. Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome. Nat. Genet. 2013, 45, 531–536. [Google Scholar] [PubMed]
- Fakhouri, F.; Scully, M.; Ardissino, G.; Al-Dakkak, I.; Miller, B.; Rondeau, E. Pregnancy-triggered atypical hemolytic uremic syndrome (aHUS): A Global aHUS Registry analysis. J. Nephrol. 2021, 34, 1581–1590. [Google Scholar]
- Caprioli, J.; Noris, M.; Brioschi, S.; Pianetti, G.; Castelletti, F.; Bettinaglio, P.; Mele, C.; Bresin, E.; Cassis, L.; Gamba, S.; et al. Genetics of HUS: The impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood 2006, 108, 1267–1279. [Google Scholar]
- Fremeaux-Bacchi, V. The development of atypical haemolytic-uraemic syndrome is influenced by susceptibility factors in factor H and membrane cofactor protein: Evidence from two independent cohorts. J. Med. Genet. 2005, 42, 852–856. [Google Scholar]
- Dixon, B.P.; Gruppo, R.A. Atypical Hemolytic Uremic Syndrome. Pediatr. Clin. N. Am. 2018, 65, 509–525. [Google Scholar]
- Marinozzi, M.C.; Vergoz, L.; Rybkine, T.; Ngo, S.; Bettoni, S.; Pashov, A.; Cayla, M.; Tabarin, F.; Jablonski, M.; Hue, C.; et al. Complement Factor B Mutations in Atypical Hemolytic Uremic Syndrome—Disease-Relevant or Benign? J. Am. Soc. Nephrol. 2014, 25, 2053–2065. [Google Scholar]
- Delvaeye, M.; Noris, M.; De Vriese, A.; Esmon, C.T.; Esmon, N.L.; Ferrell, G.; Del-Favero, J.; Plaisance, S.; Claes, B.; Lambrechts, D.; et al. Thrombomodulin Mutations in Atypical Hemolytic–Uremic Syndrome. N. Engl. J. Med. 2009, 361, 345–357. [Google Scholar]
- Tasaki, Y.; Tsujimoto, H.; Yokoyama, T.; Sugimoto, N.; Kitajima, S.; Fujii, H.; Hidaka, Y.; Kato, N.; Maruyama, S.; Inoue, N.; et al. Case report: A family of atypical hemolytic uremic syndrome involving a CFH::CFHR1 fusion gene and CFHR3-1-4-2 gene duplication. Front. Immunol. 2024, 15, 1360855. [Google Scholar]
- Pickering, M.C.; De Jorge, E.G.; Martinez-Barricarte, R.; Recalde, S.; Garcia-Layana, A.; Rose, K.L.; Moss, J.; Walport, M.J.; Cook, H.T.; de Córdoba, S.R.; et al. Spontaneous hemolytic uremic syndrome triggered by complement factor H lacking surface recognition domains. J. Exp. Med. 2007, 204, 1249–1256. [Google Scholar]
- Martín Merinero, H.; Zhang, Y.; Arjona, E.; Del Angel, G.; Goodfellow, R.; Gomez-Rubio, E.; Ji, R.-R.; Michelena, M.; Smith, R.J.H.; de Córdoba, S.R. Functional characterization of 105 factor H variants associated with aHUS: Lessons for variant classification. Blood 2021, 138, 2185–2201. [Google Scholar]
- Zipfel, P.F.; Edey, M.; Heinen, S.; Józsi, M.; Richter, H.; Misselwitz, J.; Hoppe, B.; Routledge, D.; Strain, L.; Hughes, A.E.; et al. Deletion of Complement Factor H–Related Genes CFHR1 and CFHR3 Is Associated with Atypical Hemolytic Uremic Syndrome. Roopenian DC, editor. PLoS Genet. 2007, 3, e41. [Google Scholar]
- Sinha, A.; Gulati, A.; Saini, S.; Blanc, C.; Gupta, A.; Gurjar, B.S.; Saini, H.; Kotresh, S.T.; Ali, U.; Bhatia, D.; et al. Prompt plasma exchanges and immunosuppressive treatment improves the outcomes of anti-factor H autoantibody-associated hemolytic uremic syndrome in children. Kidney Int. 2014, 85, 1151–1160. [Google Scholar] [PubMed]
- Puraswani, M.; Khandelwal, P.; Saini, H.; Saini, S.; Gurjar, B.S.; Sinha, A.; Shende, R.P.; Maiti, T.K.; Singh, A.K.; Kanga, U.; et al. Clinical and Immunological Profile of Anti-factor H Antibody Associated Atypical Hemolytic Uremic Syndrome: A Nationwide Database. Front. Immunol. 2019, 10, 1282. [Google Scholar]
- Durey, M.A.D.; Sinha, A.; Togarsimalemath, S.K.; Bagga, A. Anti-complement-factor H-associated glomerulopathies. Nat. Rev. Nephrol. 2016, 12, 563–578. [Google Scholar]
- Damoiseaux, J. The perspective on standardisation and harmonisation: The viewpoint of the EASI president. Autoimmun. Highlights 2020, 11, 4. [Google Scholar] [CrossRef]
- Nugteren, S.; Wang, H.; Van Kooten, C.; Gelderman, K.A.; Trouw, L.A. Autoantibodies and therapeutic antibodies against complement Factor H. Immunol. Lett. 2025, 274, 107002. [Google Scholar]
- Dragon-Durey, M.A.G.A.; Loirat, C.; Cloarec, S.; Macher, M.A.; Blouin, J.; Nivet, H.; Weiss, L.; Fridman, W.H.; Frémeaux-Bacchi, V. Anti–Factor H Autoantibodies Associated with Atypical Hemolytic Uremic Syndrome. J. Am. Soc. Nephrol. 2005, 16, 555–563. [Google Scholar]
- Rodríguez De Córdoba, S.; Reparaz, A.; Sanchez, D.; Pinto, S.; Juana Lopez, L.; Martin Merinero, H.; Calvete, I.; Perez-Perez, J.; Jellison, S.S.; Zhang, Y.; et al. Novel immunochromatographic test for rapid detection of anti-factor H autoantibodies with an assessment of its clinical relevance. Front. Immunol. 2025, 15, 1527016. [Google Scholar]
- Cugno, M.; Mancuso, M.C.; Depetri, F.; Peyvandi, F.; Ardissino, G. IgM autoantibodies to complement factor H in C3 glomerulopathy. J. Nephrol. 2024, 37, 1415–1416. [Google Scholar] [PubMed]
- Cugno, M.; Berra, S.; Depetri, F.; Tedeschi, S.; Griffini, S.; Grovetti, E.; Caccia, S.; Cresseri, D.; Messa, P.; Testa, S.; et al. IgM Autoantibodies to Complement Factor H in Atypical Hemolytic Uremic Syndrome. J. Am. Soc. Nephrol. 2021, 32, 1227–1235. [Google Scholar] [PubMed]
- Legendre, C.M.; Licht, C.; Muus, P.; Greenbaum, L.A.; Babu, S.; Bedrosian, C.; Cohen, D.J.; Delmas, Y.; Douglas, K.; Eitner, F.; et al. Terminal Complement Inhibitor Eculizumab in Atypical Hemolytic–Uremic Syndrome. N. Engl. J. Med. 2013, 368, 2169–2181. [Google Scholar]
- Goodship, T.H.J.; Cook, H.T.; Fakhouri, F.; Fervenza, F.C.; Frémeaux-Bacchi, V.; Kavanagh, D.; Nester, C.M.; Noris, M.; Pickering, M.C.; de Córdoba, S.R.; et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: Conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int. 2017, 91, 539–551. [Google Scholar]
- Fidan, K.; Göknar, N.; Gülhan, B.; Melek, E.; Yıldırım, Z.Y.; Baskın, E.; Hayran, M.; Gülleroglu, K.; Özçakar, Z.B.; Ozaltin, F.; et al. Extra-Renal manifestations of atypical hemolytic uremic syndrome in children. Pediatr. Nephrol. 2018, 33, 1395–1403. [Google Scholar] [PubMed]
- Noris, M.; Remuzzi, G. Cardiovascular complications in atypical haemolytic uraemic syndrome. Nat. Rev. Nephrol. 2014, 10, 174–180. [Google Scholar]
- Neuhaus, T.J.; Calonder, S.; Leumann, E.P. Heterogeneity of atypical haemolytic uraemic syndromes. Arch. Dis. Child. 1997, 76, 518–521. [Google Scholar]
- Besbas, N.; Gulhan, B.; Soylemezoglu, O.; Ozcakar, Z.B.; Korkmaz, E.; Hayran, M.; Ozaltin, F. Turkish pediatric atypical hemolytic uremic syndrome registry: Initial analysis of 146 patients. BMC Nephrol. 2017, 18, 6. [Google Scholar]
- Vilalta, R.; Lara, E.; Madrid, A.; Chocron, S.; Muñoz, M.; Casquero, A.; Nieto, J. Long-term eculizumab improves clinical outcomes in atypical hemolytic uremic syndrome. Pediatr. Nephrol. 2012, 27, 2323–2326. [Google Scholar]
- Tanné, C.; Javouhey, E.; Boyer, O.; Recher, M.; Allain-Launay, E.; Monet-Didailler, C.; Rouset-Rouvière, C.; Ryckewaert, A.; Nobili, F.; Gindre, F.A.; et al. Cardiac involvement in pediatric hemolytic uremic syndrome. Pediatr. Nephrol. 2022, 37, 3215–3221. [Google Scholar]
- Mallett, A.; Hughes, P.; Szer, J.; Tuckfield, A.; Van Eps, C.; Cambell, S.B.; Hawley, C.; Burke, J.; Kausman, J.; Hewitt, I.; et al. Atypical haemolytic uraemic syndrome treated with the complement inhibitor eculizumab: The experience of the Australian compassionate access cohort. Intern. Med. J. 2015, 45, 1054–1065. [Google Scholar] [PubMed]
- Johnson, S.; Stojanovic, J.; Ariceta, G.; Bitzan, M.; Besbas, N.; Frieling, M.; Karpman, D.; Landau, D.; Langman, C.; Licht, C.; et al. An audit analysis of a guideline for the investigation and initial therapy of diarrhea negative (atypical) hemolytic uremic syndrome. Pediatr. Nephrol. 2014, 29, 1967–1978. [Google Scholar] [PubMed]
- Román-Ortiz, E.; Mendizabal Oteiza, S.; Pinto, S.; López-Trascasa, M.; Sánchez-Corral, P.; Rodríguez De Cordoba, S. Eculizumab long-term therapy for pediatric renal transplant in aHUS with CFH/CFHR1 hybrid gene. Pediatr. Nephrol. 2014, 29, 149–153. [Google Scholar] [PubMed]
- Sellier-Leclerc, A.L.; Fremeaux-Bacchi, V.; Dragon-Durey, M.A.; Macher, M.A.; Niaudet, P.; Guest, G.; Boudailliez, B.; Bouissou, F.C.; Deschenes, G.; Gie, S.; et al. Differential Impact of Complement Mutations on Clinical Characteristics in Atypical Hemolytic Uremic Syndrome. J. Am. Soc. Nephrol. 2007, 18, 2392–2400. [Google Scholar]
- Scully, M.; Johnson, S.; Kupelian, V.; Greenbaum, L.A. Systemic Involvement at Entry into the Global Atypical Hemolytic Uremic Syndrome (aHUS) Registry. Blood 2016, 128, 3729. [Google Scholar]
- Ardissino, G.; Tel, F.; Testa, S.; Marzano, A.V.; Lazzari, R.; Salardi, S.; Edefonti, A. Skin Involvement in Atypical Hemolytic Uremic Syndrome. Am. J. Kidney Dis. 2014, 63, 652–655. [Google Scholar]
- Kaplan, B.S.; Garcia, C.D.; Chesney, R.W.; Segar, W.E.; Giugno, K.; Chem, R. Peripheral gangrene complicating idiopathic and recessive hemolytic uremic syndromes. Pediatr. Nephrol. 2000, 14, 985–989. [Google Scholar]
- Özel, A.; Çalışkan, Ü.; Gücer, Ş. Peripheral gangrene complicating hemolytic uremic syndrome in a child. Pediatr. Nephrol. 2003, 18, 465–467. [Google Scholar]
- Malina, M.; Gulati, A.; Bagga, A.; Majid, M.A.; Simkova, E.; Schaefer, F. Peripheral Gangrene in Children with Atypical Hemolytic Uremic Syndrome. Pediatrics 2013, 131, e331–e335. [Google Scholar]
- Zheng, X.; Gorovoy, I.R.; Mao, J.; Jin, J.; Chen, X.; Cui, Q.N. Recurrent Ocular Involvement in Pediatric Atypical Hemolytic Uremic Syndrome. J. Pediatr. Ophthalmol. Strabismus 2014, 51, e62–e65. [Google Scholar] [CrossRef]
- Greenwood, G. Case report of atypical hemolytic uremic syndrome with retinal arterial and venous occlusion treated with eculizumab. Int. Med. Case Rep. J. 2015, 8, 235–239. [Google Scholar]
- Ramos De Carvalho, J.E.; Schlingemann, R.O.; Oranje, M.; Bemelman, F.J.; Van Schooneveld, M.J. Reversal of threatening blindness after initiation of eculizumab in Purtscher-like retinopathy secondary to atypical hemolytic uremic syndrome. Int. Ophthalmol. 2017, 38, 399–407. [Google Scholar] [CrossRef]
- Brocklebank, V.; Johnson, S.; Sheerin, T.P.; Marks, S.D.; Gilbert, R.D.; Tyerman, K.; Kinoshita, M.; Awan, A.; Kaur, A.; Webb, N.; et al. Factor H autoantibody is associated with atypical hemolytic uremic syndrome in children in the United Kingdom and Ireland. Kidney Int. 2017, 92, 1261–1271. [Google Scholar] [CrossRef] [PubMed]
- Dragon-Durey, M.A.; Sethi, S.K.; Bagga, A.; Blanc, C.; Blouin, J.; Ranchin, B.; André, J.-L.; Takagi, N.; Cheong, H.; Hari, P.; et al. Clinical Features of Anti-Factor H Autoantibody–Associated Hemolytic Uremic Syndrome. J. Am. Soc. Nephrol. 2010, 21, 2180–2187. [Google Scholar] [PubMed]
- De Yao, J.; Kaplan, R.; Magro, C. An Atypical Case of Atypical Hemolytic Uremic Syndrome: Predominant Gastrointestinal Involvement, Intact Renal Function, and C5b-9 Deposition in Colon and Skin. J. Hematol. 2015, 4, 193–195. [Google Scholar]
- The European Paediatric Study Group for HUS; Ariceta, G.; Besbas, N.; Johnson, S.; Karpman, D.; Landau, D.; Licht, C.; Loirat, C.; Pecoraro, C.; Taylor, C.M.; et al. Guideline for the investigation and initial therapy of diarrhea-negative hemolytic uremic syndrome. Pediatr. Nephrol. 2009, 24, 687–696. [Google Scholar]
- Laurence, J.; Haller, H.; Mannucci, P.M.; Nangaku, M.; Praga, M.; Rodriguez de Cordoba, S. Atypical hemolytic uremic syndrome (aHUS): Essential aspects of an accurate diagnosis. Clin. Adv. Hematol. Oncol. HO 2016, 14 (Suppl. S11), 2–15. [Google Scholar]
- Vaisbich, M.H.; De Andrade, L.G.M.; De Menezes Neves, P.D.M.; Palma, L.M.P.; De Castro, M.C.R.; Silva, C.A.B.; Barbosa, M.I.N.d.H.; Penido, M.G.M.G.; Neto, O.Â.F.; Sobral, R.M.L.; et al. Baseline characteristics and evolution of Brazilian patients with atypical hemolytic uremic syndrome: First report of the Brazilian aHUS Registry. Clin. Kidney J. 2022, 15, 1601–1611. [Google Scholar]
- Addad, V.V.; Palma, L.M.P.; Vaisbich, M.H.; Pacheco Barbosa, A.M.; Da Rocha, N.C.; De Almeida Cardoso, M.M.; de Almeida, J.T.C.; Sordi, M.A.d.P.d.; Machado-Rugolo, J.; Arantes, L.F.; et al. A comprehensive model for assessing and classifying patients with thrombotic microangiopathy: The TMA-INSIGHT score. Thromb. J. 2023, 21, 119. [Google Scholar] [CrossRef]
- Vaisbich, M.H. Recomendações para diagnóstico e tratamento da Síndrome Hemolítico-Urêmica Atípica (SHUa): Uma declaração de consenso de especialistas do Comitê de Doenças Raras da Sociedade Brasileira de Nefrologia (COMDORA-SBN). Braz. J. Nephrol. 2025, 47, e20240087. [Google Scholar]
- Giannubilo, S.R.; Marzioni, D.; Tossetta, G.; Ciavattini, A. HELLP Syndrome and Differential Diagnosis with Other Thrombotic Microangiopathies in Pregnancy. Diagnostics 2024, 14, 352. [Google Scholar] [CrossRef]
- Song, Y.; Lee, S.Y.; Chee, Y.L.; Jen, W.Y. Hypertensive Emergency with Thrombotic Microangiopathy or TTP? A Case Series and Literature Review. J. Clin. Med. 2024, 13, 1880. [Google Scholar] [CrossRef] [PubMed]
- Bogdał, A.; Badeński, A.; Pac, M.; Wójcicka, A.; Badeńska, M.; Didyk, A.; Trembecka-Dubel, E.; Dąbrowska-Leonik, N.; Walaszczyk, M.; Matysiak, N.; et al. Atypical Hemolytic Uremic Syndrome (aHUS) and Adenosine Deaminase (ADA)-Deficient Severe Combined Immunodeficiency (SCID)—Two Diseases That Exacerbate Each Other: Case Report. Int. J. Mol. Sci. 2021, 22, 9479. [Google Scholar] [CrossRef] [PubMed]
- Burguet, L.; Taton, B.; Prezelin-Reydit, M.; Rubin, S.; Picard, W.; Gruson, D.; Ryman, A.; Contin-Bordes, C.; Coppo, P.; Combe, C.; et al. Urine Protein/Creatinine Ratio in Thrombotic Microangiopathies: A Simple Test to Facilitate Thrombotic Thrombocytopenic Purpura and Hemolytic and Uremic Syndrome Diagnosis. J. Clin. Med. 2022, 11, 648. [Google Scholar] [CrossRef]
- Mocanu, A.; Bogos, R.A.; Lazaruc, T.I.; Cianga, A.L.; Lupu, V.V.; Ioniuc, I.; Alecsa, M.; Lupu, A.; Ivanov, A.V.; Miron, I.C.; et al. Pitfalls of Thrombotic Microangiopathies in Children: Two Case Reports and Literature Review. Diagnostics 2023, 13, 1228. [Google Scholar] [CrossRef]
- Yenerel, M.N. Atypical Hemolytic Uremic Syndrome: Differential Diagnosis from TTP/HUS and Management. Turk. J. Hematol. 2014, 31, 216–225. [Google Scholar] [CrossRef]
- Ramos Mayordomo, P.; Capilla Díez, M.; Ticona Espinoza, D.A.; Torres Jaramillo, M.V.; Martínez Tejeda, N.; Ticona Espinoza, T.G.; Calleja, C.C.; Gutiérrez, V.F. Thrombotic microangiopathy (TMA) associated with pregnancy: Role of the clinical laboratory in differential diagnosis. Adv. Lab. Med. Av. En. Med. Lab. 2024, 5, 340–344. [Google Scholar]
- Yamane, R.; Yasuda, Y.; Oshima, A.; Suzuki, Y.; Kojima, H.; Kim, H.; Fukui, S.; Maruyama, S.; Ito, Y.; Mizuno, M. Serum and plasma levels of Ba, but not those of soluble C5b-9, might be affected by renal function in chronic kidney disease patients. BMC Nephrol. 2023, 24, 26. [Google Scholar]
- Volokhina, E.; Westra, D.; Xue, X.; Gros, P.; Van De Kar, N.; Van Den Heuvel, L. Novel C3 mutation p.Lys65Gln in aHUS affects complement factor H binding. Pediatr. Nephrol. 2012, 27, 1519–1524. [Google Scholar]
- Afshar-Kharghan, V. Atypical hemolytic uremic syndrome. Hematology 2016, 2016, 217–225. [Google Scholar] [CrossRef]
- Nester, C.M.; Barbour, T.; De Cordoba, S.R.; Dragon-Durey, M.A.; Fremeaux-Bacchi, V.; Goodship, T.H.J.; Kavanagh, D.; Noris, M.; Pickering, M.; Sanchez-Corral, P.; et al. Atypical aHUS: State of the art. Mol. Immunol. 2015, 67, 31–42. [Google Scholar]
- Schapkaitz, E.; Mezgebe, M.H. The Clinical Significance of Schistocytes: A Prospective Evaluation of the International Council for Standardization in Hematology Schistocyte Guidelines. Turk. J. Hematol. 2017, 34, 59–63. [Google Scholar]
- Loirat, C.; Noris, M.; Fremeaux-Bacchi, V. Complement and the atypical hemolytic uremic syndrome in children. Pediatr. Nephrol. 2008, 23, 1957–1972. [Google Scholar] [CrossRef] [PubMed]
- Ruggenenti, P.; Noris, M.; Remuzzi, G. Thrombotic microangiopathy, hemolytic uremic syndrome, and thrombotic thrombocytopenic purpura. Kidney Int. 2001, 60, 831–846. [Google Scholar] [CrossRef]
- Cofiell, R.; Kukreja, A.; Bedard, K.; Yan, Y.; Mickle, A.P.; Ogawa, M.; Bedrosian, C.L.; Faas, S.J. Eculizumab reduces complement activation, inflammation, endothelial damage, thrombosis, and renal injury markers in aHUS. Blood 2015, 125, 3253–3262. [Google Scholar] [CrossRef]
- Gruppo, R.A.; Rother, R.P. Eculizumab for Congenital Atypical Hemolytic–Uremic Syndrome. N. Engl. J. Med. 2009, 360, 544–546. [Google Scholar] [CrossRef] [PubMed]
- Campistol, J.M.; Arias, M.; Ariceta, G.; Blasco, M.; Espinosa, L.; Espinosa, M.; Grinyóf, J.M.; Macíag, M.; Mendizábalh, S.; Praga, M.; et al. Actualización en síndrome hemolítico urémico atípico: Diagnóstico y tratamiento. Documento de consenso. Nefrología 2015, 35, 421–447. [Google Scholar] [CrossRef]
- Mbaeyi, S.A.; Bozio, C.H.; Duffy, J.; Rubin, L.G.; Hariri, S.; Stephens, D.S.; MacNeil, J.R. Meningococcal Vaccination: Recommendations of the Advisory Committee on Immunization Practices, United States, 2020. MMWR. Recomm. Rep. 2020, 69, 1–41. [Google Scholar]
- Schönfelder, K.; Kühne, L.; Schulte-Kemna, L.; Kaufeld, J.; Rohn, H.; Kribben, A.; Schröppel, B.; Brinkkötter, P.T.; Gäckler, A. Clinical efficacy and safety of switching from eculizumab to ravulizumab in adult patients with aHUS– real-world data. BMC Nephrol. 2024, 25, 202. [Google Scholar] [CrossRef]
- Rondeau, E.; Scully, M.; Ariceta, G.; Barbour, T.; Cataland, S.; Heyne, N.; Miyakawa, Y.; Ortiz, S.; Swenson, E.; Vallee, M.; et al. The long-acting C5 inhibitor, Ravulizumab, is effective and safe in adult patients with atypical hemolytic uremic syndrome naïve to complement inhibitor treatment. Kidney Int. 2020, 97, 1287–1296. [Google Scholar] [PubMed]
- Lee, J.W.; Sicre De Fontbrune, F.; Wong Lee Lee, L.; Pessoa, V.; Gualandro, S.; Füreder, W.; Ptushkin, V.; Rottinghaus, S.T.; Volles, L.; Shafner, L.; et al. Ravulizumab (ALXN1210) vs eculizumab in adult patients with PNH naive to complement inhibitors: The 301 study. Blood 2019, 133, 530–539. [Google Scholar] [PubMed]
- Meena, P.; Gala, R.; Das, R.R.; Bhargava, V.; Saivani, Y.; Panda, S.; Mantri, A.M.; Agrawaal, K.K.M. Kidney and pregnancy outcomes in pregnancy-associated atypical hemolytic uremic syndrome: A systematic review and meta-analysis. Medicine 2025, 104, e41403. [Google Scholar]
- Gentile, M.; Manenti, L. Targeted Complement Treatments in Glomerulopathies: A Comprehensive Review. J. Clin. Med. 2025, 14, 702. [Google Scholar] [CrossRef] [PubMed]
- Barbour, T.; Scully, M.; Ariceta, G.; Cataland, S.; Garlo, K.; Heyne, N.; Luque, Y.; Menne, J.; Miyakawa, Y.; Yoon, S.-S.; et al. Long-Term Efficacy and Safety of the Long-Acting Complement C5 Inhibitor Ravulizumab for the Treatment of Atypical Hemolytic Uremic Syndrome in Adults. Kidney Int. Rep. 2021, 6, 1603–1613. [Google Scholar]
- Benamu, E.; Montoya, J.G. Infections associated with the use of eculizumab: Recommendations for prevention and prophylaxis. Curr. Opin. Infect. Dis. 2016, 29, 319–329. [Google Scholar]
- Ariceta, G.; Dixon, B.P.; Kim, S.H.; Kapur, G.; Mauch, T.; Ortiz, S.; Vallee, M.; Denker, A.E.; Kang, H.G.; Greenbaum, L.A.; et al. Corrigendum to “The long-acting C5 inhibitor, ravulizumab, is effective and safe in pediatric patients with atypical hemolytic uremic syndrome naïve to complement inhibitor treatment”. Kidney Int. 2021, 100, 225–237. [Google Scholar]
- Tanaka, K.; Adams, B.; Aris, A.M.; Fujita, N.; Ogawa, M.; Ortiz, S.; Vallee, M. The long-acting C5 inhibitor, ravulizumab, is efficacious and safe in pediatric patients with atypical hemolytic uremic syndrome previously treated with eculizumab. Pediatr. Nephrol. 2021, 36, 889–898. [Google Scholar]
- Nowicki, M.; Printza, N. Ravulizumab in adults and children with atypical hemolytic uremic syndrome: A plain language summary of three studies. J. Comp. Eff. Res. 2024, 13, e240103. [Google Scholar]
- Bleathman, F.; Kausman, J.Y.; Hosking, L.M.; Forbes, T.A. Ravulizumab facilitates reduced burden of vascular access, a major benefit in paediatric atypical haemolytic uraemic syndrome. J. Paediatr. Child. Health 2024, 60, 183–187. [Google Scholar]
- Gupta, M.; Govindappagari, S.; Burwick, R.M. Pregnancy-Associated Atypical Hemolytic Uremic Syndrome: A Systematic Review. Obstet. Gynecol. 2020, 135, 46–58. [Google Scholar] [PubMed]
- Ardissino, G.; Testa, S.; Possenti, I.; Tel, F.; Paglialonga, F.; Salardi, S.; Tedeschi, S.; Belingheri, M.; Cugno, M. Discontinuation of Eculizumab Maintenance Treatment for Atypical Hemolytic Uremic Syndrome: A Report of 10 Cases. Am. J. Kidney Dis. 2014, 64, 633–637. [Google Scholar] [PubMed]
- Macia, M.; De Alvaro Moreno, F.; Dutt, T.; Fehrman, I.; Hadaya, K.; Gasteyger, C.; Heyne, N. Current evidence on the discontinuation of eculizumab in patients with atypical haemolytic uraemic syndrome. Clin. Kidney J. 2017, 10, 310–319. [Google Scholar]
- Merrill, S.A.; Brittingham, Z.D.; Yuan, X.; Moliterno, A.R.; Sperati, C.J.; Brodsky, R.A. Eculizumab cessation in atypical hemolytic uremic syndrome. Blood 2017, 130, 368–372. [Google Scholar]
- Germeni, E.; Cooper, J.; Briggs, A.; Laurence, J. Treatment discontinuation in adults with atypical hemolytic uremic syndrome (aHUS): A qualitative study of international experts’ perspectives with associated cost-consequence analysis. BMC Nephrol. 2024, 25, 411. [Google Scholar]
- Menne, J.; Delmas, Y.; Fakhouri, F.; Licht, C.; Lommelé, Å.; Minetti, E.E.; Provôt, F.; Rondeau, E.; Sheerin, N.S.; Wang, J.; et al. Outcomes in patients with atypical hemolytic uremic syndrome treated with eculizumab in a long-term observational study. BMC Nephrol. 2019, 20, 125. [Google Scholar] [CrossRef]
- Fakhouri, F.; Fila, M.; Hummel, A.; Ribes, D.; Sellier-Leclerc, A.L.; Ville, S.; Pouteil-Noble, C.; Coindre, J.-P.; Le Quintrec, M.; Rondeau, E.; et al. Eculizumab discontinuation in children and adults with atypical hemolytic-uremic syndrome: A prospective multicenter study. Blood 2021, 137, 2438–2449. [Google Scholar]
- Ueda, Y.; Miwa, T.; Gullipalli, D.; Sato, S.; Ito, D.; Kim, H.; Palmer, M.; Song, W.-C. Blocking Properdin Prevents Complement-Mediated Hemolytic Uremic Syndrome and Systemic Thrombophilia. J. Am. Soc. Nephrol. 2018, 29, 1928–1937. [Google Scholar]
- Ando, M.; Kubota, K.; Kadowaki, S.; Kawamoto, M.; Kawamoto, N.; Okamoto, H.; Nagaya, S.; Miwa, Y.; Ohnishi, H. Atypical hemolytic uremic syndrome with a C3 variant following COVID-19: A case report. Front. Pediatr. 2025, 13, 1507727. [Google Scholar]
- Abdeen, A.M.; Al-Nusair, J.; Samardali, M.; Alshal, M.; Al-Astal, A.; Khitan, Z. Complement-Mediated Hemolytic Uremic Syndrome Due to MCP/CD46 Mutation: A Case Report. J. Investig. Med. High. Impact Case Rep. 2025, 13, 23247096251316364. [Google Scholar]
- Che, M.; Moran, S.M.; Smith, R.J.; Ren, K.Y.M.; Smith, G.N.; Shamseddin, M.K.; Avila-Casado, C.; Garland, J.S. A case-based narrative review of pregnancy-associated atypical hemolytic uremic syndrome/complement-mediated thrombotic microangiopathy. Kidney Int. 2024, 105, 960–970. [Google Scholar] [PubMed]
- Gonzalez Suarez, M.L.; Thongprayoon, C.; Mao, M.A.; Leeaphorn, N.; Bathini, T.; Cheungpasitporn, W. Outcomes of Kidney Transplant Patients with Atypical Hemolytic Uremic Syndrome Treated with Eculizumab: A Systematic Review and Meta-Analysis. J. Clin. Med. 2019, 8, 919. [Google Scholar] [CrossRef]
- AlZabali, S.; AlBatati, S.; Rahim, K.; Faqeehi, H.; Osman, A.; Bamhraz, A.; Saleh, M.A.; Kari, J.A.; Aloufi, M.; Eid, L.; et al. A Multicenter Study Evaluating the Discontinuation of Eculizumab Therapy in Children with Atypical Hemolytic Uremic Syndrome. Children 2022, 9, 1734. [Google Scholar] [CrossRef] [PubMed]
- Spasiano, A.; Palazzetti, D.; Dimartino, L.; Bruno, F.; Baccaro, R.; Pesce, F.; Grandaliano, G. Underlying Genetics of aHUS: Which Connection with Outcome and Treatment Discontinuation? Int. J. Mol. Sci. 2023, 24, 14496. [Google Scholar] [CrossRef]
- Wang, Y.; Johnston, K.; Popoff, E.; Myren, K.J.; Cheung, A.; Faria, C.; Tomazos, I. A US cost-minimization model comparing ravulizumab versus eculizumab for the treatment of atypical hemolytic uremic syndrome. J. Med. Econ. 2020, 23, 1503–1515. [Google Scholar] [PubMed]
- Wang, X.F.; Bao, L.R.; Hu, T.L.; Xu, R.F.; Gao, W.N.; Wang, J.Y.; Zhao, J.-R.; Fu, Z.-L.; Wang, S.-F.; Meng, Y. Adverse drug events (ADEs) risk signal mining related to eculizumab based on the FARES database. Front. Pharmacol. 2025, 15, 1440907. [Google Scholar]
- Mauch, T.J.; Chladek, M.R.; Cataland, S.; Chaturvedi, S.; Dixon, B.P.; Garlo, K.; Gasteyger, C.; Java, A.; Leguizamo, J.; Lloyd-Price, L.; et al. Treatment preference and quality of life impact: Ravulizumab vs eculizumab for atypical hemolytic uremic syndrome. J. Comp. Eff. Res. 2023, 12, e230036. [Google Scholar]
- Schaefer, F.; Al-Dakkak, I.; Anokhina, K.; Cohen, D.; Greenbaum, L.A.; Ariceta, G. Global aHUS Registry Analysis of Patients Switching to Ravulizumab from Eculizumab. Kidney Int. Rep. 2024, 9, 2648–2656. [Google Scholar]

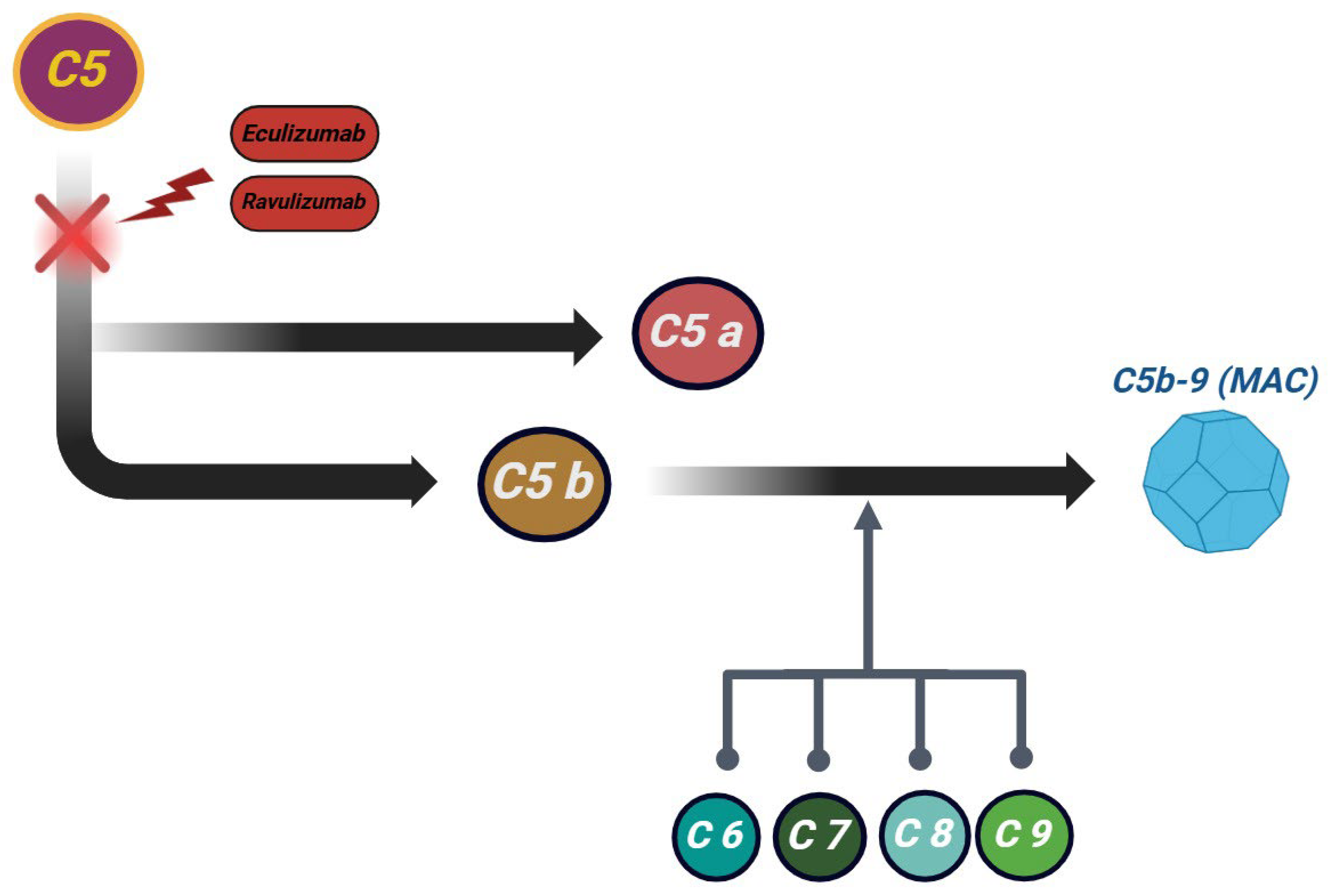

{kind=link}
{kind=link}
| Gene | Protein Affected | Mutation Type | Estimated Frequency in aHUS | Penetrance | Clinical Implications | Source |
|---|---|---|---|---|---|---|
| CFH | Complement factor H | Loss-of-function | ~20–45% | ~50% | Early onset; poor prognosis; high recurrence post-transplant | [14,38,39] |
| MCP | Membrane cofactor protein | Loss-of-function | ~10% | ~20% | Often triggers with infection; better prognosis; low recurrence after transplantation | [14,38,40] |
| CFI | Complement factor I | Loss-of-function | ~5–10% | Variable | Intermediate severity; incomplete penetrance; may coexist with other variants | [14,25] |
| C3 | Complement C3 | Gain-of-function | ~4–10% | Moderate | Severe presentation; poor prognosis; more resistant to plasma therapy | [14,41] |
| CFB | Complement factor B | Gain-of-function | <1% | Unknown | Rare; usually severe; limited data available | [14,40] |
| THBD | Thrombomodulin | Loss-of-function | ~3–5% | Low | May present with mild phenotype; data on recurrence limited | [14,42] |
| Feature | aHUS | TTP | HUS |
|---|---|---|---|
| Etiology | Inherited or acquired imbalance in the regulation of the alternative complement pathway | Severe deficiency of ADAMTS13 enzyme activity (≤10% of normal), often due to autoantibodies | Various triggers, including infections (e.g., Shiga toxin-producing E. coli), drugs or systemic diseases |
| Microangiopathic Hemolytic Anemia | Present; characterized by schistocytes on peripheral smear and elevated lactate dehydrogenase | Present; similar findings as in aHUS | Present; similar findings as in aHUS |
| Thrombocytopenia | Present; platelet count typically < 150,000/μL | Present; often severe with platelet count < 30,000/μL | Present; platelet count decreased but not as low as in TTP |
| Acute Kidney Injury | Common and often severe; elevated serum creatinine and proteinuria | Less common; renal involvement is usually mild | Prominent; often severe renal impairment |
| Neurological Symptoms | Can occur but are less frequent and less severe than in TTP | Common; may include confusion, seizures and focal deficits | Less common; when present, may include irritability and seizures |
| ADAMTS13 Activity | Typically > 10% of normal activity | Severely reduced (≤10% of normal activity) | Normal |
| Shiga Toxin Detection | Negative | Negative | May be positive if associated with E. coli infection |
| Complement Level | Often decreased (e.g., low C3 and C4) | Normal | Typically normal |
| Family History | May have a family history of similar episodes | Usually absent | Usually absent |
| Treatment Approach | Eculizumab or ravulizumab (C5 inhibitors); supportive care; vaccination prior to therapy; consider genetic testing; dialysis if needed | Urgent plasma exchange; corticosteroids; rituximab or caplacizumab in selected cases | Supportive care; avoid antibiotics and antimotility agents; dialysis if needed |
| Target in Complement Pathway | Level of Action | Examples of Complement Blockers | Mechanism of Action | Administration and Indication |
|---|---|---|---|---|
| C3 | Early stage of complement activation (alternative pathway) | Pegcetacoplan, APL-2 | Inhibits C3 cleavage, preventing complement cascade activation and C3 convertase formation. | Subcutaneous; in trial phase for complement diseases |
| C5 | Late stage of complement activation | Eculizumab, ravulizumab, crovalimab | Blocks C5 cleavage, preventing C5a (pro-inflammatory) and C5b, which leads to MAC formation. Crovalimab is an anti-C5 monoclonal antibody with subcutaneous administration. | IV (biweekly or every 8 weeks); approved in aHUS |
| Membrane Attack Complex (MAC—C5b-9) | Final stage of complement cascade | Nomacopan, zilucoplan (under investigation) | Nomacopan inhibits both C5 and leukotriene B4 (LTB4), reducing inflammation and MAC formation. Zilucoplan is a C5 inhibitor. | Under investigation |
| Factor D | Key regulator in the alternative pathway | Danicopan (ACH-4471) | Inhibits factor D, blocking C3 convertase activation and stopping the complement cascade. | Oral; in trial phase |
| Factor B | Part of the alternative pathway C3 convertase | Iptacopan (LNP023) | Inhibits factor B, preventing further complement activation. | Oral; ongoing trials |
| Hepatic C5 Synthesis | RNA interference-based inhibition | Cemdisiran | Reduces C5 production in the liver via RNA interference (siRNA), lowering circulating C5 levels and decreasing complement activation. | Subcutaneous; in development |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bogdan, R.-G.; Anderco, P.; Ichim, C.; Cimpean, A.-M.; Todor, S.B.; Glaja-Iliescu, M.; Crainiceanu, Z.P.; Popa, M.L. Atypical Hemolytic Uremic Syndrome: A Review of Complement Dysregulation, Genetic Susceptibility and Multiorgan Involvement. J. Clin. Med. 2025, 14, 2527. https://doi.org/10.3390/jcm14072527
Bogdan R-G, Anderco P, Ichim C, Cimpean A-M, Todor SB, Glaja-Iliescu M, Crainiceanu ZP, Popa ML. Atypical Hemolytic Uremic Syndrome: A Review of Complement Dysregulation, Genetic Susceptibility and Multiorgan Involvement. Journal of Clinical Medicine. 2025; 14(7):2527. https://doi.org/10.3390/jcm14072527
Chicago/Turabian StyleBogdan, Razvan-George, Paula Anderco, Cristian Ichim, Anca-Maria Cimpean, Samuel Bogdan Todor, Mihai Glaja-Iliescu, Zorin Petrisor Crainiceanu, and Mirela Livia Popa. 2025. "Atypical Hemolytic Uremic Syndrome: A Review of Complement Dysregulation, Genetic Susceptibility and Multiorgan Involvement" Journal of Clinical Medicine 14, no. 7: 2527. https://doi.org/10.3390/jcm14072527
APA StyleBogdan, R.-G., Anderco, P., Ichim, C., Cimpean, A.-M., Todor, S. B., Glaja-Iliescu, M., Crainiceanu, Z. P., & Popa, M. L. (2025). Atypical Hemolytic Uremic Syndrome: A Review of Complement Dysregulation, Genetic Susceptibility and Multiorgan Involvement. Journal of Clinical Medicine, 14(7), 2527. https://doi.org/10.3390/jcm14072527