
The Role of Inflammasomes in LPS and Gram-Negative Bacterial Sepsis

 , and
, and
Abstract
1. Introduction
2. Inflammasomes in Host Defense
3. Inflammasomes in Sepsis Induced by LPS
4. Inflammasomes in Bacterial Sepsis
5. Therapies with Immunomodulatory Effects in Sepsis
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DAMPs | Damage-associated molecular patterns |
| PAMPs | Pathogen-associated molecular patterns |
| LPS | Lipopolysaccharide |
| NETs | Neutrophil extracellular traps |
| GsdmD | Gasdermin D |
| HMGB1 | High Mobility Group Box 1 |
References
- Esposito, S.; De Simone, G.; Boccia, G.; De Caro, F.; Pagliano, P. Sepsis and septic shock: New definitions, new diagnostic and therapeutic approaches. J. Glob. Antimicrob. Resist. 2017, 10, 204–212. [Google Scholar] [CrossRef]
- World Health Organization: Sepsis. Available online: https://www.who.int/news-room/fact-sheets/detail/sepsis (accessed on 22 April 2025).
- Rudd, K.E.; Johnson, S.C.; Agesa, K.M.; Shackelford, K.A.; Tsoi, D.; Kievlan, D.R.; Colombara, D.V.; Ikuta, K.S.; Kissoon, N.; Finfer, S.; et al. Global, regional, and national sepsis incidence and mortality, 1990-2017: Analysis for the Global Burden of Disease Study. Lancet 2020, 395, 200–211. [Google Scholar] [CrossRef]
- Fleischmann-Struzek, C.; Rudd, K. Challenges of assessing the burden of sepsis. Med. Klin. Intensiv. Notfmed. 2023, 118 (Suppl. S2), 68–74. [Google Scholar] [CrossRef]
- Guarino, M.; Perna, B.; Cesaro, A.E.; Maritati, M.; Spampinato, M.D.; Contini, C.; De Giorgio, R. Update on Sepsis and Septic Shock in Adult Patients: Management in the Emergency Department. J. Clin. Med. 2023, 12, 3188. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, K. Inflammasome-associated cell death: Pyroptosis, apoptosis, and physiological implications. Microbiol. Immunol. 2020, 64, 252–269. [Google Scholar] [CrossRef]
- Wu, R.; Wang, N.; Comish, P.B.; Tang, D.; Kang, R. Inflammasome-Dependent Coagulation Activation in Sepsis. Front. Immunol. 2021, 12, 641750. [Google Scholar] [CrossRef] [PubMed]
- Gyawali, B.; Ramakrishna, K.; Dhamoon, A.S. Sepsis: The evolution in definition, pathophysiology, and management. SAGE Open Med. 2019, 7, 2050312119835043. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Moldawer, L.L.; Opal, S.M.; Reinhart, K.; Turnbull, I.R.; Vincent, J.L. Sepsis and septic shock. Nat. Rev. Dis. Primers. 2016, 2, 16045. [Google Scholar] [CrossRef]
- Chen, R.; Zou, J.; Chen, J.; Zhong, X.; Kang, R.; Tang, D. Pattern recognition receptors: Function, regulation and therapeutic potential. Signal Transduct. Target. Ther. 2025, 10, 216. [Google Scholar] [CrossRef]
- Yu, G.; Choi, Y.K.; Lee, S. Inflammasome diversity: Exploring novel frontiers in the innate immune response. Trends Immunol. 2024, 45, 248–258. [Google Scholar] [CrossRef]
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Kamada, N.; Nakamura, Y.; Burberry, A.; Kuffa, P.; Suzuki, S.; Shaw, M.H.; Kim, Y.G.; Núñez, G. NLRC4-driven production of IL-1β discriminates between pathogenic and commensal bacteria and promotes host intestinal defense. Nat. Immunol. 2012, 13, 449–456. [Google Scholar] [CrossRef]
- Miao, E.A.; Mao, D.P.; Yudkovsky, N.; Bonneau, R.; Lorang, C.G.; Warren, S.E.; Leaf, I.A.; Aderem, A. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc. Natl. Acad. Sci. USA 2010, 107, 3076–3080. [Google Scholar] [CrossRef]
- Kofoed, E.M.; Vance, R.E. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 2011, 477, 592–595. [Google Scholar] [CrossRef]
- Zhao, Y.; Yang, J.; Shi, J.; Gong, Y.N.; Lu, Q.; Xu, H.; Liu, L.; Shao, F. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 2011, 477, 596–600. [Google Scholar] [CrossRef]
- Paik, S.; Kim, J.K.; Shin, H.J.; Park, E.J.; Kim, I.S.; Jo, E.K. Updated insights into the molecular networks for NLRP3 inflammasome activation. Cell. Mol. Immunol. 2025, 22, 563–596. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Li, Q.; Xu, G.; Xiao, X.; Bai, Z. The mechanism of NLRP3 inflammasome activation and its pharmacological inhibitors. Front. Immunol. 2023, 13, 1109938. [Google Scholar] [CrossRef]
- Feng, S.; Wierzbowski, M.C.; Hrovat-Schaale, K.; Dumortier, A.; Zhang, Y.; Zyulina, M.; Baker, P.J.; Reygaerts, T.; Steiner, A.; De Nardo, D.; et al. Mechanisms of NLRP3 activation and inhibition elucidated by functional analysis of disease-associated variants. Nat. Immunol. 2025, 26, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Vande Walle, L.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef]
- Aachoui, Y.; Leaf, I.A.; Hagar, J.A.; Fontana, M.F.; Campos, C.G.; Zak, D.E.; Tan, M.H.; Cotter, P.A.; Vance, R.E.; Aderem, A.; et al. Caspase-11 protects against bacteria that escape the vacuole. Science 2013, 339, 975–978. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, D.; Guo, Y.; Kamada, N. Interaction between the inflammasome and commensal microorganisms in gastrointestinal health and disease. EMBO Mol. Med. 2021, 13, e13452. [Google Scholar] [CrossRef]
- Romberg, N.; Al Moussawi, K.; Nelson-Williams, C.; Stiegler, A.L.; Loring, E.; Choi, M.; Overton, J.; Meffre, E.; Khokha, M.K.; Huttner, A.J.; et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat. Genet. 2014, 46, 1135–1139. [Google Scholar] [CrossRef]
- Dai, Y.; Zhou, J.; Shi, C. Inflammasome: Structure, biological functions, and therapeutic targets. MedComm 2023, 4, e391. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef]
- Nadiri, A.; Wolinski, M.K.; Saleh, M. The inflammatory caspases: Key players in the host response to pathogenic invasion and sepsis. J. Immunol. 2006, 177, 4239–4245. [Google Scholar] [CrossRef] [PubMed]
- Lagrange, B.; Benaoudia, S.; Wallet, P.; Magnotti, F.; Provost, A.; Michal, F.; Martin, A.; Di Lorenzo, F.; Py, B.F.; Molinaro, A.; et al. Human caspase-4 detects tetra-acylated LPS and cytosolic Francisella and functions differently from murine caspase-11. Nat. Commun. 2018, 9, 242. [Google Scholar] [CrossRef]
- Shi, X.; Sun, Q.; Hou, Y.; Zeng, H.; Cao, Y.; Dong, M.; Ding, J.; Shao, F. Recognition and maturation of IL-18 by caspase-4 noncanonical inflammasome. Nature 2023, 624, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.W.; Monteleone, M.; Boucher, D.; Sollberger, G.; Ramnath, D.; Condon, N.D.; von Pein, J.B.; Broz, P.; Sweet, M.J.; Schroder, K. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6676. [Google Scholar] [CrossRef]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116. [Google Scholar] [CrossRef]
- Miao, E.A.; Leaf, I.A.; Treuting, P.M.; Mao, D.P.; Dors, M.; Sarkar, A.; Warren, S.E.; Wewers, M.D.; Aderem, A. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol. 2010, 11, 1136–1142. [Google Scholar] [CrossRef]
- Wang, J.; Deobald, K.; Re, F. Gasdermin D Protects from Melioidosis through Pyroptosis and Direct Killing of Bacteria. J. Immunol. 2019, 202, 3468–3473. [Google Scholar] [CrossRef]
- Hoshino, K.; Takeuchi, O.; Kawai, T.; Sanjo, H.; Ogawa, T.; Takeda, Y.; Takeda, K.; Akira, S. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: Evidence for TLR4 as the Lps gene product. J. Immunol. 1999, 162, 3749–3752. [Google Scholar] [CrossRef]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.Y.; Van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef] [PubMed]
- Molteni, M.; Gemma, S.; Rossetti, C. The Role of Toll-Like Receptor 4 in Infectious and Noninfectious Inflammation. Mediat. Inflamm. 2016, 2016, 6978936. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Allen, H.; Banerjee, S.; Franklin, S.; Herzog, L.; Johnston, C.; McDowell, J.; Paskind, M.; Rodman, L.; Salfeld, J. Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell 1995, 80, 401–411. [Google Scholar] [CrossRef]
- Weber, P.; Wang, P.; Maddens, S.; Wang, P.S.; Wu, R.; Miksa, M.; Dong, W.; Mortimore, M.; Golec, J.M.; Charlton, P. VX-166: A novel potent small molecule caspase inhibitor as a potential therapy for sepsis. Crit. Care. 2009, 13, R146. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Newton, K.; Monack, D.M.; Vucic, D.; French, D.M.; Lee, W.P.; Roose-Girma, M.; Erickson, S.; Dixit, V.M. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 2004, 430, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Miura, M.; Jung, Y.K.; Zhu, H.; Li, E.; Yuan, J. Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell 1998, 92, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic LPS activates caspase-11: Implications in TLR4-independent endotoxic shock. Science 2013, 341, 1250–1253. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; Miyake, K.; Zhang, J.; Lee, W.P.; Muszyński, A.; et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 2013, 341, 1246–1249. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, K.; Hosojima, S.; Hara, H.; Kushiyama, H.; Mahib, M.R.; Kinoshita, T.; Suda, T. Gasdermin D mediates the maturation and release of IL-1α downstream of inflammasomes. Cell Rep. 2021, 34, 108887. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Sarkar, A.; Vande Walle, L.; Vitari, A.C.; Amer, A.O.; Wewers, M.D.; Tracey, K.J.; Kanneganti, T.D.; Dixit, V.M. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J. Immunol. 2010, 185, 4385–4392. [Google Scholar] [CrossRef]
- Ohlsson, K.; Björk, P.; Bergenfeldt, M.; Hageman, R.; Thompson, R.C. Interleukin-1 receptor antagonist reduces mortality from endotoxin shock. Nature 1990, 348, 550–552. [Google Scholar] [CrossRef] [PubMed]
- Glaccum, M.B.; Stocking, K.L.; Charrier, K.; Smith, J.L.; Willis, C.R.; Maliszewski, C.; Livingston, D.J.; Peschon, J.J.; Morrissey, P.J. Phenotypic and functional characterization of mice that lack the type I receptor for IL-1. J. Immunol. 1997, 159, 3364–3371. [Google Scholar] [CrossRef] [PubMed]
- Joosten, L.A.; Van De Veerdonk, F.L.; Vonk, A.G.; Boerman, O.C.; Keuter, M.; Fantuzzi, G.; Verschueren, I.; Van Der Poll, T.; Dinarello, C.A.; Kullberg, B.J.; et al. Differential susceptibility to lethal endotoxaemia in mice deficient in IL-1α, IL-1β or IL-1 receptor type I. APMIS 2010, 118, 1000–1007. [Google Scholar] [CrossRef]
- Fantuzzi, G.; Zheng, H.; Faggioni, R.; Benigni, F.; Ghezzi, P.; Sipe, J.D.; Shaw, A.R.; Dinarello, C.A. Effect of endotoxin in IL-1 beta-deficient mice. J. Immunol. 1996, 157, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.; Tang, Y.; Li, W.; Wang, X.; Zhang, R.; Zhang, X.; Zhao, X.; Liu, J.; Tang, C.; Liu, Z.; et al. The Endotoxin Delivery Protein HMGB1 Mediates Caspase-11-Dependent Lethality in Sepsis. Immunity 2018, 49, 740–753.e7. [Google Scholar] [CrossRef]
- Liaw, P.C.; Ito, T.; Iba, T.; Thachil, J.; Zeerleder, S. DAMP and DIC: The role of extracellular DNA and DNA-binding proteins in the pathogenesis of DIC. Blood Rev. 2016, 30, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, X.; Pelayo, R.; Monestier, M.; Ammollo, C.T.; Semeraro, F.; Taylor, F.B.; Esmon, N.L.; Lupu, F.; Esmon, C.T. Extracellular histones are major mediators of death in sepsis. Nat. Med. 2009, 15, 1318–1321. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef]
- McDonald, B.; Davis, R.P.; Kim, S.J.; Tse, M.; Esmon, C.T.; Kolaczkowska, E.; Jenne, C.N. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 2017, 129, 1357–1367. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Cheng, X.; Tang, Y.; Qiu, X.; Wang, Y.; Kang, H.; Wu, J.; Wang, Z.; Liu, Y.; Chen, F.; et al. Bacterial Endotoxin Activates the Coagulation Cascade through Gasdermin D-Dependent Phosphatidylserine Exposure. Immunity 2019, 51, 983–996.e6. [Google Scholar] [CrossRef]
- Zhang, Y.; Meng, H.; Ma, R.; He, Z.; Wu, X.; Cao, M.; Yao, Z.; Zhao, L.; Li, T.; Deng, R.; et al. Circulating microparticles, blood cells, and endothelium induce procoagulant activity in sepsis through phosphatidylserine exposure. Shock 2016, 45, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Lu, W.; Zhang, Y.; Zhang, G.; Shi, X.; Hisada, Y.; Grover, S.P.; Zhang, X.; Li, L.; Xiang, B.; et al. Inflammasome Activation Triggers Blood Clotting and Host Death through Pyroptosis. Immunity 2019, 50, 1401–1411.e4. [Google Scholar] [CrossRef] [PubMed]
- Dackiw, A.P.; McGilvray, I.D.; Woodside, M.; Nathens, A.B.; Marshall, J.C.; Rotstein, O.D. Prevention of endotoxin-induced mortality by antitissue factor immunization. Arch. Surg. 1996, 131, 1273–1279. [Google Scholar] [CrossRef] [PubMed]
- Fischer, E.; Marano, M.A.; Van Zee, K.J.; Rock, C.S.; Hawes, A.S.; Thompson, W.A.; DeForge, L.; Kenney, J.S.; Remick, D.G.; Bloedow, D.C. Interleukin-1 receptor blockade improves survival and hemodynamic performance in Escherichia coli septic shock, but fails to alter host responses to sublethal endotoxemia. J. Clin. Invest. 1992, 89, 1551–1557. [Google Scholar] [CrossRef]
- Jin, L.; Batra, S.; Jeyaseelan, S. Deletion of Nlrp3 Augments Survival during Polymicrobial Sepsis by Decreasing Autophagy and Enhancing Phagocytosis. J. Immunol. 2017, 198, 1253–1262. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Hall, M.W.; Exline, M.; Hart, J.; Knatz, N.; Gatson, N.T.; Wewers, M.D. Caspase-1 regulates Escherichia coli sepsis and splenic B cell apoptosis independently of interleukin-1beta and interleukin-18. Am. J. Respir. Crit. Care Med. 2006, 174, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Chang, K.C.; Swanson, P.E.; Tinsley, K.W.; Hui, J.J.; Klender, P.; Xanthoudakis, S.; Roy, S.; Black, C.; Grimm, E.; et al. Caspase inhibitors improve survival in sepsis: A critical role of the lymphocyte. Nat. Immunol. 2000, 1, 496–501. [Google Scholar] [CrossRef]
- Jiang, C.; Chen, J.; Xu, J.; Chen, C.; Zhu, H.; Xu, Y.; Zhao, H.; Chen, J. Integrated analysis reveals NLRC4 as a potential biomarker in sepsis pathogenesis. Genes Immun. 2024, 25, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Ayres, J.S.; Trinidad, N.J.; Vance, R.E. Lethal inflammasome activation by a multidrug-resistant pathobiont upon antibiotic disruption of the microbiota. Nat. Med. 2012, 18, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.S.; Yan, C.S.; Luo, J.M. NLRC4 gene silencing-dependent blockade of NOD-like receptor pathway inhibits inflammation, reduces proliferation and increases apoptosis of dendritic cells in mice with septic shock. Aging 2021, 13, 1440–1457. [Google Scholar] [CrossRef]
- Taylor, F.B., Jr.; Chang, A.; Ruf, W.; Morrissey, J.H.; Hinshaw, L.; Catlett, R.; Blick, K.; Edgington, T.S. Lethal E. coli septic shock is prevented by blocking tissue factor with monoclonal antibody. Circ. Shock. 1991, 33, 127–134. [Google Scholar] [PubMed]
- Shah, K.G.; Wu, R.; Jacob, A.; Molmenti, E.P.; Nicastro, J.; Coppa, G.F.; Wang, P. Recombinant human milk fat globule-EGF factor 8 produces dose-dependent benefits in sepsis. Intensive Care Med. 2012, 38, 128–136. [Google Scholar] [CrossRef]
- Abraham, E.; Anzueto, A.; Gutierrez, G.; Tessler, S.; San Pedro, G.; Wunderink, R.; Dal Nogare, A.; Nasraway, S.; Berman, S.; Cooney, R.; et al. Double-blind randomised controlled trial of monoclonal antibody to human tumour necrosis factor in treatment of septic shock. NORASEPT II Study Group. Lancet 1998, 351, 929–933. [Google Scholar] [CrossRef]
- Cohen, J.; Carlet, J. INTERSEPT: An international, multicenter, placebo-controlled trial of monoclonal antibody to human tumor necrosis factor-alpha in patients with sepsis. International Sepsis Trial Study Group. Crit. Care Med. 1996, 24, 1431–1440. [Google Scholar] [CrossRef] [PubMed]
- Lv, S.; Han, M.; Yi, R.; Kwon, S.; Dai, C.; Wang, R. Anti-TNF-α therapy for patients with sepsis: A systematic meta-analysis. Int. J. Clin. Pract. 2014, 68, 520–528. [Google Scholar] [CrossRef]
- Minneci, P.C.; Deans, K.J.; Banks, S.M.; Eichacker, P.Q.; Natanson, C. Should we continue to target the platelet-activating factor pathway in septic patients? Crit. Care Med. 2004, 32, 585–588. [Google Scholar] [CrossRef] [PubMed]
- Shakoory, B.; Carcillo, J.A.; Chatham, W.W.; Amdur, R.L.; Zhao, H.; Dinarello, C.A.; Cron, R.Q.; Opal, S.M. Interleukin-1 Receptor Blockade Is Associated with Reduced Mortality in Sepsis Patients with Features of Macrophage Activation Syndrome: Reanalysis of a Prior Phase III Trial. Crit. Care Med. 2016, 44, 275–281. [Google Scholar] [CrossRef]
- Opal, S.M.; Fisher, C.J., Jr.; Dhainaut, J.F.; Vincent, J.L.; Brase, R.; Lowry, S.F.; Sadoff, J.C.; Slotman, G.J.; Levy, H.; Balk, R.A.; et al. Confirmatory interleukin-1 receptor antagonist trial in severe sepsis: A phase III, randomized, double-blind, placebo-controlled, multicenter trial. The Interleukin-1 Receptor Antagonist Sepsis Investigator Group. Crit. Care Med. 1997, 25, 1115–1124. [Google Scholar] [CrossRef]
- Robey, R.C.; Logue, C.; Caird, C.A.; Hansel, J.; Hellyer, T.P.; Simpson, J.; Dark, P.; Mathioudakis, A.G.; Felton, T. Immunomodulatory drugs in sepsis: A systematic review and meta-analysis. Anaesthesia 2024, 79, 869–879. [Google Scholar] [CrossRef]
- François, B.; Lambden, S.; Fivez, T.; Gibot, S.; Derive, M.; Grouin, J.M.; Salcedo-Magguilli, M.; Lemarié, J.; De Schryver, N.; Jalkanen, V.; et al. Prospective evaluation of the efficacy, safety, and optimal biomarker enrichment strategy for nangibotide, a TREM-1 inhibitor, in patients with septic shock (ASTONISH): A double-blind, randomised, controlled, phase 2b trial. Lancet. Respir. Med. 2023, 11, 894–904. [Google Scholar] [CrossRef]
- Liu, F.; Wang, H.M.; Wang, T.; Zhang, Y.M.; Zhu, X. The efficacy of thymosin α1 as immunomodulatory treatment for sepsis: A systematic review of randomized controlled trials. BMC Infect. Dis. 2016, 16, 488. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
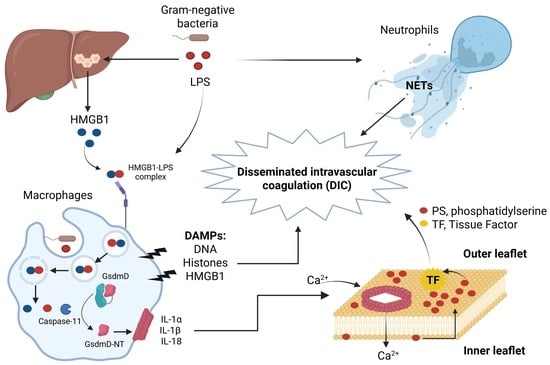
| Inflammasome-Derived DAMPs | Action in LPS and Bacterial Sepsis Leading to Lethality |
|---|---|
| Hepatocyte-derived HMGB1 | Promotes internalization of LPS into macrophages and endothelial cells via RAGE receptor, activation of caspase-11, and lethality in mice. Neutralizing HMGB1 or hmgb1-deficient mice were protected from polymicrobial sepsis and endotoxemia [51]. HMGB1 induces DIC [52]. |
| Histones | Induce endothelial dysfunction, hemorrhage, and thrombosis in LPS and polymicrobial sepsis. Lethality was reduced in mice by neutralizing antibodies to H4 or activated protein C (APC) [52,53]. |
| NETs | LPS and bacteria induce NETs extrusion in neutrophils. NETs cause platelet- and erythrocyte-aggregation, fibrin deposition, and thrombosis leading to DIC in mice. Removal of NETs with a DNase infusion coagulation and improved tissue perfusion [52,54,55]. |
| Tissue Factor (TF) | LPS induces non-canonical inflammasome activation and phosphatidylserine (PS) exposure, resulting in TF activation and DIC. Neutralization of TF and PS prevents LPS-induced DIC and reduces mortality in mice [56,57,58,59]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernández-Cuellar, E.; Tsuchiya, K.; Medina-Contreras, O.; Valle-Ríos, R. The Role of Inflammasomes in LPS and Gram-Negative Bacterial Sepsis. J. Clin. Med. 2025, 14, 7102. https://doi.org/10.3390/jcm14197102
Hernández-Cuellar E, Tsuchiya K, Medina-Contreras O, Valle-Ríos R. The Role of Inflammasomes in LPS and Gram-Negative Bacterial Sepsis. Journal of Clinical Medicine. 2025; 14(19):7102. https://doi.org/10.3390/jcm14197102
Chicago/Turabian StyleHernández-Cuellar, Eduardo, Kohsuke Tsuchiya, Oscar Medina-Contreras, and Ricardo Valle-Ríos. 2025. "The Role of Inflammasomes in LPS and Gram-Negative Bacterial Sepsis" Journal of Clinical Medicine 14, no. 19: 7102. https://doi.org/10.3390/jcm14197102
APA StyleHernández-Cuellar, E., Tsuchiya, K., Medina-Contreras, O., & Valle-Ríos, R. (2025). The Role of Inflammasomes in LPS and Gram-Negative Bacterial Sepsis. Journal of Clinical Medicine, 14(19), 7102. https://doi.org/10.3390/jcm14197102