
A Case of Horseshoe Kidney and Autosomal Dominant Polycystic Kidney Disease with PKD1 Gene Mutation



Abstract
1. Introduction
2. Case Description
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PKD1 | polycystin 1 |
| ADPKD | autosomal dominant polycystic kidney disease |
| PKD | polycystic kidney disease |
| HSK | horseshoe kidney |
References
- Raina, R.; Chakraborty, R.; Sethi, S.K.; Kumar, D.; Gibson, K.; Bergmann, C. Diagnosis and Management of Renal Cystic Disease of the Newborn: Core Curriculum 2021. Am. J. Kidney Dis. 2021, 78, 125–141. [Google Scholar] [CrossRef] [PubMed]
- Burgmaier, K.; Broekaert, I.J.; Liebau, M.C. Autosomal Recessive Polycystic Kidney Disease: Diagnosis, Prognosis, and Management. Adv. Kidney Dis. Health 2023, 30, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Econimo, L.; Schaeffer, C.; Zeni, L.; Cortinovis, R.; Alberici, F.; Rampoldi, L.; Scolari, F.; Izzi, C. Autosomal Dominant Tubulointerstitial Kidney Disease: An Emerging Cause of Genetic CKD. Kidney Int. Rep. 2022, 7, 2332–2344. [Google Scholar] [CrossRef]
- McConnachie, D.J.; Stow, J.L.; Mallett, A.J. Ciliopathies and the Kidney: A Review. Am. J. Kidney Dis. 2021, 77, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Goggolidou, P.; Richards, T. The genetics of Autosomal Recessive Polycystic Kidney Disease (ARPKD). Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166348. [Google Scholar] [CrossRef]
- Srivastava, S.; Molinari, E.; Raman, S.; Sayer, J.A. Many Genes-One Disease? Genetics of Nephronophthisis (NPHP) and NPHP-Associated Disorders. Front. Pediatr. 2017, 5, 287. [Google Scholar] [CrossRef]
- Dalgaard, O.Z. Bilateral polycystic disease of the kidneys; a follow-up of 284 patients and their families. Dan. Med. Bull. 1957, 4, 128–133. [Google Scholar]
- Iglesias, C.G.; Torres, V.E.; Offord, K.P.; Holley, K.E.; Beard, C.M.; Kurland, L.T. Epidemiology of adult polycystic kidney disease, Olmsted County, Minnesota: 1935–1980. Am. J. Kidney Dis. 1983, 2, 630–639. [Google Scholar] [CrossRef]
- Peters, D.J.; Sandkuijl, L.A. Genetic heterogeneity of polycystic kidney disease in Europe. Contrib. Nephrol. 1992, 97, 128–139. [Google Scholar]
- Torres, V.E.; Harris, P.C. Autosomal dominant polycystic kidney disease: The last 3 years. Kidney Int. 2009, 76, 149–168. [Google Scholar] [CrossRef]
- Nechita, O.C.; Badescu, D.; Popescu, R.I.; Rascu, S.; Petca, R.C.; Aurelian, J.; Constantin, T.; Toma, C.V.; Jinga, V.; Geavlete, B. Reviewing the complexities of horseshoe kidney: Insights into embryogenesis and surgical considerations. J. Med. Life 2025, 18, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Houat, A.P.; Guimarães, C.T.S.; Takahashi, M.S.; Rodi, G.P.; Gasparetto, T.P.D.; Blasbalg, R.; Velloni, F.G. Congenital Anomalies of the Upper Urinary Tract: A Comprehensive Review. Radiographics 2021, 41, 462–486. [Google Scholar] [CrossRef] [PubMed]
- Shah, H.U.; Ojili, V. Multimodality imaging spectrum of complications of horseshoe kidney. Indian J. Radiol. Imaging 2017, 27, 133–140. [Google Scholar] [CrossRef]
- Yoshinaga, K.; Kodama, K.; Tanii, I.; Toshimori, K. Morphological study of a horseshoe kidney with special reference to the vascular system. Anat. Sci. Int. 2002, 77, 134–139. [Google Scholar] [CrossRef]
- Alobaili, S.S.; Aljasser, S.M.; Asseri, A.S.; Alotaibi, D.A. Polycystic horseshoe kidney case report: Genetically reviewed. Saudi J. Kidney Dis. Transpl. 2021, 32, 574–578. [Google Scholar] [CrossRef]
- Chikkannaiah, P.; Kangle, R.; Bali, A.; Honawad, M.N. Polycystic horseshoe kidney. Med. J. Armed. Forces India 2015, 71, S181–S183. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Humphries, A.; Speroni, S.; Eden, K.; Nolan, M.; Gilbert, C.; McNamara, J. Horseshoe kidney: Morphologic features, embryologic and genetic etiologies, and surgical implications. Clin. Anat. 2023, 36, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Nagendra, V.; Phatak, S.V.; Singh, R.K.; Pandey, S.S.; Gupta, R. Rare presentation of autosomal dominant polycystic kidney disease in horseshoe kidney ultrasound evaluation: A case report. Pan. Afr. Med. J. 2022, 42, 116. [Google Scholar] [CrossRef]
- Sun, Y.; Zhou, H.; Yang, B.X. Drug discovery for polycystic kidney disease. Acta Pharmacol. Sin. 2011, 32, 805–816. [Google Scholar] [CrossRef]
- Chauvet, V.; Qian, F.; Boute, N.; Cai, Y.; Phakdeekitacharoen, B.; Onuchic, L.F.; Attié-Bitach, T.; Guicharnaud, L.; Devuyst, O.; Germino, G.G.; et al. Expression of PKD1 and PKD2 transcripts and proteins in human embryo and during normal kidney development. Am. J. Pathol. 2002, 160, 973–983. [Google Scholar] [CrossRef]
- Bergmann, C. Genetics of Autosomal Recessive Polycystic Kidney Disease and Its Differential Diagnoses. Front. Pediatr. 2017, 5, 221. [Google Scholar] [CrossRef] [PubMed]
- Arogundade, F.A.; Akinbodewa, A.A.; Sanusi, A.A.; Okunola, O.; Hassan, M.O.; Akinsola, A. Clinical presentation and outcome of autosomal dominant polycystic kidney disease in Nigeria. Afr. Health Sci. 2018, 18, 671–680. [Google Scholar] [CrossRef]
- Reed, B.; McFann, K.; Kimberling, W.J.; Pei, Y.; Gabow, P.A.; Christopher, K.; Petersen, E.; Kelleher, C.; Fain, P.R.; Johnson, A.; et al. Presence of de novo mutations in autosomal dominant polycystic kidney disease patients without family history. Am. J. Kidney Dis. 2008, 52, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Bhandarkar, K.P.; Kittur, D.H.; Patil, S.V.; Jadhav, S.S. Horseshoe kidney and associated anomalies: Single institutional review of 20 cases. Afr. J. Paediatr. Surg. 2018, 15, 104–107. [Google Scholar] [CrossRef]
- Isaksen, C.V.; Eik-Nes, S.H.; Blaas, H.G.; Torp, S.H. Fetuses and infants with congenital urinary system anomalies: Correlation between prenatal ultrasound and postmortem findings. Ultrasound Obstet. Gynecol. 2000, 15, 177–185. [Google Scholar] [CrossRef]
- Ram, R.; Swarnalatha, G.; Bantwal Hebbalsinhakatte, S.P.; Dakshinamurty, K.V. Polycystic horseshoe kidney. Clin. Kidney J. 2013, 6, 103–104. [Google Scholar] [CrossRef]
- Rasouly, H.M.; Lu, W. Lower urinary tract development and disease. Wiley Interdiscip. Rev. Syst. Biol. Med. 2013, 5, 307–342. [Google Scholar] [CrossRef]
- Stonebrook, E.; Hoff, M.; Spencer, J.D. Congenital Anomalies of the Kidney and Urinary Tract: A Clinical Review. Curr. Treat. Options. Pediatr. 2019, 5, 223–235. [Google Scholar] [CrossRef]
- Chern, L.C.; Albert, C.M.O. Autosomal dominant polycystic kidney disease. Clin. Med. 2009, 9, 278–283. [Google Scholar]
- Franceschini, N.; Feldman, D.L.; Berg, J.S.; Besse, W.; Chang, A.R.; Dahl, N.K.; Gbadegesin, R.; Pollak, M.R.; Rasouly., H.M.; Smith, R.J.H.; et al. Advancing Genetic Testing in Kidney Diseases: Report From a National Kidney Foundation Working Group. Am. J. Kidney Dis. 2024, 84, 751–766. [Google Scholar] [CrossRef]
- Stein, Q.; Westemeyer, M.; Darwish, T.; Pitman, T.; Hager, M.; Tabriziani, H.; Curry, K.; Collett, K.; Raible, D.; Hendricks, E. Genetic Counseling in Kidney Disease: A Perspective. Kidney Med. 2023, 5, 100668. [Google Scholar] [CrossRef]
- Mahmoud, A.H.; Talaat, I.M.; Tlili, A.; Hamoudi, R. Congenital anomalies of the kidney and urinary tract. Front. Med. 2024, 11, 1384676. [Google Scholar] [CrossRef]


{kind=link}
| Parameter | Details |
|---|---|
| Age | 24 years |
| Gender | Male |
| Family history | Positive for ADPKD (father and uncle affected) The other five male brothers of the patient’s father were not affected |
| Symptoms | No flank pain No gross hematuria, proteinuria, or albuminuria Hypertension |
| Blood pressure | 150/90 mmHg |
| Physical examination | No palpable abdominal kidney |
| Laboratory examination | Serum creatinine 0.85 mg/dL |
| Estimated GFR 122 mL/min/1.7 m2 Creatinine clearance 104 mL/min Urea clearance 101 mL/min Blood urea nitrogen 16 mg/dL Aspartate aminotransferase 28 U/L Alanine aminotransferase 38 U/L Total bilirubin 0.75 mg/dL Albumin 4.9 g/dL Hb 15.0 g/dL U/A RBC 0-2/HPF, Protein (-), Urine protein-to-creatinine ratio 79 mg/g Urine albumin-to-creatinine ratio 14 mg/g | |
| Kidney volume | Right 542 mL, Left 440 mL |
| Genetic testing | PKD1 mutation (c.165_171del (p.Leu56ArgfsTer15)) |
| Management plan Pedigree | Lifestyle modifications Antihypertensive medications with telmisartan 40 mg Regular monitoring of blood pressure and kidney function every 6 months 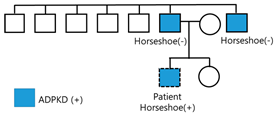 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.; Lee, S.J.; Kim, W. A Case of Horseshoe Kidney and Autosomal Dominant Polycystic Kidney Disease with PKD1 Gene Mutation. J. Clin. Med. 2025, 14, 4008. https://doi.org/10.3390/jcm14114008
Kim H, Lee SJ, Kim W. A Case of Horseshoe Kidney and Autosomal Dominant Polycystic Kidney Disease with PKD1 Gene Mutation. Journal of Clinical Medicine. 2025; 14(11):4008. https://doi.org/10.3390/jcm14114008
Chicago/Turabian StyleKim, Hyeongwan, Soo Jin Lee, and Won Kim. 2025. "A Case of Horseshoe Kidney and Autosomal Dominant Polycystic Kidney Disease with PKD1 Gene Mutation" Journal of Clinical Medicine 14, no. 11: 4008. https://doi.org/10.3390/jcm14114008
APA StyleKim, H., Lee, S. J., & Kim, W. (2025). A Case of Horseshoe Kidney and Autosomal Dominant Polycystic Kidney Disease with PKD1 Gene Mutation. Journal of Clinical Medicine, 14(11), 4008. https://doi.org/10.3390/jcm14114008